
Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
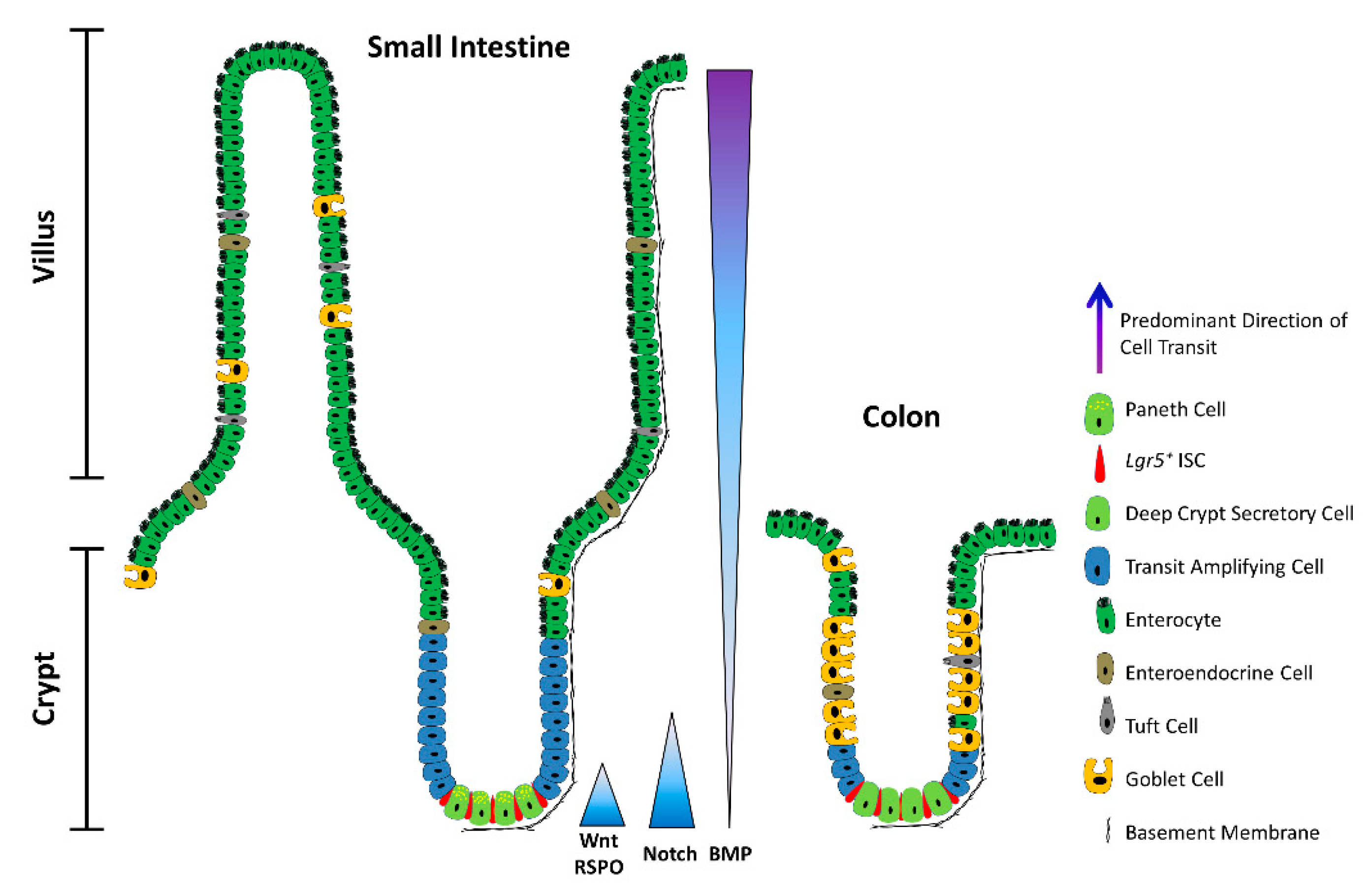
2. ISCs in a Nutshell
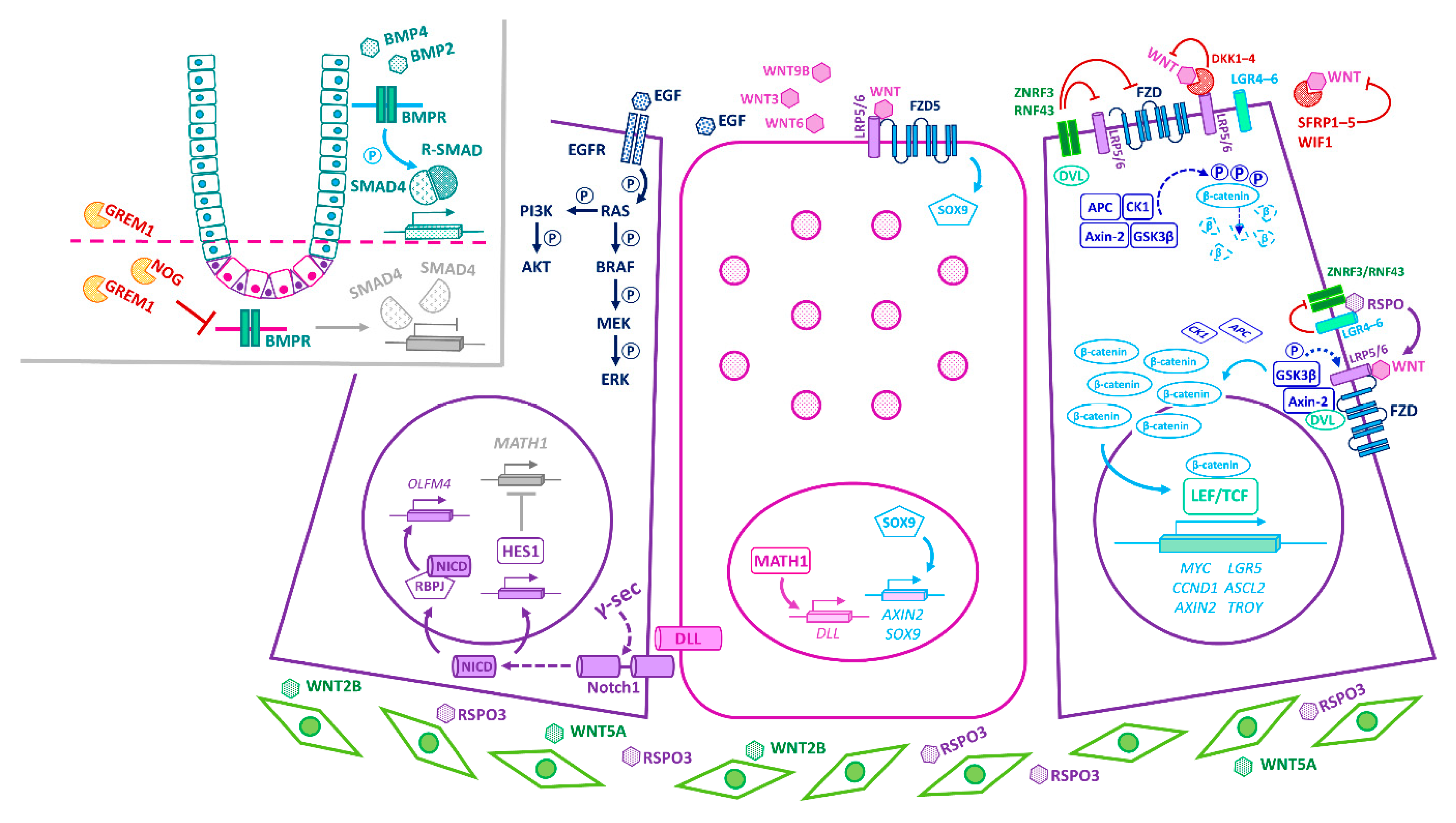
3. Principal Niche-Signalling Pathways
4. Convergence of Niche-Signalling Pathways in Homeostasis
5. Cellular Circuitries
6. The Immune Niche
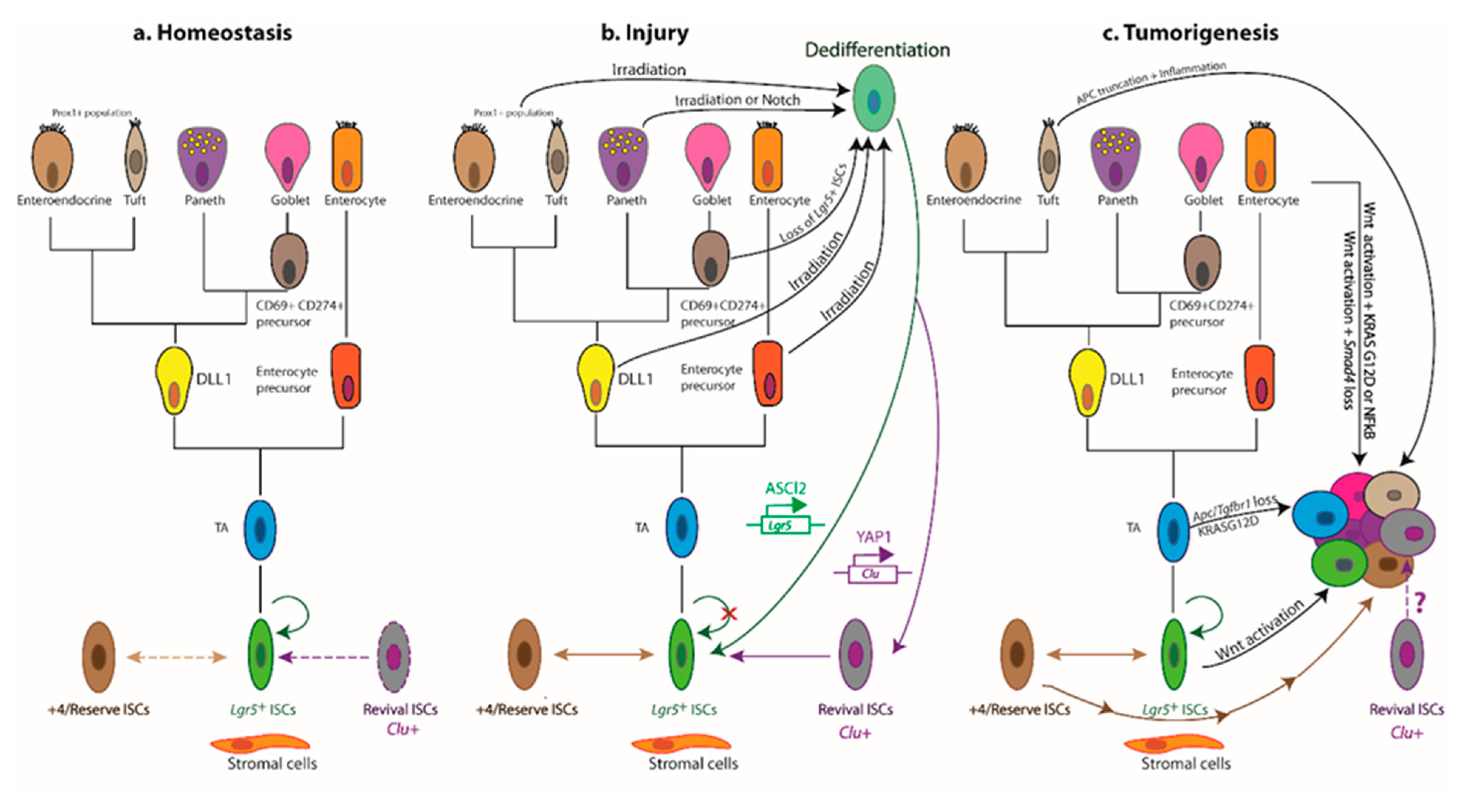
7. Niche Remodelling Post Injury
8. At the Crossroads of Intestinal Regeneration and Tumorigenesis—The YAP-Driven Foetal-Like Signature
9. Nutritional Cues and ISC Function
10. Microbiota-Derived Metabolites and ISC Function
11. Microbiota Contributions to the Niche
12. ISC Dynamics in the Niche: “Winner Takes All”
13. Cells-of-Origin
14. All Roads Lead through LGR5
15. Microenvironmental Influences on Tumour Cell Plasticity
16. CRC Subtypes and Niche-Signalling Pathways
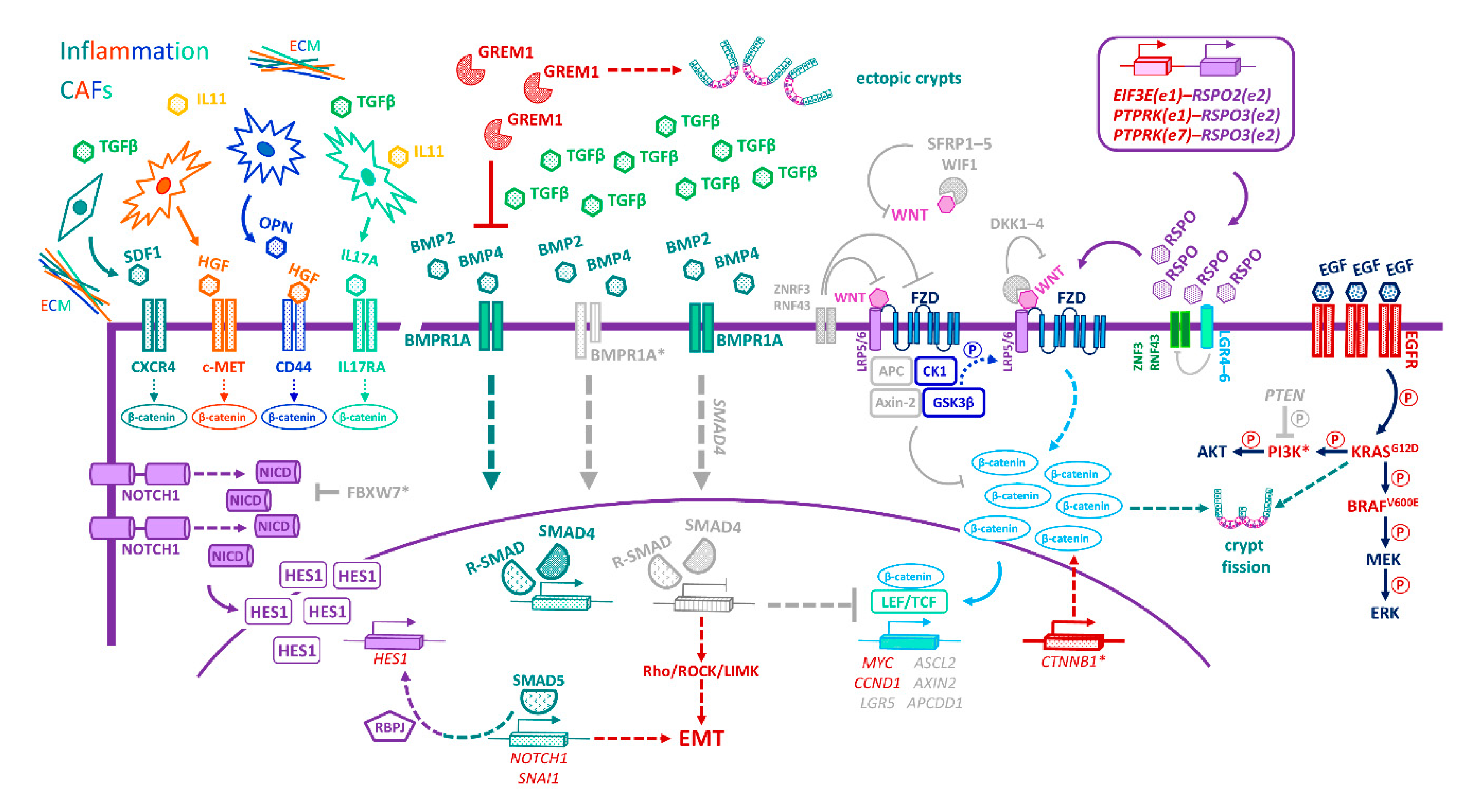
17. Niche-Emancipating Mutations, Tumour Progression, and Therapeutic Implications
18. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beumer, J.; Clevers, H. Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell Biol. 2021, 22, 39–53. [Google Scholar] [CrossRef]
- Cheng, H.; Leblond, C.P. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types. Am. J. Anat. 1974, 141, 537–561. [Google Scholar] [CrossRef]
- Sasaki, N.; Sachs, N.; Wiebrands, K.; Ellenbroek, S.I.; Fumagalli, A.; Lyubimova, A.; Begthel, H.; van den Born, M.; van Es, J.H.; Karthaus, W.R.; et al. Reg4+ deep crypt secretory cells function as epithelial niche for Lgr5+ stem cells in colon. Proc. Natl. Acad. Sci. USA 2016, 113, E5399–E5407. [Google Scholar] [CrossRef]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Tian, H.; Biehs, B.; Warming, S.; Leong, K.G.; Rangell, L.; Klein, O.D.; de Sauvage, F.J. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 2011, 478, 255–259. [Google Scholar] [CrossRef]
- Metcalfe, C.; Kljavin, N.M.; Ybarra, R.; de Sauvage, F.J. Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell 2014, 14, 149–159. [Google Scholar] [CrossRef]
- Potten, C.S.; Hume, W.J.; Reid, P.; Cairns, J. The segregation of DNA in epithelial stem cells. Cell 1978, 15, 899–906. [Google Scholar] [CrossRef]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.K.; Carlone, D.L.; Richmond, C.A.; Farilla, L.; Kranendonk, M.E.; Henderson, D.E.; Baffour-Awuah, N.Y.; Ambruzs, D.M.; Fogli, L.K.; Algra, S.; et al. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc. Natl. Acad Sci. USA 2011, 108, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Jain, R.; LeBoeuf, M.R.; Wang, Q.; Lu, M.M.; Epstein, J.A. Interconversion between intestinal stem cell populations in distinct niches. Science 2011, 334, 1420–1424. [Google Scholar] [CrossRef]
- Powell, A.E.; Wang, Y.; Li, Y.; Poulin, E.J.; Means, A.L.; Washington, M.K.; Higginbotham, J.N.; Juchheim, A.; Prasad, N.; Levy, S.E.; et al. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 2012, 149, 146–158. [Google Scholar] [CrossRef]
- Li, N.; Yousefi, M.; Nakauka-Ddamba, A.; Jain, R.; Tobias, J.; Epstein, J.A.; Jensen, S.T.; Lengner, C.J. Single-cell analysis of proxy reporter allele-marked epithelial cells establishes intestinal stem cell hierarchy. Stem Cell Rep. 2014, 3, 876–891. [Google Scholar] [CrossRef]
- Yan, K.S.; Chia, L.A.; Li, X.; Ootani, A.; Su, J.; Lee, J.Y.; Su, N.; Luo, Y.; Heilshorn, S.C.; Amieva, M.R.; et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc. Natl. Acad. Sci. USA 2012, 109, 466–471. [Google Scholar] [CrossRef]
- Asfaha, S.; Hayakawa, Y.; Muley, A.; Stokes, S.; Graham, T.A.; Ericksen, R.E.; Westphalen, C.B.; von Burstin, J.; Mastracci, T.L.; Worthley, D.L.; et al. Krt19(+)/Lgr5(-) cells are radioresistant cancer-initiating stem cells in the colon and intestine. Cell Stem Cell 2015, 16, 627–638. [Google Scholar] [CrossRef]
- Bankaitis, E.D.; Ha, A.; Kuo, C.J.; Magness, S.T. Reserve stem cells in intestinal homeostasis and injury. Gastroenterology 2018, 155, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Tetteh, P.W.; Basak, O.; Farin, H.F.; Wiebrands, K.; Kretzschmar, K.; Begthel, H.; van den Born, M.; Korving, J.; de Sauvage, F.; van Es, J.H.; et al. Replacement of lost Lgr5-positive stem cells through plasticity of their enterocyte-lineage daughters. Cell Stem Cell 2016, 18, 203–213. [Google Scholar] [CrossRef]
- Yan, K.S.; Gevaert, O.; Zheng, G.X.Y.; Anchang, B.; Probert, C.S.; Larkin, K.A.; Davies, P.S.; Cheng, Z.F.; Kaddis, J.S.; Han, A.; et al. Intestinal enteroendocrine lineage cells possess homeostatic and injury-inducible stem cell activity. Cell Stem Cell 2017, 21, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Schonhoff, S.E.; Giel-Moloney, M.; Leiter, A.B. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev. Biol. 2004, 270, 443–454. [Google Scholar] [CrossRef]
- Gross, S.; Balderes, D.; Liu, J.; Asfaha, S.; Gu, G.; Wang, T.C.; Sussel, L. Nkx2.2 is expressed in a subset of enteroendocrine cells with expanded lineage potential. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G975–G987. [Google Scholar] [CrossRef] [PubMed]
- Sei, Y.; Lu, X.; Liou, A.; Zhao, X.; Wank, S.A. A stem cell marker-expressing subset of enteroendocrine cells resides at the crypt base in the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G345–G356. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, U.; Saxena, M.; O’Neill, N.K.; Saadatpour, A.; Yuan, G.C.; Herbert, Z.; Murata, K.; Shivdasani, R.A. Dynamic reorganization of chromatin accessibility signatures during dedifferentiation of secretory precursors into Lgr5+ intestinal stem cells. Cell Stem Cell 2017, 21, 65–77. [Google Scholar] [CrossRef]
- Van Es, J.H.; Sato, T.; van de Wetering, M.; Lyubimova, A.; Yee Nee, A.N.; Gregorieff, A.; Sasaki, N.; Zeinstra, L.; van den Born, M.; Korving, J.; et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat. Cell Biol. 2012, 14, 1099–1104. [Google Scholar] [CrossRef]
- Castillo-Azofeifa, D.; Fazio, E.N.; Nattiv, R.; Good, H.J.; Wald, T.; Pest, M.A.; de Sauvage, F.J.; Klein, O.D.; Asfaha, S. Atoh1(+) secretory progenitors possess renewal capacity independent of Lgr5(+) cells during colonic regeneration. EMBO J. 2019, 38, e99984. [Google Scholar] [CrossRef]
- Ishibashi, F.; Shimizu, H.; Nakata, T.; Fujii, S.; Suzuki, K.; Kawamoto, A.; Anzai, S.; Kuno, R.; Nagata, S.; Ito, G.; et al. Contribution of ATOH1(+) cells to the homeostasis, repair, and tumorigenesis of the colonic epithelium. Stem Cell Rep. 2018, 10, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Tomic, G.; Morrissey, E.; Kozar, S.; Ben-Moshe, S.; Hoyle, A.; Azzarelli, R.; Kemp, R.; Chilamakuri, C.S.R.; Itzkovitz, S.; Philpott, A.; et al. Phospho-regulation of ATOH1 is required for plasticity of secretory progenitors and tissue regeneration. Cell Stem Cell 2018, 23, 436–443. [Google Scholar] [CrossRef]
- Buczacki, S.J.; Zecchini, H.I.; Nicholson, A.M.; Russell, R.; Vermeulen, L.; Kemp, R.; Winton, D.J. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 2013, 495, 65–69. [Google Scholar] [CrossRef]
- Harnack, C.; Berger, H.; Antanaviciute, A.; Vidal, R.; Sauer, S.; Simmons, A.; Meyer, T.F.; Sigal, M. R-spondin 3 promotes stem cell recovery and epithelial regeneration in the colon. Nat. Commun. 2019, 10, 4368. [Google Scholar] [CrossRef]
- Westphalen, C.B.; Asfaha, S.; Hayakawa, Y.; Takemoto, Y.; Lukin, D.J.; Nuber, A.H.; Brandtner, A.; Setlik, W.; Remotti, H.; Muley, A.; et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Investig. 2014, 124, 1283–1295. [Google Scholar] [CrossRef]
- Sei, Y.; Feng, J.; Samsel, L.; White, A.; Zhao, X.; Yun, S.; Citrin, D.; McCoy, J.P.; Sundaresan, S.; Hayes, M.M.; et al. Mature enteroendocrine cells contribute to basal and pathological stem cell dynamics in the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G495–G510. [Google Scholar] [CrossRef]
- Schmitt, M.; Schewe, M.; Sacchetti, A.; Feijtel, D.; van de Geer, W.S.; Teeuwssen, M.; Sleddens, H.F.; Joosten, R.; van Royen, M.E.; van de Werken, H.J.G.; et al. Paneth cells respond to inflammation and contribute to tissue regeneration by acquiring stem-like features through SCF/c-Kit signaling. Cell Rep. 2018, 24, 2312–2328. [Google Scholar] [CrossRef]
- Yu, S.; Tong, K.; Zhao, Y.; Balasubramanian, I.; Yap, G.S.; Ferraris, R.P.; Bonder, E.M.; Verzi, M.P.; Gao, N. Paneth Cell Multipotency Induced by Notch Activation following Injury. Cell Stem Cell 2018, 23, 46–59. [Google Scholar] [CrossRef]
- Jones, J.C.; Brindley, C.D.; Elder, N.H.; Myers, M.G., Jr.; Rajala, M.W.; Dekaney, C.M.; McNamee, E.N.; Frey, M.R.; Shroyer, N.F.; Dempsey, P.J. Cellular plasticity of Defa4(Cre)-expressing Paneth cells in response to Notch activation and intestinal injury. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 533–554. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; van Es, J.H.; van den Born, M.; Begthel, H.; Stange, D.E.; Barker, N.; Clevers, H. Prominin-1/CD133 marks stem cells and early progenitors in mouse small intestine. Gastroenterology 2009, 136, 2187–2194. [Google Scholar] [CrossRef]
- Itzkovitz, S.; Lyubimova, A.; Blat, I.C.; Maynard, M.; van Es, J.; Lees, J.; Jacks, T.; Clevers, H.; van Oudenaarden, A. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat. Cell Biol. 2011, 14, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.; Stange, D.E.; Schepers, A.G.; van de Wetering, M.; Koo, B.K.; Itzkovitz, S.; Volckmann, R.; Kung, K.S.; Koster, J.; Radulescu, S.; et al. The Lgr5 intestinal stem cell signature: Robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012, 31, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Tang, D.; Morita, Y.; Sperka, T.; Omrani, O.; Lechel, A.; Sakk, V.; Kraus, J.; Kestler, H.A.; Kuhl, M.; et al. Wnt activity and basal niche position sensitize intestinal stem and progenitor cells to DNA damage. EMBO J. 2015, 34, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Yousefi, M.; Nakauka-Ddamba, A.; Tobias, J.W.; Jensen, S.T.; Morrisey, E.E.; Lengner, C.J. Heterogeneity in readouts of canonical wnt pathway activity within intestinal crypts. Dev. Dyn. 2016, 245, 822–833. [Google Scholar] [CrossRef]
- Li, N.; Nakauka-Ddamba, A.; Tobias, J.; Jensen, S.T.; Lengner, C.J. Mouse Label-Retaining Cells Are Molecularly and Functionally Distinct from Reserve Intestinal Stem Cells. Gastroenterology 2016, 151, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Barriga, F.M.; Montagni, E.; Mana, M.; Mendez-Lago, M.; Hernando-Momblona, X.; Sevillano, M.; Guillaumet-Adkins, A.; Rodriguez-Esteban, G.; Buczacki, S.J.A.; Gut, M.; et al. Mex3a marks a slowly dividing subpopulation of Lgr5+ intestinal stem cells. Cell Stem Cell 2017, 20, 801–816. [Google Scholar] [CrossRef] [PubMed]
- Giroux, V.; Stephan, J.; Chatterji, P.; Rhoades, B.; Wileyto, E.P.; Klein-Szanto, A.J.; Lengner, C.J.; Hamilton, K.E.; Rustgi, A.K. Mouse intestinal Krt15+ crypt cells are radio-resistant and tumor initiating. Stem Cell Rep. 2018, 10, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Lin, Z.; Lv, C.; Shao, C.; Bi, X.; Deng, M.; Xu, J.; Guerrero-Juarez, C.F.; Li, M.; Wu, X.; et al. Cycling Stem Cells Are Radioresistant and Regenerate the Intestine. Cell Rep. 2020, 32, 107952. [Google Scholar] [CrossRef]
- Li, L.; Clevers, H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef]
- Kim, T.H.; Saadatpour, A.; Guo, G.; Saxena, M.; Cavazza, A.; Desai, N.; Jadhav, U.; Jiang, L.; Rivera, M.N.; Orkin, S.H.; et al. Single-cell transcript profiles reveal multilineage priming in early progenitors derived from Lgr5(+) intestinal stem cells. Cell Rep. 2016, 16, 2053–2060. [Google Scholar] [CrossRef]
- Baulies, A.; Angelis, N.; Foglizzo, V.; Danielsen, E.T.; Patel, H.; Novellasdemunt, L.; Kucharska, A.; Carvalho, J.; Nye, E.; De Coppi, P.; et al. The transcription co-repressors MTG8 and MTG16 regulate exit of intestinal stem cells from their niche and differentiation into enterocyte vs secretory lineages. Gastroenterology 2020, 159, 1328–1341. [Google Scholar] [CrossRef]
- Leedham, S.J. Reserving the right to change the intestinal stem cell model. Cell Stem Cell 2020, 26, 301–302. [Google Scholar] [CrossRef]
- Murata, K.; Jadhav, U.; Madha, S.; van Es, J.; Dean, J.; Cavazza, A.; Wucherpfennig, K.; Michor, F.; Clevers, H.; Shivdasani, R.A. Ascl2-dependent cell dedifferentiation drives regeneration of ablated intestinal stem cells. Cell Stem Cell 2020, 26, 377–390. [Google Scholar] [CrossRef]
- Kim, T.H.; Li, F.; Ferreiro-Neira, I.; Ho, L.L.; Luyten, A.; Nalapareddy, K.; Long, H.; Verzi, M.; Shivdasani, R.A. Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature 2014, 506, 511–515. [Google Scholar] [CrossRef]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Ireland, H.; Kemp, R.; Houghton, C.; Howard, L.; Clarke, A.R.; Sansom, O.J.; Winton, D.J. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: Effect of loss of beta-catenin. Gastroenterology 2004, 126, 1236–1246. [Google Scholar] [CrossRef]
- Pinto, D.; Gregorieff, A.; Begthel, H.; Clevers, H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003, 17, 1709–1713. [Google Scholar] [CrossRef]
- Fevr, T.; Robine, S.; Louvard, D.; Huelsken, J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol. Cell. Biol. 2007, 27, 7551–7559. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; Haegebarth, A.; Kujala, P.; Itzkovitz, S.; Koo, B.K.; Boj, S.F.; Korving, J.; van den Born, M.; van Oudenaarden, A.; Robine, S.; et al. A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol. Cell. Biol. 2012, 32, 1918–1927. [Google Scholar] [CrossRef]
- Flanagan, D.J.; Phesse, T.J.; Barker, N.; Schwab, R.H.; Amin, N.; Malaterre, J.; Stange, D.E.; Nowell, C.J.; Currie, S.A.; Saw, J.T.; et al. Frizzled7 functions as a Wnt receptor in intestinal epithelial Lgr5(+) stem cells. Stem Cell Rep. 2015, 4, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Degirmenci, B.; Moor, A.E.; Herr, P.; Zimmerli, D.; Moor, M.B.; Hausmann, G.; Cantu, C.; Aguet, M.; Basler, K. Wnt ligands secreted by subepithelial mesenchymal cells are essential for the survival of intestinal stem cells and gut homeostasis. Cell Rep. 2016, 15, 911–918. [Google Scholar] [CrossRef]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Powell, S.M.; Zilz, N.; Beazer-Barclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, C.; Breukel, C.; van der Luijt, R.; Fidalgo, P.; Lage, P.; Slors, F.J.; Leitao, C.N.; Fodde, R.; Smits, R. The ‘just-right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Hum. Mol. Genet. 2002, 11, 1549–1560. [Google Scholar] [CrossRef]
- Leedham, S.J.; Rodenas-Cuadrado, P.; Howarth, K.; Lewis, A.; Mallappa, S.; Segditsas, S.; Davis, H.; Jeffery, R.; Rodriguez-Justo, M.; Keshav, S.; et al. A basal gradient of Wnt and stem-cell number influences regional tumour distribution in human and mouse intestinal tracts. Gut 2013, 62, 83–93. [Google Scholar] [CrossRef]
- Huels, D.J.; Bruens, L.; Hodder, M.C.; Cammareri, P.; Campbell, A.D.; Ridgway, R.A.; Gay, D.M.; Solar-Abboud, M.; Faller, W.J.; Nixon, C.; et al. Wnt ligands influence tumour initiation by controlling the number of intestinal stem cells. Nat. Commun. 2018, 9, 1132. [Google Scholar] [CrossRef]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef]
- Yan, K.S.; Janda, C.Y.; Chang, J.; Zheng, G.X.Y.; Larkin, K.A.; Luca, V.C.; Chia, L.A.; Mah, A.T.; Han, A.; Terry, J.M.; et al. Non-equivalence of Wnt and R-spondin ligands during Lgr5(+) intestinal stem-cell self-renewal. Nature 2017, 545, 238–242. [Google Scholar] [CrossRef]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef]
- Kosinski, C.; Li, V.S.; Chan, A.S.; Zhang, J.; Ho, C.; Tsui, W.Y.; Chan, T.L.; Mifflin, R.C.; Powell, D.W.; Yuen, S.T.; et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc. Natl. Acad. Sci. USA 2007, 104, 15418–15423. [Google Scholar] [CrossRef]
- Van der Flier, L.G.; Sabates-Bellver, J.; Oving, I.; Haegebarth, A.; De Palo, M.; Anti, M.; Van Gijn, M.E.; Suijkerbuijk, S.; Van de Wetering, M.; Marra, G.; et al. The intestinal Wnt/TCF signature. Gastroenterology 2007, 132, 628–632. [Google Scholar] [CrossRef]
- van der Flier, L.G.; van Gijn, M.E.; Hatzis, P.; Kujala, P.; Haegebarth, A.; Stange, D.E.; Begthel, H.; van den Born, M.; Guryev, V.; Oving, I.; et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell 2009, 136, 903–912. [Google Scholar] [CrossRef]
- Schuijers, J.; Junker, J.P.; Mokry, M.; Hatzis, P.; Koo, B.K.; Sasselli, V.; van der Flier, L.G.; Cuppen, E.; van Oudenaarden, A.; Clevers, H. Ascl2 acts as an R-spondin/Wnt-responsive switch to control stemness in intestinal crypts. Cell Stem Cell 2015, 16, 158–170. [Google Scholar] [CrossRef]
- Van Es, J.H.; van Gijn, M.E.; Riccio, O.; van den Born, M.; Vooijs, M.; Begthel, H.; Cozijnsen, M.; Robine, S.; Winton, D.J.; Radtke, F.; et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005, 435, 959–963. [Google Scholar] [CrossRef]
- Fre, S.; Huyghe, M.; Mourikis, P.; Robine, S.; Louvard, D.; Artavanis-Tsakonas, S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 2005, 435, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; de Geest, N.; van de Born, M.; Clevers, H.; Hassan, B.A. Intestinal stem cells lacking the Math1 tumour suppressor are refractory to Notch inhibitors. Nat. Commun. 2010, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Sancho, R.; Cremona, C.A.; Behrens, A. Stem cell and progenitor fate in the mammalian intestine: Notch and lateral inhibition in homeostasis and disease. EMBO Rep. 2015, 16, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Milano, J.; McKay, J.; Dagenais, C.; Foster-Brown, L.; Pognan, F.; Gadient, R.; Jacobs, R.T.; Zacco, A.; Greenberg, B.; Ciaccio, P.J. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol. Sci. 2004, 82, 341–358. [Google Scholar] [CrossRef] [PubMed]
- VanDussen, K.L.; Samuelson, L.C. Mouse atonal homolog 1 directs intestinal progenitors to secretory cell rather than absorptive cell fate. Dev. Biol 2010, 346, 215–223. [Google Scholar] [CrossRef]
- Yang, Q.; Bermingham, N.A.; Finegold, M.J.; Zoghbi, H.Y. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 2001, 294, 2155–2158. [Google Scholar] [CrossRef]
- Shroyer, N.F.; Helmrath, M.A.; Wang, V.Y.; Antalffy, B.; Henning, S.J.; Zoghbi, H.Y. Intestine-specific ablation of mouse atonal homolog 1 (Math1) reveals a role in cellular homeostasis. Gastroenterology 2007, 132, 2478–2488. [Google Scholar] [CrossRef]
- Pellegrinet, L.; Rodilla, V.; Liu, Z.; Chen, S.; Koch, U.; Espinosa, L.; Kaestner, K.H.; Kopan, R.; Lewis, J.; Radtke, F. Dll1- and dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology 2011, 140, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Fre, S.; Hannezo, E.; Sale, S.; Huyghe, M.; Lafkas, D.; Kissel, H.; Louvi, A.; Greve, J.; Louvard, D.; Artavanis-Tsakonas, S. Notch lineages and activity in intestinal stem cells determined by a new set of knock-in mice. PLoS ONE 2011, 6, e25785. [Google Scholar] [CrossRef] [PubMed]
- VanDussen, K.L.; Carulli, A.J.; Keeley, T.M.; Patel, S.R.; Puthoff, B.J.; Magness, S.T.; Tran, I.T.; Maillard, I.; Siebel, C.; Kolterud, A.; et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 2012, 139, 488–497. [Google Scholar] [CrossRef]
- Hardwick, J.C.; Van Den Brink, G.R.; Bleuming, S.A.; Ballester, I.; Van Den Brande, J.M.; Keller, J.J.; Offerhaus, G.J.; Van Deventer, S.J.; Peppelenbosch, M.P. Bone morphogenetic protein 2 is expressed by, and acts upon, mature epithelial cells in the colon. Gastroenterology 2004, 126, 111–121. [Google Scholar] [CrossRef]
- Davis, H.; Raja, E.; Miyazono, K.; Tsubakihara, Y.; Moustakas, A. Mechanisms of action of bone morphogenetic proteins in cancer. Cytokine Growth Factor Rev. 2016, 27, 81–92. [Google Scholar] [CrossRef]
- Haramis, A.P.; Begthel, H.; van den Born, M.; van Es, J.; Jonkheer, S.; Offerhaus, G.J.; Clevers, H. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science 2004, 303, 1684–1686. [Google Scholar] [CrossRef]
- He, X.C.; Zhang, J.; Tong, W.G.; Tawfik, O.; Ross, J.; Scoville, D.H.; Tian, Q.; Zeng, X.; He, X.; Wiedemann, L.M.; et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat. Genet. 2004, 36, 1117–1121. [Google Scholar] [CrossRef]
- Aoki, R.; Shoshkes-Carmel, M.; Gao, N.; Shin, S.; May, C.L.; Golson, M.L.; Zahm, A.M.; Ray, M.; Wiser, C.L.; Wright, C.V.; et al. Foxl1-expressing mesenchymal cells constitute the intestinal stem cell niche. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 175–188. [Google Scholar] [CrossRef]
- Reynolds, A.; Wharton, N.; Parris, A.; Mitchell, E.; Sobolewski, A.; Kam, C.; Bigwood, L.; El Hadi, A.; Munsterberg, A.; Lewis, M.; et al. Canonical Wnt signals combined with suppressed TGFbeta/BMP pathways promote renewal of the native human colonic epithelium. Gut 2014, 63, 610–621. [Google Scholar] [CrossRef]
- Stzepourginski, I.; Nigro, G.; Jacob, J.M.; Dulauroy, S.; Sansonetti, P.J.; Eberl, G.; Peduto, L. CD34+ mesenchymal cells are a major component of the intestinal stem cells niche at homeostasis and after injury. Proc. Natl. Acad. Sci. USA 2017, 114, E506–E513. [Google Scholar] [CrossRef]
- McCarthy, N.; Manieri, E.; Storm, E.E.; Saadatpour, A.; Luoma, A.M.; Kapoor, V.N.; Madha, S.; Gaynor, L.T.; Cox, C.; Keerthivasan, S.; et al. Distinct mesenchymal cell populations generate the essential intestinal BMP signaling gradient. Cell Stem Cell 2020, 26, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.C.; Jahns, H.; Mthunzi, L.; Piouceau, L.; Cornwell, J.; Doody, R.; Frohlich, S.; Callanan, J.J.; McLoughlin, P. Gremlin 1 depletion in vivo causes severe enteropathy and bone marrow failure. J. Pathol. 2020, 251, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.; Irshad, S.; Bansal, M.; Rafferty, H.; Boitsova, T.; Bardella, C.; Jaeger, E.; Lewis, A.; Freeman-Mills, L.; Giner, F.C.; et al. Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat. Med. 2015, 21, 62–70. [Google Scholar] [CrossRef]
- Qi, Z.; Li, Y.; Zhao, B.; Xu, C.; Liu, Y.; Li, H.; Zhang, B.; Wang, X.; Yang, X.; Xie, W.; et al. BMP restricts stemness of intestinal Lgr5(+) stem cells by directly suppressing their signature genes. Nat. Commun. 2017, 8, 13824. [Google Scholar] [CrossRef]
- Howe, J.R.; Bair, J.L.; Sayed, M.G.; Anderson, M.E.; Mitros, F.A.; Petersen, G.M.; Velculescu, V.E.; Traverso, G.; Vogelstein, B. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet. 2001, 28, 184–187. [Google Scholar] [CrossRef]
- Auclair, B.A.; Benoit, Y.D.; Rivard, N.; Mishina, Y.; Perreault, N. Bone morphogenetic protein signaling is essential for terminal differentiation of the intestinal secretory cell lineage. Gastroenterology 2007, 133, 887–896. [Google Scholar] [CrossRef]
- Tian, H.; Biehs, B.; Chiu, C.; Siebel, C.W.; Wu, Y.; Costa, M.; de Sauvage, F.J.; Klein, O.D. Opposing activities of Notch and Wnt signaling regulate intestinal stem cells and gut homeostasis. Cell Rep. 2015, 11, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The intestinal crypt, a prototype stem cell compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef]
- Basak, O.; Beumer, J.; Wiebrands, K.; Seno, H.; van Oudenaarden, A.; Clevers, H. Induced quiescence of Lgr5+ stem cells in intestinal organoids enables differentiation of hormone-producing enteroendocrine cells. Cell Stem Cell 2017, 20, 177–190. [Google Scholar] [CrossRef]
- Kabiri, Z.; Greicius, G.; Zaribafzadeh, H.; Hemmerich, A.; Counter, C.M.; Virshup, D.M. Wnt signaling suppresses MAPK-driven proliferation of intestinal stem cells. J. Clin. Investig. 2018, 128, 3806–3812. [Google Scholar] [CrossRef]
- Van der Flier, L.G.; Haegebarth, A.; Stange, D.E.; van de Wetering, M.; Clevers, H. OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology 2009, 137, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, H.; Hong, S.H.; Piszczek, G.P.; Chen, W.; Rodgers, G.P. Olfactomedin 4 deletion induces colon adenocarcinoma in Apc(Min/+) mice. Oncogene 2016, 35, 5237–5247. [Google Scholar] [CrossRef]
- Gregorieff, A.; Pinto, D.; Begthel, H.; Destree, O.; Kielman, M.; Clevers, H. Expression pattern of Wnt signaling components in the adult intestine. Gastroenterology 2005, 129, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Shoshkes-Carmel, M.; Wang, Y.J.; Wangensteen, K.J.; Toth, B.; Kondo, A.; Massasa, E.E.; Itzkovitz, S.; Kaestner, K.H. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 2018, 557, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Pentinmikko, N.; Iqbal, S.; Mana, M.; Andersson, S.; Cognetta, A.B., 3rd; Suciu, R.M.; Roper, J.; Luopajarvi, K.; Markelin, E.; Gopalakrishnan, S.; et al. Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature 2019, 571, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Kakugawa, S.; Langton, P.F.; Zebisch, M.; Howell, S.; Chang, T.H.; Liu, Y.; Feizi, T.; Bineva, G.; O’Reilly, N.; Snijders, A.P.; et al. Notum deacylates Wnt proteins to suppress signalling activity. Nature 2015, 519, 187–192. [Google Scholar] [CrossRef]
- Pentinmikko, N.; Katajisto, P. The role of stem cell niche in intestinal aging. Mech. Ageing Dev. 2020, 191, 111330. [Google Scholar] [CrossRef]
- Farin, H.F.; Van Es, J.H.; Clevers, H. Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 2012, 143, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; Jay, P.; Gregorieff, A.; van Gijn, M.E.; Jonkheer, S.; Hatzis, P.; Thiele, A.; van den Born, M.; Begthel, H.; Brabletz, T.; et al. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat. Cell Biol. 2005, 7, 381–386. [Google Scholar] [CrossRef]
- Mori-Akiyama, Y.; van den Born, M.; van Es, J.H.; Hamilton, S.R.; Adams, H.P.; Zhang, J.; Clevers, H.; de Crombrugghe, B. SOX9 is required for the differentiation of Paneth cells in the intestinal epithelium. Gastroenterology 2007, 133, 539–546. [Google Scholar] [CrossRef]
- Bastide, P.; Darido, C.; Pannequin, J.; Kist, R.; Robine, S.; Marty-Double, C.; Bibeau, F.; Scherer, G.; Joubert, D.; Hollande, F.; et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J. Cell Biol 2007, 178, 635–648. [Google Scholar] [CrossRef]
- Andreu, P.; Peignon, G.; Slomianny, C.; Taketo, M.M.; Colnot, S.; Robine, S.; Lamarque, D.; Laurent-Puig, P.; Perret, C.; Romagnolo, B. A genetic study of the role of the Wnt/beta-catenin signalling in Paneth cell differentiation. Dev. Biol. 2008, 324, 288–296. [Google Scholar] [CrossRef]
- Batlle, E.; Henderson, J.T.; Beghtel, H.; van den Born, M.M.; Sancho, E.; Huls, G.; Meeldijk, J.; Robertson, J.; van de Wetering, M.; Pawson, T.; et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 2002, 111, 251–263. [Google Scholar] [CrossRef]
- Rodriguez-Colman, M.J.; Schewe, M.; Meerlo, M.; Stigter, E.; Gerrits, J.; Pras-Raves, M.; Sacchetti, A.; Hornsveld, M.; Oost, K.C.; Snippert, H.J.; et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 2017, 543, 424–427. [Google Scholar] [CrossRef]
- Rothenberg, M.E.; Nusse, Y.; Kalisky, T.; Lee, J.J.; Dalerba, P.; Scheeren, F.; Lobo, N.; Kulkarni, S.; Sim, S.; Qian, D.; et al. Identification of a cKit(+) colonic crypt base secretory cell that supports Lgr5(+) stem cells in mice. Gastroenterology 2012, 142, 1195–1205. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Kabiri, Z.; Greicius, G.; Madan, B.; Biechele, S.; Zhong, Z.; Zaribafzadeh, H.; Edison; Aliyev, J.; Wu, Y.; Bunte, R.; et al. Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Development 2014, 141, 2206–2215. [Google Scholar] [CrossRef]
- San Roman, A.K.; Jayewickreme, C.D.; Murtaugh, L.C.; Shivdasani, R.A. Wnt secretion from epithelial cells and subepithelial myofibroblasts is not required in the mouse intestinal stem cell niche in vivo. Stem Cell Rep. 2014, 2, 127–134. [Google Scholar] [CrossRef]
- Durand, A.; Donahue, B.; Peignon, G.; Letourneur, F.; Cagnard, N.; Slomianny, C.; Perret, C.; Shroyer, N.F.; Romagnolo, B. Functional intestinal stem cells after Paneth cell ablation induced by the loss of transcription factor Math1 (Atoh1). Proc. Natl. Acad. Sci. USA 2012, 109, 8965–8970. [Google Scholar] [CrossRef]
- Kim, T.H.; Escudero, S.; Shivdasani, R.A. Intact function of Lgr5 receptor-expressing intestinal stem cells in the absence of Paneth cells. Proc. Natl. Acad. Sci. USA 2012, 109, 3932–3937. [Google Scholar] [CrossRef]
- Van Es, J.H.; Wiebrands, K.; Lopez-Iglesias, C.; van de Wetering, M.; Zeinstra, L.; van den Born, M.; Korving, J.; Sasaki, N.; Peters, P.J.; van Oudenaarden, A.; et al. Enteroendocrine and tuft cells support Lgr5 stem cells on Paneth cell depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 26599–26605. [Google Scholar] [CrossRef]
- Zou, W.Y.; Blutt, S.E.; Zeng, X.L.; Chen, M.S.; Lo, Y.H.; Castillo-Azofeifa, D.; Klein, O.D.; Shroyer, N.F.; Donowitz, M.; Estes, M.K. Epithelial WNT ligands are essential drivers of intestinal stem cell activation. Cell Rep. 2018, 22, 1003–1015. [Google Scholar] [CrossRef]
- Degirmenci, B.; Valenta, T.; Dimitrieva, S.; Hausmann, G.; Basler, K. GLI1-expressing mesenchymal cells form the essential Wnt-secreting niche for colon stem cells. Nature 2018, 558, 449–453. [Google Scholar] [CrossRef]
- Greicius, G.; Kabiri, Z.; Sigmundsson, K.; Liang, C.; Bunte, R.; Singh, M.K.; Virshup, D.M. PDGFRalpha(+) pericryptal stromal cells are the critical source of Wnts and RSPO3 for murine intestinal stem cells in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E3173–E3181. [Google Scholar] [CrossRef]
- Israel, D.I.; Nove, J.; Kerns, K.M.; Kaufman, R.J.; Rosen, V.; Cox, K.A.; Wozney, J.M. Heterodimeric bone morphogenetic proteins show enhanced activity in vitro and in vivo. Growth Factors 1996, 13, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Bahar Halpern, K.; Massalha, H.; Zwick, R.K.; Moor, A.E.; Castillo-Azofeifa, D.; Rozenberg, M.; Farack, L.; Egozi, A.; Miller, D.R.; Averbukh, I.; et al. Lgr5+ telocytes are a signaling source at the intestinal villus tip. Nat. Commun. 2020, 11, 1936. [Google Scholar] [CrossRef]
- McCarthy, N.; Kraiczy, J.; Shivdasani, R.A. Cellular and molecular architecture of the intestinal stem cell niche. Nat. Cell Biol. 2020, 22, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Ruder, B.; Becker, C. At the forefront of the mucosal barrier: The role of macrophages in the intestine. Cells 2020, 9, 2162. [Google Scholar] [CrossRef]
- Sehgal, A.; Donaldson, D.S.; Pridans, C.; Sauter, K.A.; Hume, D.A.; Mabbott, N.A. The role of CSF1R-dependent macrophages in control of the intestinal stem-cell niche. Nat. Commun. 2018, 9, 1272. [Google Scholar] [CrossRef]
- Saha, S.; Aranda, E.; Hayakawa, Y.; Bhanja, P.; Atay, S.; Brodin, N.P.; Li, J.; Asfaha, S.; Liu, L.; Tailor, Y.; et al. Macrophage-derived extracellular vesicle-packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nat. Commun. 2016, 7, 13096. [Google Scholar] [CrossRef] [PubMed]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef]
- von Moltke, J.; Ji, M.; Liang, H.E.; Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016, 529, 221–225. [Google Scholar] [CrossRef]
- Gronke, K.; Hernandez, P.P.; Zimmermann, J.; Klose, C.S.N.; Kofoed-Branzk, M.; Guendel, F.; Witkowski, M.; Tizian, C.; Amann, L.; Schumacher, F.; et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 2019, 566, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Biton, M.; Haber, A.L.; Rogel, N.; Burgin, G.; Beyaz, S.; Schnell, A.; Ashenberg, O.; Su, C.W.; Smillie, C.; Shekhar, K.; et al. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 2018, 175, 1307–1320. [Google Scholar] [CrossRef]
- Cosovanu, C.; Neumann, C. The many functions of Foxp3(+) regulatory T cells in the intestine. Front. Immunol. 2020, 11, 600973. [Google Scholar] [CrossRef]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Gerbe, F.; Sidot, E.; Smyth, D.J.; Ohmoto, M.; Matsumoto, I.; Dardalhon, V.; Cesses, P.; Garnier, L.; Pouzolles, M.; Brulin, B.; et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016, 529, 226–230. [Google Scholar] [CrossRef]
- Howitt, M.R.; Lavoie, S.; Michaud, M.; Blum, A.M.; Tran, S.V.; Weinstock, J.V.; Gallini, C.A.; Redding, K.; Margolskee, R.F.; Osborne, L.C.; et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016, 351, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; O’Leary, C.E.; von Moltke, J.; Liang, H.E.; Ang, Q.Y.; Turnbaugh, P.J.; Radhakrishnan, S.; Pellizzon, M.; Ma, A.; Locksley, R.M. A metabolite-triggered tuft cell-ILC2 circuit drives small intestinal remodeling. Cell 2018, 174, 271–284. [Google Scholar] [CrossRef]
- Bohin, N.; Keeley, T.M.; Carulli, A.J.; Walker, E.M.; Carlson, E.A.; Gao, J.; Aifantis, I.; Siebel, C.W.; Rajala, M.W.; Myers, M.G., Jr.; et al. Rapid crypt cell remodeling regenerates the intestinal stem cell niche after Notch inhibition. Stem Cell Rep. 2020, 15, 156–170. [Google Scholar] [CrossRef]
- Jardé, T.; Chan, W.H.; Rossello, F.J.; Kaur Kahlon, T.; Theocharous, M.; Kurian Arackal, T.; Flores, T.; Giraud, M.; Richards, E.; Chan, E.; et al. Mesenchymal niche-derived neuregulin-1 drives intestinal stem cell proliferation and regeneration of damaged epithelium. Cell Stem Cell 2020, 27, 646–662. [Google Scholar] [CrossRef]
- Bigaeva, E.; Uniken Venema, W.T.C.; Weersma, R.K.; Festen, E.A.M. Understanding human gut diseases at single-cell resolution. Hum. Mol. Genet. 2020, 29, R51–R58. [Google Scholar] [CrossRef] [PubMed]
- Kinchen, J.; Chen, H.H.; Parikh, K.; Antanaviciute, A.; Jagielowicz, M.; Fawkner-Corbett, D.; Ashley, N.; Cubitt, L.; Mellado-Gomez, E.; Attar, M.; et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell 2018, 175, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 2019, 178, 714–730. [Google Scholar] [CrossRef]
- Parikh, K.; Antanaviciute, A.; Fawkner-Corbett, D.; Jagielowicz, M.; Aulicino, A.; Lagerholm, C.; Davis, S.; Kinchen, J.; Chen, H.H.; Alham, N.K.; et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019, 567, 49–55. [Google Scholar] [CrossRef]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Görtz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Nusse, Y.M.; Savage, A.K.; Marangoni, P.; Rosendahl-Huber, A.K.M.; Landman, T.A.; de Sauvage, F.J.; Locksley, R.M.; Klein, O.D. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 2018, 559, 109–113. [Google Scholar] [CrossRef]
- Yui, S.; Azzolin, L.; Maimets, M.; Pedersen, M.T.; Fordham, R.P.; Hansen, S.L.; Larsen, H.L.; Guiu, J.; Alves, M.R.P.; Rundsten, C.F.; et al. YAP/TAZ-dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell 2018, 22, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Mustata, R.C.; Vasile, G.; Fernandez-Vallone, V.; Strollo, S.; Lefort, A.; Libert, F.; Monteyne, D.; Perez-Morga, D.; Vassart, G.; Garcia, M.I. Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep. 2013, 5, 421–432. [Google Scholar] [CrossRef]
- Gregorieff, A.; Wrana, J.L. Hippo signalling in intestinal regeneration and cancer. Curr. Opin. Cell Biol. 2017, 48, 17–25. [Google Scholar] [CrossRef]
- Gregorieff, A.; Liu, Y.; Inanlou, M.R.; Khomchuk, Y.; Wrana, J.L. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 2015, 526, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Morikawa, T.; Butler, B.L.; Shrestha, K.; de la Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 2013, 493, 106–110. [Google Scholar] [CrossRef]
- Li, Q.; Sun, Y.; Jarugumilli, G.K.; Liu, S.; Dang, K.; Cotton, J.L.; Xiol, J.; Chan, P.Y.; DeRan, M.; Ma, L.; et al. Lats1/2 sustain intestinal stem cells and Wnt activation through TEAD-dependent and independent transcription. Cell Stem Cell 2020, 26, 675–692. [Google Scholar] [CrossRef]
- Cheung, P.; Xiol, J.; Dill, M.T.; Yuan, W.C.; Panero, R.; Roper, J.; Osorio, F.G.; Maglic, D.; Li, Q.; Gurung, B.; et al. Regenerative Reprogramming of the intestinal stem cell state via Hippo signaling suppresses metastatic colorectal cancer. Cell Stem Cell 2020, 27, 590–604. [Google Scholar] [CrossRef] [PubMed]
- Ayyaz, A.; Kumar, S.; Sangiorgi, B.; Ghoshal, B.; Gosio, J.; Ouladan, S.; Fink, M.; Barutcu, S.; Trcka, D.; Shen, J.; et al. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 2019, 569, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.R.; Spitofsky, N.; Zamani, A.; Hood, R.; Boggs, A.; Li, X.; Li, M.; Reiner, E.; Ayyaz, A.; Etwebi, Z.; et al. TNFAIP8 controls murine intestinal stem cell homeostasis and regeneration by regulating microbiome-induced Akt signaling. Nat. Commun. 2020, 11, 2591. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Maitra, A.; Anders, R.A.; Taketo, M.M.; Pan, D. β-Catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015, 29, 1493–1506. [Google Scholar] [CrossRef]
- Roulis, M.; Kaklamanos, A.; Schernthanner, M.; Bielecki, P.; Zhao, J.; Kaffe, E.; Frommelt, L.S.; Qu, R.; Knapp, M.S.; Henriques, A.; et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature 2020, 580, 524–529. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, Y.; Wu, H.; Barry, E.; Yin, Y.; Lawrence, E.; Dawson, D.; Willis, J.E.; Markowitz, S.D.; Camargo, F.D.; et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. USA 2011, 108, E1312–E1320. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef]
- Reischmann, N.; Andrieux, G.; Griffin, R.; Reinheckel, T.; Boerries, M.; Brummer, T. BRAF(V600E) drives dedifferentiation in small intestinal and colonic organoids and cooperates with mutant p53 and Apc loss in transformation. Oncogene 2020, 39, 6053–6070. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Goswami, S.; Hu, Y.; Tang, F.; Zafra, M.P.; Murphy, C.; Cao, Z.; Poirier, J.T.; Khurana, E.; Elemento, O.; et al. Lineage Reversion Drives WNT Independence in Intestinal Cancer. Cancer Discov. 2020, 10, 1590–1609. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.B.; Kim, M.; Park, Y.S.; Park, I.; Kim, T.; Yang, S.Y.; Cho, C.J.; Hwang, D.; Jung, J.H.; Markowitz, S.D.; et al. Prostaglandin E(2) activates YAP and a positive-signaling loop to promote colon regeneration after colitis but also carcinogenesis in mice. Gastroenterology 2017, 152, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Koulis, C.; Yap, R.; Engel, R.; Jardé, T.; Wilkins, S.; Solon, G.; Shapiro, J.D.; Abud, H.; McMurrick, P. Personalized medicine-current and emerging predictive and prognostic biomarkers in colorectal cancer. Cancers 2020, 12, 812. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Basnet, H.; Kaygusuz, Y.; Laughney, A.M.; He, L.; Sharma, R.; O’Rourke, K.P.; Reuter, V.P.; Huang, Y.H.; Turkekul, M.; et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nat. Cancer 2020, 1, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The intestinal microbiota in colorectal cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef]
- Francescangeli, F.; De Angelis, M.L.; Zeuner, A. Dietary factors in the control of gut homeostasis, intestinal stem cells, and colorectal cancer. Nutrients 2019, 11, 2936. [Google Scholar] [CrossRef]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef]
- Xing, P.Y.; Pettersson, S.; Kundu, P. Microbial metabolites & intestinal stem cells tune intestinal homeostasis. Proteomics 2020, 20, e1800419. [Google Scholar] [CrossRef]
- Goyal, S.; Tsang, D.K.L.; Maisonneuve, C.; Girardin, S.E. Sending signals—The microbiota’s contribution to intestinal epithelial homeostasis. Microbes Infect. 2020. [Google Scholar] [CrossRef]
- Janney, A.; Powrie, F.; Mann, E.H. Host-microbiota maladaptation in colorectal cancer. Nature 2020, 585, 509–517. [Google Scholar] [CrossRef]
- Calibasi-Kocal, G.; Mashinchian, O.; Basbinar, Y.; Ellidokuz, E.; Cheng, C.W.; Yilmaz, Ö.H. Nutritional control of intestinal stem cells in homeostasis and tumorigenesis. Trends Endocrinol. Metab. 2021, 32, 20–35. [Google Scholar] [CrossRef]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef]
- Wang, B.; Rong, X.; Palladino, E.N.D.; Wang, J.; Fogelman, A.M.; Martin, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell 2018, 22, 206–220. [Google Scholar] [CrossRef]
- Cheng, C.W.; Biton, M.; Haber, A.L.; Gunduz, N.; Eng, G.; Gaynor, L.T.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, V.L.; et al. Ketone body signaling mediates intestinal stem cell homeostasis and adaptation to diet. Cell 2019, 178, 1115–1131. [Google Scholar] [CrossRef]
- Li, W.; Zimmerman, S.E.; Peregrina, K.; Houston, M.; Mayoral, J.; Zhang, J.; Maqbool, S.; Zhang, Z.; Cai, Y.; Ye, K.; et al. The nutritional environment determines which and how intestinal stem cells contribute to homeostasis and tumorigenesis. Carcinogenesis 2019, 40, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Newmark, H.L.; Lipkin, M.; Maheshwari, N. Colonic hyperproliferation induced in rats and mice by nutritional-stress diets containing four components of a human Western-style diet (series 2). Am. J. Clin. Nutr 1991, 54, 209S–214S. [Google Scholar] [CrossRef] [PubMed]
- Newmark, H.L.; Yang, K.; Lipkin, M.; Kopelovich, L.; Liu, Y.; Fan, K.; Shinozaki, H. A Western-style diet induces benign and malignant neoplasms in the colon of normal C57Bl/6 mice. Carcinogenesis 2001, 22, 1871–1875. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, K.; Kurihara, N.; Fan, K.; Newmark, H.; Rigas, B.; Bancroft, L.; Corner, G.; Livote, E.; Lesser, M.; Edelmann, W.; et al. Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res. 2008, 68, 7803–7810. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Peregrina, K.; Houston, M.; Augenlicht, L.H. Vitamin D and the nutritional environment in functions of intestinal stem cells: Implications for tumorigenesis and prevention. J. Steroid Biochem. Mol. Biol. 2020, 198, 105556. [Google Scholar] [CrossRef]
- Wang, D.; Peregrina, K.; Dhima, E.; Lin, E.Y.; Mariadason, J.M.; Augenlicht, L.H. Paneth cell marker expression in intestinal villi and colon crypts characterizes dietary induced risk for mouse sporadic intestinal cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 10272–10277. [Google Scholar] [CrossRef] [PubMed]
- Peregrina, K.; Houston, M.; Daroqui, C.; Dhima, E.; Sellers, R.S.; Augenlicht, L.H. Vitamin D is a determinant of mouse intestinal Lgr5 stem cell functions. Carcinogenesis 2015, 36, 25–31. [Google Scholar] [CrossRef]
- McCullough, M.L.; Zoltick, E.S.; Weinstein, S.J.; Fedirko, V.; Wang, M.; Cook, N.R.; Eliassen, A.H.; Zeleniuch-Jacquotte, A.; Agnoli, C.; Albanes, D.; et al. Circulating vitamin D and colorectal cancer risk: An international pooling project of 17 cohorts. J. Natl. Cancer Inst. 2019, 111, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R. Use of high-fat diets to study rodent obesity as a model of human obesity. Int. J. Obes. 2019, 43, 1491–1492. [Google Scholar] [CrossRef]
- Bastías-Pérez, M.; Serra, D.; Herrero, L. Dietary options for rodents in the study of obesity. Nutrients 2020, 12, 3234. [Google Scholar] [CrossRef]
- Li, W.; Houston, M.; Peregrina, K.; Ye, K.; Augenlicht, L.H. Effects of diet choice on stem cell function necessitate clarity in selection and reporting. Cell Stem Cell 2020, 27, 11–12. [Google Scholar] [CrossRef]
- Weindruch, R.; Walford, R.L.; Fligiel, S.; Guthrie, D. The retardation of aging in mice by dietary restriction: Longevity, cancer, immunity and lifetime energy intake. J. Nutr. 1986, 116, 641–654. [Google Scholar] [CrossRef]
- Yilmaz, O.H.; Katajisto, P.; Lamming, D.W.; Gultekin, Y.; Bauer-Rowe, K.E.; Sengupta, S.; Birsoy, K.; Dursun, A.; Yilmaz, V.O.; Selig, M.; et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 2012, 486, 490–495. [Google Scholar] [CrossRef]
- Richmond, C.A.; Shah, M.S.; Deary, L.T.; Trotier, D.C.; Thomas, H.; Ambruzs, D.M.; Jiang, L.; Whiles, B.B.; Rickner, H.D.; Montgomery, R.K.; et al. Dormant intestinal stem cells are regulated by PTEN and nutritional status. Cell Rep. 2015, 13, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Tinkum, K.L.; Stemler, K.M.; White, L.S.; Loza, A.J.; Jeter-Jones, S.; Michalski, B.M.; Kuzmicki, C.; Pless, R.; Stappenbeck, T.S.; Piwnica-Worms, D.; et al. Fasting protects mice from lethal DNA damage by promoting small intestinal epithelial stem cell survival. Proc. Natl. Acad. Sci. USA 2015, 112, E7148–E7154. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Cheng, C.W.; Cao, A.Q.; Tripathi, S.; Mana, M.D.; Bauer-Rowe, K.E.; Abu-Remaileh, M.; Clavain, L.; Erdemir, A.; Lewis, C.A.; et al. Fasting activates fatty acid oxidation to enhance intestinal stem cell function during homeostasis and aging. Cell Stem Cell 2018, 22, 769–778. [Google Scholar] [CrossRef]
- Beyaz, S.; Yilmaz, Ö.H. Molecular pathways: Dietary regulation of stemness and tumor initiation by the PPAR-δ pathway. Clin. Cancer Res. 2016, 22, 5636–5641. [Google Scholar] [CrossRef]
- Wang, D.; Fu, L.; Wei, J.; Xiong, Y.; DuBois, R.N. PPARδ mediates the effect of dietary fat in promoting colorectal cancer metastasis. Cancer Res. 2019, 79, 4480–4490. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Zeng, Z.L.; Lu, J.; Wang, Y.; Liu, Z.X.; He, M.M.; Zhao, Q.; Wang, Z.X.; Li, T.; Lu, Y.X.; et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 2018, 37, 6025–6040. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Kaiko, G.E.; Ryu, S.H.; Koues, O.I.; Collins, P.L.; Solnica-Krezel, L.; Pearce, E.J.; Pearce, E.L.; Oltz, E.M.; Stappenbeck, T.S. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell 2016, 165, 1708–1720. [Google Scholar] [CrossRef]
- Bultman, S.J. Molecular pathways: Gene-environment interactions regulating dietary fiber induction of proliferation and apoptosis via butyrate for cancer prevention. Clin. Cancer Res. 2014, 20, 799–803. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef]
- Bultman, S.J.; Jobin, C. Microbial-derived butyrate: An oncometabolite or tumor-suppressive metabolite? Cell Host Microbe 2014, 16, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Yeoh, B.S.; Vijay-Kumar, M. Feed your gut with caution! Transl. Cancer Res. 2016, 5, S507–S513. [Google Scholar] [CrossRef]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR regulates intestinal cancer stem cell proliferation. Cell 2019, 176, 1098–1112. [Google Scholar] [CrossRef]
- Sorrentino, G.; Perino, A.; Yildiz, E.; El Alam, G.; Bou Sleiman, M.; Gioiello, A.; Pellicciari, R.; Schoonjans, K. Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology 2020, 159, 956–968. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; DeLany, J.P.; Zhang, M.; Sharma, S.; O’Keefe, S.J. Association between low colonic short-chain fatty acids and high bile acids in high colon cancer risk populations. Nutr. Cancer 2012, 64, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Carbonero, F.; Zoetendal, E.G.; DeLany, J.P.; Wang, M.; Newton, K.; Gaskins, H.R.; O’Keefe, S.J. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am. J. Clin. Nutr. 2013, 98, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, T.Y.; Kim, Y.; Lee, S.H.; Kim, S.; Kang, S.W.; Yang, J.Y.; Baek, I.J.; Sung, Y.H.; Park, Y.Y.; et al. Microbiota-derived lactate accelerates intestinal stem-cell-mediated epithelial development. Cell Host Microbe 2018, 24, 833–846. [Google Scholar] [CrossRef]
- Das, U.N. Arachidonic acid and other unsaturated fatty acids and some of their metabolites function as endogenous antimicrobial molecules: A review. J. Adv. Res. 2018, 11, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lin, Y.; Sheng, X.; Xu, J.; Hou, X.; Li, Y.; Zhang, H.; Guo, H.; Yu, Z.; Ren, F. Arachidonic acid promotes intestinal regeneration by activating WNT signaling. Stem Cell Rep. 2020, 15, 374–388. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z.; et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Nava, G.M.; Friedrichsen, H.J.; Stappenbeck, T.S. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J. 2011, 5, 627–638. [Google Scholar] [CrossRef]
- Pédron, T.; Mulet, C.; Dauga, C.; Frangeul, L.; Chervaux, C.; Grompone, G.; Sansonetti, P.J. A crypt-specific core microbiota resides in the mouse colon. mBio 2012, 3, e00116-12. [Google Scholar] [CrossRef]
- Naito, T.; Mulet, C.; De Castro, C.; Molinaro, A.; Saffarian, A.; Nigro, G.; Bérard, M.; Clerc, M.; Pedersen, A.B.; Sansonetti, P.J.; et al. Lipopolysaccharide from crypt-specific core microbiota modulates the colonic epithelial proliferation-to-differentiation balance. mBio 2017, 8, e01680-17. [Google Scholar] [CrossRef] [PubMed]
- Saffarian, A.; Mulet, C.; Regnault, B.; Amiot, A.; Tran-Van-Nhieu, J.; Ravel, J.; Sobhani, I.; Sansonetti, P.J.; Pédron, T. Crypt- and mucosa-associated core microbiotas in humans and their alteration in colon cancer patients. mBio 2019, 10, e01315-19. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.D.; Sodhi, C.P.; Jia, H.; Dyer, M.; Egan, C.E.; Yazji, I.; Good, M.; Afrazi, A.; Marino, R.; Slagle, D.; et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J. Biol. Chem. 2012, 287, 37296–37308. [Google Scholar] [CrossRef]
- Nigro, G.; Rossi, R.; Commere, P.H.; Jay, P.; Sansonetti, P.J. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe 2014, 15, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Stedman, A.; Deutsch, E.; Donnadieu, F.; Virgin, H.W.; Sansonetti, P.J.; Nigro, G. Innate immune receptor NOD2 mediates LGR5(+) intestinal stem cell protection against ROS cytotoxicity via mitophagy stimulation. Proc. Natl. Acad. Sci. USA 2020, 117, 1994–2003. [Google Scholar] [CrossRef]
- Lee, C.; Choi, C.; Kang, H.S.; Shin, S.W.; Kim, S.Y.; Park, H.C.; Hong, S.N. NOD2 supports crypt survival and epithelial regeneration after radiation-induced injury. Int. J. Mol. Sci. 2019, 20, 4297. [Google Scholar] [CrossRef]
- Hou, Q.; Ye, L.; Liu, H.; Huang, L.; Yang, Q.; Turner, J.R.; Yu, Q. Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ. 2018, 25, 1657–1670. [Google Scholar] [CrossRef]
- Wu, H.; Xie, S.; Miao, J.; Li, Y.; Wang, Z.; Wang, M.; Yu, Q. Lactobacillus reuteri maintains intestinal epithelial regeneration and repairs damaged intestinal mucosa. Gut Microbes 2020, 11, 997–1014. [Google Scholar] [CrossRef]
- Mileto, S.J.; Jardé, T.; Childress, K.O.; Jensen, J.L.; Rogers, A.P.; Kerr, G.; Hutton, M.L.; Sheedlo, M.J.; Bloch, S.C.; Shupe, J.A.; et al. Clostridioides difficile infection damages colonic stem cells via TcdB, impairing epithelial repair and recovery from disease. Proc. Natl. Acad. Sci. USA 2020, 117, 8064–8073. [Google Scholar] [CrossRef]
- Jones, R.M.; Wu, H.; Wentworth, C.; Luo, L.; Collier-Hyams, L.; Neish, A.S. Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe 2008, 3, 233–244. [Google Scholar] [CrossRef]
- Liu, X.; Lu, R.; Wu, S.; Sun, J. Salmonella regulation of intestinal stem cells through the Wnt/beta-catenin pathway. FEBS Lett. 2010, 584, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Wu, S.; Zhang, Y.G.; Xia, Y.; Liu, X.; Zheng, Y.; Chen, H.; Schaefer, K.L.; Zhou, Z.; Bissonnette, M.; et al. Enteric bacterial protein AvrA promotes colonic tumorigenesis and activates colonic beta-catenin signaling pathway. Oncogenesis 2014, 3, e105. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Purcell, R.V.; Visnovska, M.; Biggs, P.J.; Schmeier, S.; Frizelle, F.A. Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci. Rep. 2017, 7, 11590. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef]
- Kang, X.; Zhang, R.; Kwong, T.N.; Lui, R.N.; Wu, W.K.; Sung, J.J.; Yu, J.; Wong, S.H. Serrated neoplasia in the colorectum: Gut microbiota and molecular pathways. Gut Microbes 2021, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Diaz-Meco, M.T.; Moscat, J. Serrated colorectal cancer: The road less travelled? Trends Cancer 2019, 5, 742–754. [Google Scholar] [CrossRef]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Weisburger, J.H.; Narisawa, T.; Wynder, E.L. Colon carcinogenesis in germ-free rats with 1,2-dimethylhydrazine and N-methyl-n′-nitro-N-nitrosoguanidine. Cancer Res. 1974, 34, 2368–2372. [Google Scholar]
- Vannucci, L.; Stepankova, R.; Kozakova, H.; Fiserova, A.; Rossmann, P.; Tlaskalova-Hogenova, H. Colorectal carcinogenesis in germ-free and conventionally reared rats: Different intestinal environments affect the systemic immunity. Int. J. Oncol. 2008, 32, 609–617. [Google Scholar] [CrossRef]
- Uronis, J.M.; Mühlbauer, M.; Herfarth, H.H.; Rubinas, T.C.; Jones, G.S.; Jobin, C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS ONE 2009, 4, e6026. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. . Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Mima, K.; Sukawa, Y.; Nishihara, R.; Qian, Z.R.; Yamauchi, M.; Inamura, K.; Kim, S.A.; Masuda, A.; Nowak, J.A.; Nosho, K.; et al. Fusobacterium nucleatum and T Cells in colorectal carcinoma. JAMA Oncol. 2015, 1, 653–661. [Google Scholar] [CrossRef]
- Chung, L.; Thiele Orberg, E.; Geis, A.L.; Chan, J.L.; Fu, K.; DeStefano Shields, C.E.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe 2018, 23, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017, 170, 548–563. [Google Scholar] [CrossRef]
- Man, S.M.; Zhu, Q.; Zhu, L.; Liu, Z.; Karki, R.; Malik, A.; Sharma, D.; Li, L.; Malireddi, R.K.; Gurung, P.; et al. Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell 2015, 162, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Kadosh, E.; Snir-Alkalay, I.; Venkatachalam, A.; May, S.; Lasry, A.; Elyada, E.; Zinger, A.; Shaham, M.; Vaalani, G.; Mernberger, M.; et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020, 586, 133–138. [Google Scholar] [CrossRef]
- Slowicka, K.; Petta, I.; Blancke, G.; Hoste, E.; Dumas, E.; Sze, M.; Vikkula, H.; Radaelli, E.; Haigh, J.J.; Jonckheere, S.; et al. Zeb2 drives invasive and microbiota-dependent colon carcinoma. Nat. Cancer 2020, 1, 620–634. [Google Scholar] [CrossRef]
- Lopez-Garcia, C.; Klein, A.M.; Simons, B.D.; Winton, D.J. Intestinal stem cell replacement follows a pattern of neutral drift. Science 2010, 330, 822–825. [Google Scholar] [CrossRef]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef]
- Ritsma, L.; Ellenbroek, S.I.J.; Zomer, A.; Snippert, H.J.; de Sauvage, F.J.; Simons, B.D.; Clevers, H.; van Rheenen, J. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature 2014, 507, 362–365. [Google Scholar] [CrossRef]
- Farin, H.F.; Jordens, I.; Mosa, M.H.; Basak, O.; Korving, J.; Tauriello, D.V.; de Punder, K.; Angers, S.; Peters, P.J.; Maurice, M.M.; et al. Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature 2016, 530, 340–343. [Google Scholar] [CrossRef]
- Kozar, S.; Morrissey, E.; Nicholson, A.M.; van der Heijden, M.; Zecchini, H.I.; Kemp, R.; Tavare, S.; Vermeulen, L.; Winton, D.J. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell 2013, 13, 626–633. [Google Scholar] [CrossRef]
- Vermeulen, L.; Morrissey, E.; van der Heijden, M.; Nicholson, A.M.; Sottoriva, A.; Buczacki, S.; Kemp, R.; Tavare, S.; Winton, D.J. Defining stem cell dynamics in models of intestinal tumor initiation. Science 2013, 342, 995–998. [Google Scholar] [CrossRef]
- Baker, A.M.; Cereser, B.; Melton, S.; Fletcher, A.G.; Rodriguez-Justo, M.; Tadrous, P.J.; Humphries, A.; Elia, G.; McDonald, S.A.; Wright, N.A.; et al. Quantification of crypt and stem cell evolution in the normal and neoplastic human colon. Cell Rep. 2014, 8, 940–947. [Google Scholar] [CrossRef]
- Nicholson, A.M.; Olpe, C.; Hoyle, A.; Thorsen, A.S.; Rus, T.; Colombe, M.; Brunton-Sim, R.; Kemp, R.; Marks, K.; Quirke, P.; et al. Fixation and spread of somatic mutations in adult human colonic epithelium. Cell Stem Cell 2018, 22, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Stamp, C.; Zupanic, A.; Sachdeva, A.; Stoll, E.A.; Shanley, D.P.; Mathers, J.C.; Kirkwood, T.B.L.; Heer, R.; Simons, B.D.; Turnbull, D.M.; et al. Predominant Asymmetrical stem cell fate outcome limits the rate of niche succession in human colonic crypts. EBioMedicine 2018, 31, 166–173. [Google Scholar] [CrossRef]
- Sugimoto, S.; Ohta, Y.; Fujii, M.; Matano, M.; Shimokawa, M.; Nanki, K.; Date, S.; Nishikori, S.; Nakazato, Y.; Nakamura, T.; et al. Reconstruction of the human colon epithelium in vivo. Cell Stem Cell 2018, 22, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Hodder, M.C.; Flanagan, D.J.; Sansom, O.J. Intestinal stem cell dynamics: A story of mice and humans. Cell Stem Cell 2018, 22, 785–787. [Google Scholar] [CrossRef] [PubMed]
- Mai, V.; Colbert, L.H.; Berrigan, D.; Perkins, S.N.; Pfeiffer, R.; Lavigne, J.A.; Lanza, E.; Haines, D.C.; Schatzkin, A.; Hursting, S.D. Calorie restriction and diet composition modulate spontaneous intestinal tumorigenesis in Apc(Min) mice through different mechanisms. Cancer Res. 2003, 63, 1752–1755. [Google Scholar] [PubMed]
- Bruens, L.; Ellenbroek, S.I.J.; Suijkerbuijk, S.J.E.; Azkanaz, M.; Hale, A.J.; Toonen, P.; Flanagan, D.J.; Sansom, O.J.; Snippert, H.J.; van Rheenen, J. Calorie restriction increases the number of competing stem cells and decreases mutation retention in the intestine. Cell Rep. 2020, 32, 107937. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; Schepers, A.G.; van Es, J.H.; Simons, B.D.; Clevers, H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 2014, 15, 62–69. [Google Scholar] [CrossRef]
- Vermeulen, L.; Snippert, H.J. Stem cell dynamics in homeostasis and cancer of the intestine. Nat. Rev. Cancer 2014, 14, 468–480. [Google Scholar] [CrossRef]
- Fischer, J.M.; Schepers, A.G.; Clevers, H.; Shibata, D.; Liskay, R.M. Occult progression by Apc-deficient intestinal crypts as a target for chemoprevention. Carcinogenesis 2014, 35, 237–246. [Google Scholar] [CrossRef][Green Version]
- Preston, S.L.; Wong, W.M.; Chan, A.O.; Poulsom, R.; Jeffery, R.; Goodlad, R.A.; Mandir, N.; Elia, G.; Novelli, M.; Bodmer, W.F.; et al. Bottom-up histogenesis of colorectal adenomas: Origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 2003, 63, 3819–3825. [Google Scholar] [PubMed]
- Cernat, L.; Blaj, C.; Jackstadt, R.; Brandl, L.; Engel, J.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Colorectal cancers mimic structural organization of normal colonic crypts. PLoS ONE 2014, 9, e104284. [Google Scholar] [CrossRef] [PubMed]
- Zeuner, A.; Todaro, M.; Stassi, G.; De Maria, R. Colorectal cancer stem cells: From the crypt to the clinic. Cell Stem Cell 2014, 15, 692–705. [Google Scholar] [CrossRef]
- Wahab, S.M.R.; Islam, F.; Gopalan, V.; Lam, A.K. The identifications and clinical implications of cancer stem cells in colorectal cancer. Clin. Colorectal Cancer 2017, 16, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2018, 9, 33403–33415. [Google Scholar] [CrossRef] [PubMed]
- Munro, M.J.; Wickremesekera, S.K.; Peng, L.; Tan, S.T.; Itinteang, T. Cancer stem cells in colorectal cancer: A review. J. Clin. Pathol. 2018, 71, 110–116. [Google Scholar] [CrossRef]
- Hirata, A.; Hatano, Y.; Niwa, M.; Hara, A.; Tomita, H. Heterogeneity in colorectal cancer stem cells. Cancer Prev. Res. 2019, 12, 413–420. [Google Scholar] [CrossRef]
- Das, P.K.; Islam, F.; Lam, A.K. The roles of cancer stem cells and therapy resistance in colorectal carcinoma. Cells 2020, 9, 1392. [Google Scholar] [CrossRef]
- Shih, I.M.; Wang, T.L.; Traverso, G.; Romans, K.; Hamilton, S.R.; Ben-Sasson, S.; Kinzler, K.W.; Vogelstein, B. Top-down morphogenesis of colorectal tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 2640–2645. [Google Scholar] [CrossRef]
- Hilkens, J.; Timmer, N.C.; Boer, M.; Ikink, G.J.; Schewe, M.; Sacchetti, A.; Koppens, M.A.J.; Song, J.Y.; Bakker, E.R.M. RSPO3 expands intestinal stem cell and niche compartments and drives tumorigenesis. Gut 2017, 66, 1095–1105. [Google Scholar] [CrossRef]
- Zhu, L.; Gibson, P.; Currle, D.S.; Tong, Y.; Richardson, R.J.; Bayazitov, I.T.; Poppleton, H.; Zakharenko, S.; Ellison, D.W.; Gilbertson, R.J. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 2009, 457, 603–607. [Google Scholar] [CrossRef]
- Powell, A.E.; Vlacich, G.; Zhao, Z.Y.; McKinley, E.T.; Washington, M.K.; Manning, H.C.; Coffey, R.J. Inducible loss of one Apc allele in Lrig1-expressing progenitor cells results in multiple distal colonic tumors with features of familial adenomatous polyposis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G16–G23. [Google Scholar] [CrossRef]
- Cammareri, P.; Vincent, D.F.; Hodder, M.C.; Ridgway, R.A.; Murgia, C.; Nobis, M.; Campbell, A.D.; Varga, J.; Huels, D.J.; Subramani, C.; et al. TGFbeta pathway limits dedifferentiation following WNT and MAPK pathway activation to suppress intestinal tumourigenesis. Cell Death Differ. 2017, 24, 1681–1693. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Goktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Tetteh, P.W.; Kretzschmar, K.; Begthel, H.; van den Born, M.; Korving, J.; Morsink, F.; Farin, H.; van Es, J.H.; Offerhaus, G.J.; Clevers, H. Generation of an inducible colon-specific Cre enzyme mouse line for colon cancer research. Proc. Natl. Acad. Sci. USA 2016, 113, 11859–11864. [Google Scholar] [CrossRef] [PubMed]
- Perekatt, A.O.; Shah, P.P.; Cheung, S.; Jariwala, N.; Wu, A.; Gandhi, V.; Kumar, N.; Feng, Q.; Patel, N.; Chen, L.; et al. SMAD4 suppresses WNT-driven dedifferentiation and oncogenesis in the differentiated gut epithelium. Cancer Res. 2018, 78, 4878–4890. [Google Scholar] [CrossRef] [PubMed]
- Means, A.L.; Freeman, T.J.; Zhu, J.; Woodbury, L.G.; Marincola-Smith, P.; Wu, C.; Meyer, A.R.; Weaver, C.J.; Padmanabhan, C.; An, H.; et al. Epithelial Smad4 deletion up-regulates inflammation and promotes inflammation-associated cancer. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 257–276. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Tsuboi, M.; Asfaha, S.; Kinoshita, H.; Niikura, R.; Konishi, M.; Hata, M.; Oya, Y.; Kim, W.; Middelhoff, M.; et al. BHLHA15-positive secretory precursor cells can give rise to tumors in intestine and colon in mice. Gastroenterology 2019, 156, 1066–1081. [Google Scholar] [CrossRef]
- Herrinton, L.J.; Liu, L.; Levin, T.R.; Allison, J.E.; Lewis, J.D.; Velayos, F. Incidence and mortality of colorectal adenocarcinoma in persons with inflammatory bowel disease from 1998 to 2010. Gastroenterology 2012, 143, 382–389. [Google Scholar] [CrossRef]
- Vermeulen, L.; Todaro, M.; de Sousa Mello, F.; Sprick, M.R.; Kemper, K.; Perez Alea, M.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar] [CrossRef]
- Junttila, M.R.; Mao, W.; Wang, X.; Wang, B.E.; Pham, T.; Flygare, J.; Yu, S.F.; Yee, S.; Goldenberg, D.; Fields, C.; et al. Targeting LGR5+ cells with an antibody-drug conjugate for the treatment of colon cancer. Sci. Transl. Med. 2015, 7, 314ra186. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Prasetyanti, P.R.; De Lau, W.; Rodermond, H.; Clevers, H.; Medema, J.P. Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells. Stem Cells 2012, 30, 2378–2386. [Google Scholar] [CrossRef]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef]
- Roper, J.; Tammela, T.; Cetinbas, N.M.; Akkad, A.; Roghanian, A.; Rickelt, S.; Almeqdadi, M.; Wu, K.; Oberli, M.A.; Sanchez-Rivera, F.J.; et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 2017, 35, 569–576. [Google Scholar] [CrossRef]
- de Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Cortina, C.; Turon, G.; Stork, D.; Hernando-Momblona, X.; Sevillano, M.; Aguilera, M.; Tosi, S.; Merlos-Suarez, A.; Stephan-Otto Attolini, C.; Sancho, E.; et al. A genome editing approach to study cancer stem cells in human tumors. EMBO Mol. Med. 2017, 9, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef]
- Kobayashi, S.; Yamada-Okabe, H.; Suzuki, M.; Natori, O.; Kato, A.; Matsubara, K.; Jau Chen, Y.; Yamazaki, M.; Funahashi, S.; Yoshida, K.; et al. LGR5-positive colon cancer stem cells interconvert with drug-resistant LGR5-negative cells and are capable of tumor reconstitution. Stem Cells 2012, 30, 2631–2644. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, A.; Suijkerbuijk, S.J.E.; Begthel, H.; Beerling, E.; Oost, K.C.; Snippert, H.J.; van Rheenen, J.; Drost, J. A surgical orthotopic organoid transplantation approach in mice to visualize and study colorectal cancer progression. Nat. Protoc. 2018, 13, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, A.; Oost, K.C.; Kester, L.; Morgner, J.; Bornes, L.; Bruens, L.; Spaargaren, L.; Azkanaz, M.; Schelfhorst, T.; Beerling, E.; et al. Plasticity of Lgr5-negative cancer cells drives metastasis in colorectal cancer. Cell Stem Cell 2020, 26, 569–578. [Google Scholar] [CrossRef]
- Er, E.E.; Valiente, M.; Ganesh, K.; Zou, Y.; Agrawal, S.; Hu, J.; Griscom, B.; Rosenblum, M.; Boire, A.; Brogi, E.; et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 2018, 20, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Merlos-Suarez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Cespedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Munoz, P.; et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, S.; Schmidt, E.M.; Blaj, C.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Multicolor lineage tracing reveals clonal architecture and dynamics in colon cancer. Nat. Commun. 2017, 8, 1406. [Google Scholar] [CrossRef] [PubMed]
- Lenos, K.J.; Miedema, D.M.; Lodestijn, S.C.; Nijman, L.E.; van den Bosch, T.; Romero Ros, X.; Lourenco, F.C.; Lecca, M.C.; van der Heijden, M.; van Neerven, S.M.; et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat. Cell Biol. 2018, 20, 1193–1202. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Gavert, N.; Conacci-Sorrell, M.; Gast, D.; Schneider, A.; Altevogt, P.; Brabletz, T.; Ben-Ze’ev, A. L1, a novel target of beta-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J. Cell Biol. 2005, 168, 633–642. [Google Scholar] [CrossRef]
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A., Jr.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef]
- Baker, A.M.; Graham, T.A.; Elia, G.; Wright, N.A.; Rodriguez-Justo, M. Characterization of LGR5 stem cells in colorectal adenomas and carcinomas. Sci. Rep. 2015, 5, 8654. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fu, L.; Sun, H.; Guo, L.; DuBois, R.N. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology 2015, 149, 1884–1895. [Google Scholar] [CrossRef]
- Thanki, K.; Nicholls, M.E.; Gajjar, A.; Senagore, A.J.; Qiu, S.; Szabo, C.; Hellmich, M.R.; Chao, C. Consensus molecular subtypes of colorectal cancer and their clinical implications. Int. Biol. Biomed. J. 2017, 3, 105–111. [Google Scholar] [PubMed]
- Isella, C.; Brundu, F.; Bellomo, S.E.; Galimi, F.; Zanella, E.; Porporato, R.; Petti, C.; Fiori, A.; Orzan, F.; Senetta, R.; et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat. Commun. 2017, 8, 15107. [Google Scholar] [CrossRef] [PubMed]
- Mouillet-Richard, S.; Laurent-Puig, P. YAP/TAZ signalling in colorectal cancer: Lessons from consensus molecular subtypes. Cancers 2020, 12, 3160. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.M.F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef]
- Isella, C.; Terrasi, A.; Bellomo, S.E.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef]
- Linnekamp, J.F.; Hooff, S.R.V.; Prasetyanti, P.R.; Kandimalla, R.; Buikhuisen, J.Y.; Fessler, E.; Ramesh, P.; Lee, K.; Bochove, G.G.W.; de Jong, J.H.; et al. Consensus molecular subtypes of colorectal cancer are recapitulated in in vitro and in vivo models. Cell Death Differ. 2018, 25, 616–633. [Google Scholar] [CrossRef]
- Albuquerque, C.; Pebre Pereira, L. Wnt signalling-targeted therapy in the cms2 tumour subtype: A new paradigm in CRC Treatment? Adv. Exp. Med. Biol. 2018, 1110, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Stange, D.E.; Engel, F.; Longerich, T.; Koo, B.K.; Koch, M.; Delhomme, N.; Aigner, M.; Toedt, G.; Schirmacher, P.; Lichter, P.; et al. Expression of an ASCL2 related stem cell signature and IGF2 in colorectal cancer liver metastases with 11p15.5 gain. Gut 2010, 59, 1236–1244. [Google Scholar] [CrossRef]
- Joo, M.; Shahsafaei, A.; Odze, R.D. Paneth cell differentiation in colonic epithelial neoplasms: Evidence for the role of the Apc/beta-catenin/Tcf pathway. Hum. Pathol. 2009, 40, 872–880. [Google Scholar] [CrossRef]
- Dieter, S.M.; Ball, C.R.; Hoffmann, C.M.; Nowrouzi, A.; Herbst, F.; Zavidij, O.; Abel, U.; Arens, A.; Weichert, W.; Brand, K.; et al. Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell 2011, 9, 357–365. [Google Scholar] [CrossRef]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef]
- Morelli, M.P.; Overman, M.J.; Dasari, A.; Kazmi, S.M.; Mazard, T.; Vilar, E.; Morris, V.K.; Lee, M.S.; Herron, D.; Eng, C.; et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann. Oncol. 2015, 26, 731–736. [Google Scholar] [CrossRef]
- Giessler, K.M.; Kleinheinz, K.; Huebschmann, D.; Balasubramanian, G.P.; Dubash, T.D.; Dieter, S.M.; Siegl, C.; Herbst, F.; Weber, S.; Hoffmann, C.M.; et al. Genetic subclone architecture of tumor clone-initiating cells in colorectal cancer. J. Exp. Med. 2017, 214, 2073–2088. [Google Scholar] [CrossRef] [PubMed]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Naxerova, K.; Reiter, J.G.; Brachtel, E.; Lennerz, J.K.; van de Wetering, M.; Rowan, A.; Cai, T.; Clevers, H.; Swanton, C.; Nowak, M.A.; et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science 2017, 357, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- O’Rourke, K.P.; Loizou, E.; Livshits, G.; Schatoff, E.M.; Baslan, T.; Manchado, E.; Simon, J.; Romesser, P.B.; Leach, B.; Han, T.; et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 2017, 35, 577–582. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y.; et al. Combined mutation of Apc, Kras, and Tgfbr2 effectively drives metastasis of intestinal cancer. Cancer Res. 2018, 78, 1334–1346. [Google Scholar] [CrossRef]
- Fumagalli, A.; Drost, J.; Suijkerbuijk, S.J.; van Boxtel, R.; de Ligt, J.; Offerhaus, G.J.; Begthel, H.; Beerling, E.; Tan, E.H.; Sansom, O.J.; et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc. Natl. Acad. Sci. USA 2017, 114, E2357–E2364. [Google Scholar] [CrossRef]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef]
- Kleeman, S.O.; Koelzer, V.H.; Jones, H.J.; Vazquez, E.G.; Davis, H.; East, J.E.; Arnold, R.; Koppens, M.A.; Blake, A.; Domingo, E.; et al. Exploiting differential Wnt target gene expression to generate a molecular biomarker for colorectal cancer stratification. Gut 2020, 69, 1092–1103. [Google Scholar] [CrossRef]
- Han, T.; Schatoff, E.M.; Murphy, C.; Zafra, M.P.; Wilkinson, J.E.; Elemento, O.; Dow, L.E. R-Spondin chromosome rearrangements drive Wnt-dependent tumour initiation and maintenance in the intestine. Nat. Commun. 2017, 8, 15945. [Google Scholar] [CrossRef]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Gala, M.K.; Mizukami, Y.; Le, L.P.; Moriichi, K.; Austin, T.; Yamamoto, M.; Lauwers, G.Y.; Bardeesy, N.; Chung, D.C. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 2014, 146, 520–529. [Google Scholar] [CrossRef]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef]
- Hao, H.X.; Jiang, X.; Cong, F. Control of Wnt receptor turnover by R-spondin-ZNRF3/RNF43 signaling module and its dysregulation in cancer. Cancers 2016, 8, 54. [Google Scholar] [CrossRef]
- Storm, E.E.; Durinck, S.; de Sousa e Melo, F.; Tremayne, J.; Kljavin, N.; Tan, C.; Ye, X.; Chiu, C.; Pham, T.; Hongo, J.A.; et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature 2016, 529, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef]
- Fenderico, N.; van Scherpenzeel, R.C.; Goldflam, M.; Proverbio, D.; Jordens, I.; Kralj, T.; Stryeck, S.; Bass, T.Z.; Hermans, G.; Ullman, C.; et al. Anti-LRP5/6 VHHs promote differentiation of Wnt-hypersensitive intestinal stem cells. Nat. Commun. 2019, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; McDonald, M.J.; Foxa, G.E.; Diegel, C.R.; Williams, B.O.; Virshup, D.M. Bone loss from Wnt inhibition mitigated by concurrent alendronate therapy. Bone Res. 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.M.F.; Colak, S.; Buikhuisen, J.; Koster, J.; Cameron, K.; de Jong, J.H.; Tuynman, J.B.; Prasetyanti, P.R.; Fessler, E.; van den Bergh, S.P.; et al. Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 2011, 9, 476–485. [Google Scholar] [CrossRef]
- Varnat, F.; Siegl-Cachedenier, I.; Malerba, M.; Gervaz, P.; Ruiz i Altaba, A. Loss of WNT-TCF addiction and enhancement of HH-GLI1 signalling define the metastatic transition of human colon carcinomas. EMBO Mol. Med. 2010, 2, 440–457. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Geng, L.; Wang, D.; Yi, H.; Talmon, G.; Wang, J. R-Spondin1/LGR5 activates TGFbeta signaling and suppresses colon cancer metastasis. Cancer Res. 2017, 77, 6589–6602. [Google Scholar] [CrossRef]
- Morgan, R.G.; Mortensson, E.; Williams, A.C. Targeting LGR5 in colorectal cancer: Therapeutic gold or too plastic? Br. J. Cancer 2018, 118, 1410–1418. [Google Scholar] [CrossRef]
- Seth, C.; Ruiz i Altaba, A. Metastases and colon cancer tumor growth display divergent responses to modulation of canonical WNT signaling. PLoS ONE 2016, 11, e0150697. [Google Scholar] [CrossRef]
- Fre, S.; Pallavi, S.K.; Huyghe, M.; Lae, M.; Janssen, K.P.; Robine, S.; Artavanis-Tsakonas, S.; Louvard, D. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 6309–6314. [Google Scholar] [CrossRef]
- Reedijk, M.; Odorcic, S.; Zhang, H.; Chetty, R.; Tennert, C.; Dickson, B.C.; Lockwood, G.; Gallinger, S.; Egan, S.E. Activation of Notch signaling in human colon adenocarcinoma. Int. J. Oncol. 2008, 33, 1223–1229. [Google Scholar] [PubMed]
- Babaei-Jadidi, R.; Li, N.; Saadeddin, A.; Spencer-Dene, B.; Jandke, A.; Muhammad, B.; Ibrahim, E.E.; Muraleedharan, R.; Abuzinadah, M.; Davis, H.; et al. FBXW7 influences murine intestinal homeostasis and cancer, targeting Notch, Jun, and DEK for degradation. J. Exp. Med. 2011, 208, 295–312. [Google Scholar] [CrossRef]
- Meng, R.D.; Shelton, C.C.; Li, Y.M.; Qin, L.X.; Notterman, D.; Paty, P.B.; Schwartz, G.K. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009, 69, 573–582. [Google Scholar] [CrossRef]
- Sonoshita, M.; Aoki, M.; Fuwa, H.; Aoki, K.; Hosogi, H.; Sakai, Y.; Hashida, H.; Takabayashi, A.; Sasaki, M.; Robine, S.; et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell 2011, 19, 125–137. [Google Scholar] [CrossRef]
- Mourao, L.; Jacquemin, G.; Huyghe, M.; Nawrocki, W.J.; Menssouri, N.; Servant, N.; Fre, S. Lineage tracing of Notch1-expressing cells in intestinal tumours reveals a distinct population of cancer stem cells. Sci. Rep. 2019, 9, 888. [Google Scholar] [CrossRef]
- Srinivasan, T.; Walters, J.; Bu, P.; Than, E.B.; Tung, K.L.; Chen, K.Y.; Panarelli, N.; Milsom, J.; Augenlicht, L.; Lipkin, S.M.; et al. NOTCH signaling regulates asymmetric cell fate of fast- and slow-cycling colon cancer-initiating cells. Cancer Res. 2016, 76, 3411–3421. [Google Scholar] [CrossRef]
- Schmidt, E.M.; Lamprecht, S.; Blaj, C.; Schaaf, C.; Krebs, S.; Blum, H.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Targeting tumor cell plasticity by combined inhibition of NOTCH and MAPK signaling in colon cancer. J. Exp. Med. 2018, 215, 1693–1708. [Google Scholar] [CrossRef]
- Nakata, T.; Shimizu, H.; Nagata, S.; Ito, G.; Fujii, S.; Suzuki, K.; Kawamoto, A.; Ishibashi, F.; Kuno, R.; Anzai, S.; et al. Indispensable role of Notch ligand-dependent signaling in the proliferation and stem cell niche maintenance of APC-deficient intestinal tumors. Biochem. Biophys. Res. Commun. 2017, 482, 1296–1303. [Google Scholar] [CrossRef]
- Goto, N.; Ueo, T.; Fukuda, A.; Kawada, K.; Sakai, Y.; Miyoshi, H.; Taketo, M.M.; Chiba, T.; Seno, H. Distinct roles of HES1 in normal stem cells and tumor stem-like cells of the intestine. Cancer Res. 2017, 77, 3442–3454. [Google Scholar] [CrossRef] [PubMed]
- Dunkin, D.; Iuga, A.C.; Mimouna, S.; Harris, C.L.; Haure-Mirande, J.V.; Bozec, D.; Yeretssian, G.; Dahan, S. Intestinal epithelial Notch-1 protects from colorectal mucinous adenocarcinoma. Oncotarget 2018, 9, 33536–33548. [Google Scholar] [CrossRef][Green Version]
- Houlston, R.; Bevan, S.; Williams, A.; Young, J.; Dunlop, M.; Rozen, P.; Eng, C.; Markie, D.; Woodford-Richens, K.; Rodriguez-Bigas, M.A.; et al. Mutations in DPC4 (SMAD4) cause juvenile polyposis syndrome, but only account for a minority of cases. Hum. Mol. Genet. 1998, 7, 1907–1912. [Google Scholar] [CrossRef]
- Howe, J.R.; Roth, S.; Ringold, J.C.; Summers, R.W.; Jarvinen, H.J.; Sistonen, P.; Tomlinson, I.P.; Houlston, R.S.; Bevan, S.; Mitros, F.A.; et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998, 280, 1086–1088. [Google Scholar] [CrossRef]
- Lubbe, S.J.; Pittman, A.M.; Matijssen, C.; Twiss, P.; Olver, B.; Lloyd, A.; Qureshi, M.; Brown, N.; Nye, E.; Stamp, G.; et al. Evaluation of germline BMP4 mutation as a cause of colorectal cancer. Hum. Mutat. 2011, 32, E1928–E1938. [Google Scholar] [CrossRef] [PubMed]
- Cheah, P.Y.; Wong, Y.H.; Chau, Y.P.; Loi, C.; Lim, K.H.; Lim, J.F.; Koh, P.K.; Eu, K.W. Germline bone morphogenesis protein receptor 1A mutation causes colorectal tumorigenesis in hereditary mixed polyposis syndrome. Am. J. Gastroenterol. 2009, 104, 3027–3033. [Google Scholar] [CrossRef]
- Kodach, L.L.; Wiercinska, E.; de Miranda, N.F.; Bleuming, S.A.; Musler, A.R.; Peppelenbosch, M.P.; Dekker, E.; van den Brink, G.R.; van Noesel, C.J.; Morreau, H.; et al. The bone morphogenetic protein pathway is inactivated in the majority of sporadic colorectal cancers. Gastroenterology 2008, 134, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Kodach, L.L.; Bleuming, S.A.; Musler, A.R.; Peppelenbosch, M.P.; Hommes, D.W.; van den Brink, G.R.; van Noesel, C.J.; Offerhaus, G.J.; Hardwick, J.C. The bone morphogenetic protein pathway is active in human colon adenomas and inactivated in colorectal cancer. Cancer 2008, 112, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.; Carvajal-Carmona, L.G.; Dobbins, S.E.; Tenesa, A.; Jones, A.M.; Howarth, K.; Palles, C.; Broderick, P.; Jaeger, E.E.; Farrington, S.; et al. Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet. 2011, 7, e1002105. [Google Scholar] [CrossRef]
- Jaeger, E.; Leedham, S.; Lewis, A.; Segditsas, S.; Becker, M.; Cuadrado, P.R.; Davis, H.; Kaur, K.; Heinimann, K.; Howarth, K.; et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat. Genet. 2012, 44, 699–703. [Google Scholar] [CrossRef]
- Lewis, A.; Freeman-Mills, L.; de la Calle-Mustienes, E.; Giraldez-Perez, R.M.; Davis, H.; Jaeger, E.; Becker, M.; Hubner, N.C.; Nguyen, L.N.; Zeron-Medina, J.; et al. A polymorphic enhancer near GREM1 influences bowel cancer risk through differential CDX2 and TCF7L2 binding. Cell Rep. 2014, 8, 983–990. [Google Scholar] [CrossRef]
- Kodach, L.L.; Jacobs, R.J.; Voorneveld, P.W.; Wildenberg, M.E.; Verspaget, H.W.; van Wezel, T.; Morreau, H.; Hommes, D.W.; Peppelenbosch, M.P.; van den Brink, G.R.; et al. Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness’ via the bone morphogenetic protein pathway. Gut 2011, 60, 1544–1553. [Google Scholar] [CrossRef]
- Lombardo, Y.; Scopelliti, A.; Cammareri, P.; Todaro, M.; Iovino, F.; Ricci-Vitiani, L.; Gulotta, G.; Dieli, F.; de Maria, R.; Stassi, G. Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology 2011, 140, 297–309. [Google Scholar] [CrossRef]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; van Noesel, C.J.; Peppelenbosch, M.P.; Korkmaz, K.S.; Molendijk, I.; Dekker, E.; Morreau, H.; van Pelt, G.W.; et al. The BMP pathway either enhances or inhibits the Wnt pathway depending on the SMAD4 and p53 status in CRC. Br. J. Cancer 2015, 112, 122–130. [Google Scholar] [CrossRef]
- Freeman, T.J.; Smith, J.J.; Chen, X.; Washington, M.K.; Roland, J.T.; Means, A.L.; Eschrich, S.A.; Yeatman, T.J.; Deane, N.G.; Beauchamp, R.D. Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of beta-catenin. Gastroenterology 2012, 142, 562–571. [Google Scholar] [CrossRef]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; Liv, N.; Zonnevylle, A.C.; Hoogenboom, J.P.; Biemond, I.; Verspaget, H.W.; Hommes, D.W.; de Rooij, K.; et al. Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology 2014, 147, 196–208. [Google Scholar] [CrossRef]
- Fessler, E.; Drost, J.; van Hooff, S.R.; Linnekamp, J.F.; Wang, X.; Jansen, M.; De Sousa, E.M.F.; Prasetyanti, P.R.; JE, I.J.; Franitza, M.; et al. TGFbeta signaling directs serrated adenomas to the mesenchymal colorectal cancer subtype. EMBO Mol. Med. 2016, 8, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Irshad, S.; Bansal, M.; Guarnieri, P.; Davis, H.; Al Haj Zen, A.; Baran, B.; Pinna, C.M.A.; Rahman, H.; Biswas, S.; Bardella, C.; et al. Bone morphogenetic protein and Notch signalling crosstalk in poor-prognosis, mesenchymal-subtype colorectal cancer. J. Pathol. 2017, 242, 178–192. [Google Scholar] [CrossRef]
- Jackstadt, R.; van Hooff, S.R.; Leach, J.D.; Cortes-Lavaud, X.; Lohuis, J.O.; Ridgway, R.A.; Wouters, V.M.; Roper, J.; Kendall, T.J.; Roxburgh, C.S.; et al. Epithelial NOTCH signaling rewires the tumor microenvironment of colorectal cancer to drive poor-prognosis subtypes and metastasis. Cancer Cell 2019, 36, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Nicolas, A.; Petrocelli, V.; Pesic, M.; Mahmoud, A.; Michels, B.E.; Etlioglu, E.; Yepes, D.; Haupl, B.; Ziegler, P.K.; et al. AKT-dependent NOTCH3 activation drives tumor progression in a model of mesenchymal colorectal cancer. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, N.; Zheng, Y.; de Wilde, R.F.; Maitra, A.; Pan, D. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010, 24, 2383–2388. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103. [Google Scholar] [CrossRef]
- Goto, N.; Fukuda, A.; Yamaga, Y.; Yoshikawa, T.; Maruno, T.; Maekawa, H.; Inamoto, S.; Kawada, K.; Sakai, Y.; Miyoshi, H.; et al. Lineage tracing and targeting of IL17RB(+) tuft cell-like human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2019, 116, 12996–13005. [Google Scholar] [CrossRef]
- Gong, X.; Azhdarinia, A.; Ghosh, S.C.; Xiong, W.; An, Z.; Liu, Q.; Carmon, K.S. LGR5-targeted antibody-drug conjugate eradicates gastrointestinal tumors and prevents recurrence. Mol. Cancer Ther. 2016, 15, 1580–1590. [Google Scholar] [CrossRef]
- Trumpi, K.; Ubink, I.; Trinh, A.; Djafarihamedani, M.; Jongen, J.M.; Govaert, K.M.; Elias, S.G.; van Hooff, S.R.; Medema, J.P.; Lacle, M.M.; et al. Neoadjuvant chemotherapy affects molecular classification of colorectal tumors. Oncogenesis 2017, 6, e357. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sphyris, N.; Hodder, M.C.; Sansom, O.J. Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell. Cancers 2021, 13, 1000. https://doi.org/10.3390/cancers13051000
Sphyris N, Hodder MC, Sansom OJ. Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell. Cancers. 2021; 13(5):1000. https://doi.org/10.3390/cancers13051000
Chicago/Turabian StyleSphyris, Nathalie, Michael C. Hodder, and Owen J. Sansom. 2021. "Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell" Cancers 13, no. 5: 1000. https://doi.org/10.3390/cancers13051000
APA StyleSphyris, N., Hodder, M. C., & Sansom, O. J. (2021). Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell. Cancers, 13(5), 1000. https://doi.org/10.3390/cancers13051000