
Plasticity in Colorectal Cancer: Why Cancer Cells Differentiate


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
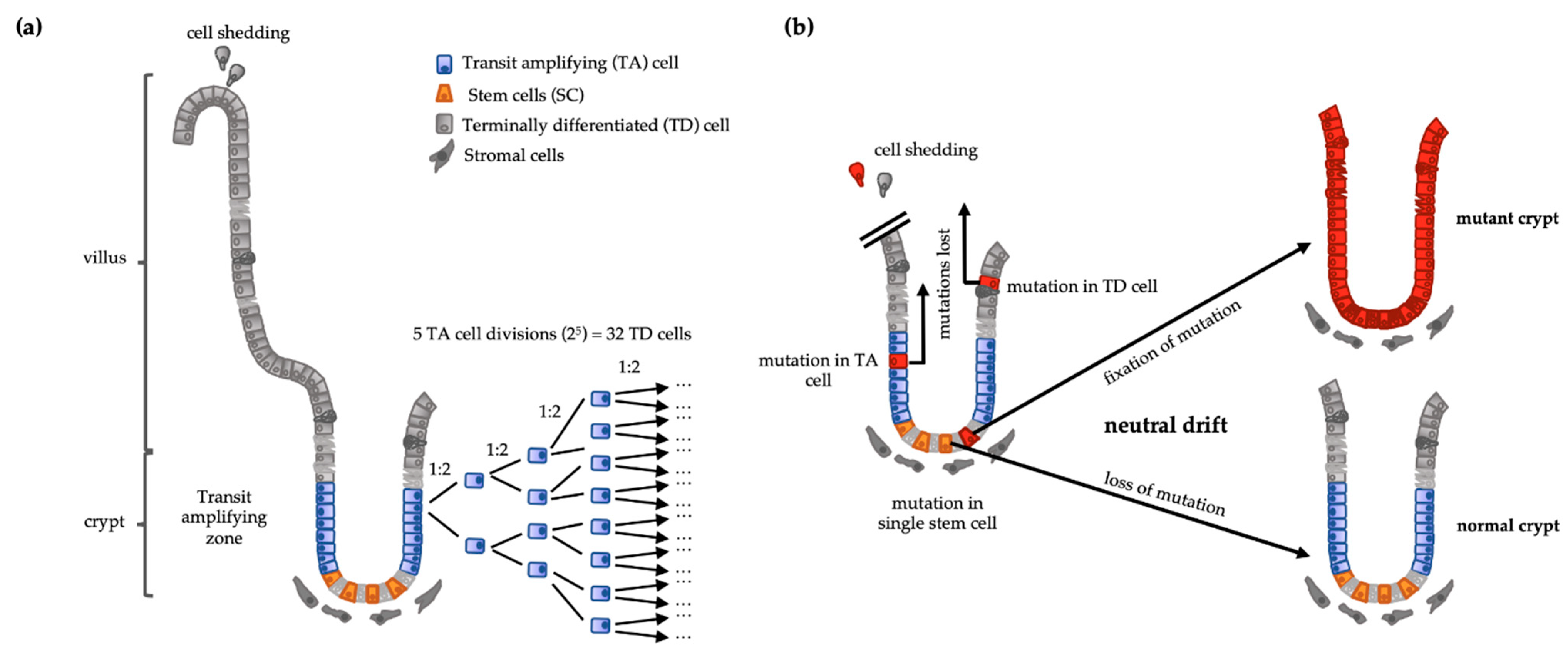
2. Cell Hierarchy Is a Tumor-Suppressor Mechanism
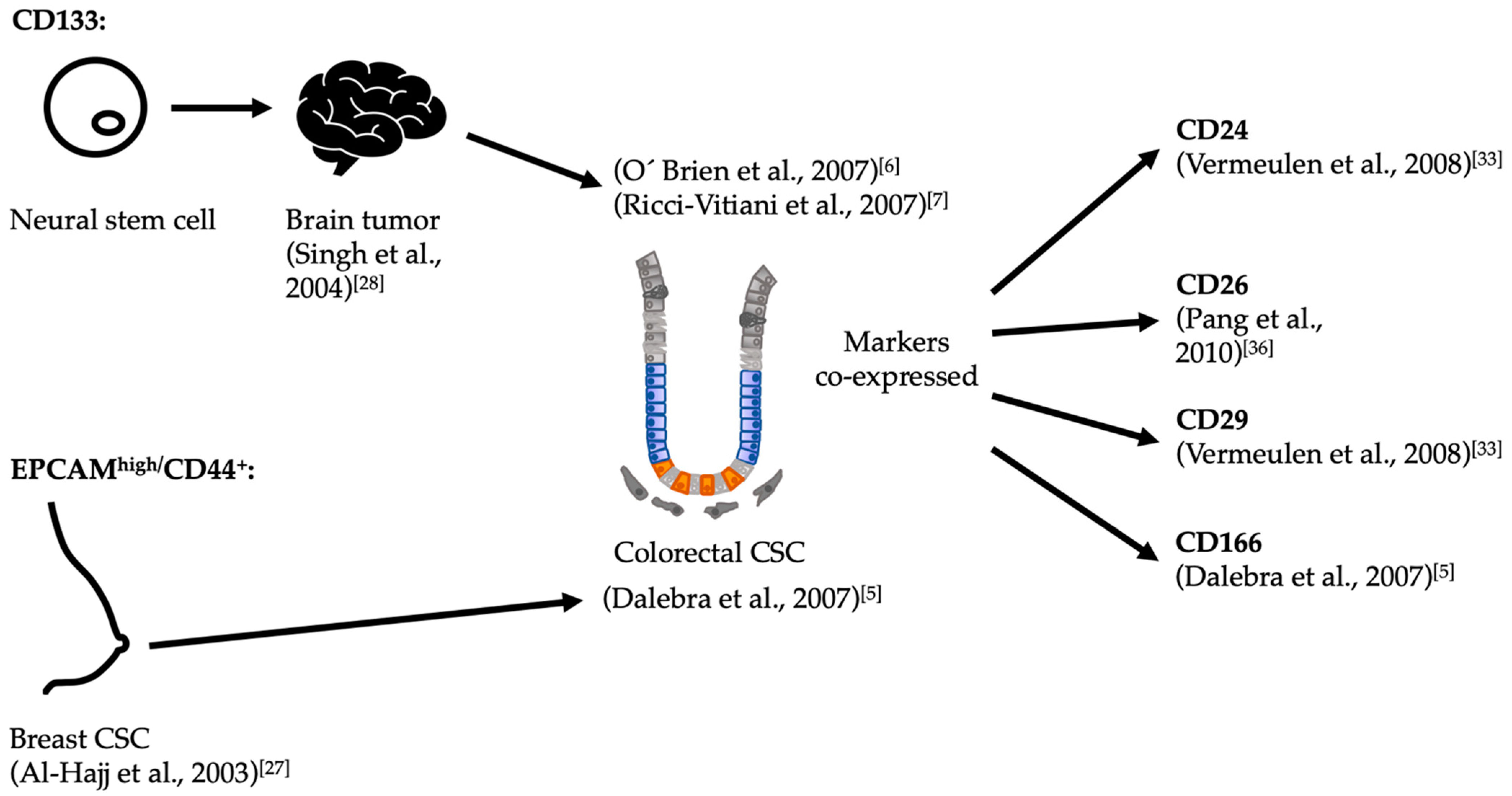
3. Cancer Stem Cells in Colorectal Tumors
3.1. Serial Transplantation and CSC Markers
3.2. Lineage Tracing and Organoids: Evidence for Cancer Stem Cells
3.3. Tumor Organoids
4. Contribution of Differentiated Cancer Cells to Tumor Progression
4.1. Cancer Cell Plasticity
4.1.1. Signaling Pathways Involved in Cancer Cell Plasticity
4.1.2. Plasticity to Overcome Metastatic Selective Bottlenecks: Differentiation States as an Adaptive Trait
4.2. Selective Advantages of Cancer Cell Phenotypic Heterogeneity
4.2.1. Variable Selection of Clones with Distinct Phenotypic Traits
4.2.2. Clonal Cooperation
4.2.3. The Role of Hierarchical Cancer Cells’ Organization in Therapeutic Resistance
5. Conclusions
Funding
Conflicts of Interest
References
- O’Brien, C.W.; Moorey, S. Outlook and adaptation in advanced cancer: A systematic review. Psychooncology 2010, 19, 1239–1249. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell. 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Dalerba, P.; Dylla, S.J.; Park, I.-K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2006, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer Stem Cells: Impact, Heterogeneity, and Uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar] [CrossRef]
- Almendro, V.; Marusyk, A.; Polyak, K. Cellular Heterogeneity and Molecular Evolution in Cancer. Annu. Rev. Pathol. 2013, 8, 277–302. [Google Scholar] [CrossRef]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage Tracing Reveals Lgr5+ Stem Cell Activity in Mouse Intestinal Adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef]
- Cortina, C.; Turon, G.; Stork, D.; Hernando-Momblona, X.; Sevillano, M.; Aguilera, M.; Tosi, S.; Merlos-Suárez, A.; Attolini, C.S.-O.; Sancho, E.; et al. A genome editing approach to study cancer stem cells in human tumors. EMBO Mol. Med. 2017, 9, 869–879. [Google Scholar] [CrossRef]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.N.S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of Lgr5+human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef]
- Fumagalli, A.; Oost, K.C.; Kester, L.; Morgner, J.; Bornes, L.; Bruens, L.; Spaargaren, L.; Azkanaz, M.; Schelfhorst, T.; Beerling, E.; et al. Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell. 2020, 26, 569–578.e7. [Google Scholar] [CrossRef]
- Merlos-Suárez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Céspedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Muñoz, P.; et al. The Intestinal Stem Cell Signature Identifies Colorectal Cancer Stem Cells and Predicts Disease Relapse. Cell Stem Cell. 2011, 8, 511–524. [Google Scholar] [CrossRef]
- Kitao, H.; Iimori, M.; Kataoka, Y.; Wakasa, T.; Tokunaga, E.; Saeki, H.; Oki, E.; Maehara, Y. DNA replication stress and cancer chemotherapy. Cancer Sci. 2018, 109, 264–271. [Google Scholar] [CrossRef]
- Lange, S.S.; Takata, K.-I.; Wood, R.D. DNA polymerases and cancer. Nat. Rev. Cancer 2011, 11, 96–110. [Google Scholar] [CrossRef]
- Umar, A.; Kunkel, T.A. DNA-replication Fidelity, Mismatch Repair and Genome Instability in Cancer Cells. Eur. J. Biochem. 1996, 238, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Pepper, J.W.; Sprouffske, K.; Maley, C.C. Animal Cell Differentiation Patterns Suppress Somatic Evolution. PLoS Comput. Biol. 2007, 3, e250. [Google Scholar] [CrossRef]
- Snippert, H.J.; Van Der Flier, L.G.; Sato, T.; Van Es, J.H.; Born, M.V.D.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; Van Rheenen, J.; Simons, B.D.; et al. Intestinal Crypt Homeostasis Results from Neutral Competition between Symmetrically Dividing Lgr5 Stem Cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef]
- Lopez-Garcia, C.; Klein, A.M.; Simons, B.D.; Winton, D.J. Intestinal Stem Cell Replacement Follows a Pattern of Neutral Drift. Science 2010, 330, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.M.; Simons, B.D. Universal patterns of stem cell fate in cycling adult tissues. Development 2011, 138, 3103–3111. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer Stem Cells and Self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef] [PubMed]
- Sprouffske, K.; Pepper, J.W.; Maley, C.C. Accurate Reconstruction of the Temporal Order of Mutations in Neoplastic Progression. Cancer Prev. Res. 2011, 4, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.S.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.-C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.-M.; Nephew, K.P. Identification and Characterization of Ovarian Cancer-Initiating Cells from Primary Human Tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer Stem Cells—Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Quintana, E.; Shackleton, M.; Foster, H.R.; Fullen, D.R.; Sabel, M.S.; Johnson, T.M.; Morrison, S.J. Phenotypic Heterogeneity among Tumorigenic Melanoma Cells from Patients that Is Reversible and Not Hierarchically Organized. Cancer Cell 2010, 18, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Todaro, M.; Melo, F.D.S.E.; Sprick, M.R.; Kemper, K.; Alea, M.P.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Sprick, M.R.; De Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G.; et al. The AC133 Epitope, but not the CD133 Protein, Is Lost upon Cancer Stem Cell Differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.B.; Blakely, K.M.; Williams, R.A.; Penttilä, P.-A.; Shukalyuk, A.I.; Osman, K.T.; Kasimer, D.; Ketela, T.; Moffat, J. CD133 Protein N-Glycosylation Processing Contributes to Cell Surface Recognition of the Primitive Cell Marker AC133 Epitope. J. Biol. Chem. 2011, 286, 41046–41056. [Google Scholar] [CrossRef]
- Pang, R.; Law, W.L.; Chu, A.C.; Poon, J.T.; Lam, C.S.; Chow, A.K.; Ng, L.; Cheung, L.W.; Lan, X.R.; Lan, H.Y.; et al. A Subpopulation of CD26+ Cancer Stem Cells with Metastatic Capacity in Human Colorectal Cancer. Cell Stem Cell. 2010, 6, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Morath, I.; Hartmann, T.; Orian-Rousseau, V. CD44: More than a mere stem cell marker. Int. J. Biochem. Cell Biol. 2016, 81, 166–173. [Google Scholar] [CrossRef]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell Stem Cell. 2014, 14, 342–356. [Google Scholar] [CrossRef]
- Medema, J.P.; Vermeulen, L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 2011, 474, 318–326. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from Hereditary Colorectal Cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Nathke, I.S.; et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Groden, J.; Thliveris, A.; Samowitz, W.; Carlson, M.; Gelbert, L.; Albertsen, H.; Joslyn, G.; Stevens, J.; Spirio, L.; Robertson, M.; et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66, 589–600. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Nilbert, M.C.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hamilton, S.R.; Hedge, P.; Markham, A.; et al. Identification of a gene located at chromosome 5q21 that is mutated in colorectal cancers. Science 1991, 251, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable -catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Melo, F.D.S.E.; Van Der Heijden, M.A.; Cameron, K.; De Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.M.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Tian, H.; Biehs, B.; Warming, S.; Leong, K.G.; Rangell, L.; Klein, O.D.; De Sauvage, F.J. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 2011, 478, 255–259. [Google Scholar] [CrossRef]
- Melo, F.D.S.E.; Colak, S.; Buikhuisen, J.; Koster, J.; Cameron, K.; de Jong, J.H.; Tuynman, J.B.; Prasetyanti, P.R.; Fessler, E.; Bergh, S.P.V.D.; et al. Methylation of Cancer-Stem-Cell-Associated Wnt Target Genes Predicts Poor Prognosis in Colorectal Cancer Patients. Cell Stem Cell. 2011, 9, 476–485. [Google Scholar] [CrossRef]
- Wang, Y.; Chiang, I.-L.; Ohara, T.E.; Fujii, S.; Cheng, J.; Muegge, B.D.; Heul, A.V.; Han, N.D.; Lu, Q.; Xiong, S.; et al. Long-Term Culture Captures Injury-Repair Cycles of Colonic Stem Cells. Cell 2019, 179, 1144–1159.e15. [Google Scholar] [CrossRef]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.M.; Olpe, C.; Hoyle, A.; Thorsen, A.-S.; Rus, T.; Colombé, M.; Brunton-Sim, R.; Kemp, R.; Marks, K.; Quirke, P.; et al. Fixation and Spread of Somatic Mutations in Adult Human Colonic Epithelium. Cell Stem Cell. 2018, 22, 909–918.e8. [Google Scholar] [CrossRef] [PubMed]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Sato, T.; Van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; Van De Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2010, 470, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Van De Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; Van Houdt, W.; Van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell. 2016, 18, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Wu, C.; O’Rourke, K.P.; Szeglin, B.C.; Zheng, Y.; Sauvé, C.-E.G.; Adileh, M.; Wasserman, I.; Marco, M.R.; Kim, A.S.; et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 2019, 25, 1607–1614. [Google Scholar] [CrossRef]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell. 2013, 13, 653–658. [Google Scholar] [CrossRef]
- Drost, J.; Van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; Van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9—Mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Kreso, A.; O’Brien, C.A.; Van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.K.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable Clonal Repopulation Dynamics Influence Chemotherapy Response in Colorectal Cancer. Science 2012, 339, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Fair, K.L.; Colquhoun, J.; Hannan, N.R.F. Intestinal organoids for modelling intestinal development and disease. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170217. [Google Scholar] [CrossRef]
- Almeqdadi, M.; Mana, M.D.; Roper, J.; Yilmaz, Ö.H. Gut organoids: Mini-tissues in culture to study intestinal physiology and disease. Am. J. Physiol. Cell Physiol. [CrossRef]
- Van Der Heijden, M.; Vermeulen, L. Stem Cells: All that Is Solid Melts into Air. Cell Stem Cell. 2017, 21, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Jain, R.; Li, D.; Li, L.; Lu, M.M.; Epstein, J.A. Lgr5 Identifies Progenitor Cells Capable of Taste Bud Regeneration after Injury. PLoS ONE 2013, 8, e66314. [Google Scholar] [CrossRef]
- Buczacki, S.J.A.; Zecchini, H.I.; Nicholson, A.M.; Russell, R.; Vermeulen, L.; Kemp, R.; Winton, D.J. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 2013, 495, 65–69. [Google Scholar] [CrossRef]
- Asfaha, S.; Hayakawa, Y.; Muley, A.; Stokes, S.; Graham, T.A.; Ericksen, R.E.; Westphalen, C.B.; Von Burstin, J.; Mastracci, T.L.; Worthley, D.L.; et al. Krt19+/Lgr5− Cells Are Radioresistant Cancer-Initiating Stem Cells in the Colon and Intestine. Cell Stem Cell. 2015, 16, 627–638. [Google Scholar] [CrossRef]
- Tetteh, P.W.; Basak, O.; Farin, H.F.; Wiebrands, K.; Kretzschmar, K.; Begthel, H.; Born, M.V.D.; Korving, J.; de Sauvage, F.; van Es, J.H.; et al. Replacement of Lost Lgr5-Positive Stem Cells through Plasticity of Their Enterocyte-Lineage Daughters. Cell Stem Cell. 2016, 18, 203–213. [Google Scholar] [CrossRef]
- Yan, K.S.; Gevaert, O.; Zheng, G.X.; Anchang, B.; Probert, C.S.; Larkin, K.A.; Davies, P.S.; Cheng, Z.-F.; Kaddis, J.S.; Han, A.; et al. Intestinal Enteroendocrine Lineage Cells Possess Homeostatic and Injury-Inducible Stem Cell Activity. Cell Stem Cell. 2017, 21, 78–90.e6. [Google Scholar] [CrossRef]
- Schmitt, M.; Schewe, M.; Sacchetti, A.; Feijtel, D.; Van De Geer, W.S.; Teeuwssen, M.; Sleddens, H.F.; Joosten, R.; Van Royen, M.E.; Van De Werken, H.J.; et al. Paneth Cells Respond to Inflammation and Contribute to Tissue Regeneration by Acquiring Stem-like Features through SCF/c-Kit Signaling. Cell Rep. 2018, 24, 2312–2328.e7. [Google Scholar] [CrossRef]
- Lenos, K.J.; Miedema, D.M.; Lodestijn, S.C.; Nijman, L.E.; Bosch, T.V.D.; Ros, X.R.; Lourenço, F.C.; Lecca, M.C.; Van Der Heijden, M.; Van Neerven, S.M.; et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat. Cell Biol. 2018, 20, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, M.; Miedema, D.M.; Waclaw, B.; Veenstra, V.L.; Lecca, M.C.; Nijman, L.E.; Van Dijk, E.; Van Neerven, S.M.; Lodestijn, S.C.; Lenos, K.J.; et al. Spatiotemporal regulation of clonogenicity in colorectal cancer xenografts. Proc. Natl. Acad. Sci. USA 2019, 116, 6140–6145. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Sleeman, J. CD44 is a Multidomain Signaling Platform that Integrates Extracellular Matrix Cues with Growth Factor and Cytokine Signals. Adv. Cancer Res. 2014, 123, 231–254. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Metzger, M.; Gradl, D.; Davidson, G.; Orian-Rousseau, V. CD44 functions in Wnt signaling by regulating LRP6 localization and activation. Cell Death Differ. 2014, 22, 677–689. [Google Scholar] [CrossRef]
- Orian-Rousseau, V.; Chen, L.; Sleeman, J.P.; Herrlich, P.; Ponta, H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 2002, 16, 3074–3086. [Google Scholar] [CrossRef] [PubMed]
- Matzke, A.; Herrlich, P.; Ponta, H.; Orian-Rousseau, V. A Five-Amino-Acid Peptide Blocks Met- and Ron-Dependent Cell Migration. Cancer Res. 2005, 65, 6105–6110. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hasenauer, S.; Malinger, D.; Koschut, D.; Pace, G.; Matzke, A.; Von Au, A.; Orian-Rousseau, V. Internalization of Met Requires the Co-Receptor CD44v6 and Its Link to ERM Proteins. PLoS ONE 2013, 8, e62357. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Zimberlin, C.D.; Bots, M.; Vermeulen, L.; Melo, F.D.S.E.; Medema, J.P. Regulation of stem cell self-renewal and differentiation by Wnt and Notch are conserved throughout the adenoma-carcinoma sequence in the colon. Mol. Cancer 2013, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Fender, A.W.; Nutter, J.M.; Fitzgerald, T.L.; Bertrand, F.E.; Sigounas, G. Notch-1 Promotes Stemness and Epithelial to Mesenchymal Transition in Colorectal Cancer. J. Cell. Biochem. 2015, 116, 2517–2527. [Google Scholar] [CrossRef]
- Sonoshita, M.; Aoki, M.; Fuwa, H.; Aoki, K.; Hosogi, H.; Sakai, Y.; Hashida, H.; Takabayashi, A.; Sasaki, M.; Robine, S.; et al. Suppression of Colon Cancer Metastasis by Aes through Inhibition of Notch Signaling. Cancer Cell 2011, 19, 125–137. [Google Scholar] [CrossRef]
- Jackstadt, R.; van Hooff, S.R.; Leach, J.D.; Cortes-Lavaud, X.; Lohuis, J.O.; Ridgway, R.A.; Wouters, V.M.; Roper, J.; Kendall, T.J.; Roxburgh, C.S.; et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell 2019, 36, 319–336.e7. [Google Scholar] [CrossRef]
- Serra, D.; Mayr, U.; Boni, A.; Lukonin, I.; Rempfler, M.; Meylan, L.C.; Stadler, M.B.; Strnad, P.; Papasaikas, P.; Vischi, D.; et al. Self-organization and symmetry breaking in intestinal organoid development. Nature 2019, 569, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Lukonin, I.; Serra, D.; Meylan, L.C.; Volkmann, K.; Baaten, J.; Zhao, R.; Meeusen, S.; Colman, K.; Maurer, F.; Stadler, M.B.; et al. Phenotypic landscape of intestinal organoid regeneration. Nature 2020, 586, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Xiol, J.; Dill, M.T.; Yuan, W.-C.; Panero, R.; Roper, J.; Osorio, F.G.; Maglic, D.; Li, Q.; Gurung, B.; et al. Regenerative Reprogramming of the Intestinal Stem Cell State via Hippo Signaling Suppresses Metastatic Colorectal Cancer. Cell Stem Cell. 2020, 27, 590–604.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Häussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Luzzi, K.J.; Macdonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep Nature of Metastatic Inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Dieter, S.M.; Ball, C.R.; Hoffmann, C.M.; Nowrouzi, A.; Herbst, F.; Zavidij, O.; Abel, U.; Arens, A.; Weichert, W.; Brand, K.; et al. Distinct Types of Tumor-Initiating Cells Form Human Colon Cancer Tumors and Metastases. Cell Stem Cell. 2011, 9, 357–365. [Google Scholar] [CrossRef]
- Cleary, A.S.; Leonard, T.L.; Gestl, S.A.; Gunther, E.J. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 2014, 508, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Tabassum, D.P.; Altrock, P.M.; Almendro, V.; Michor, F.; Polyak, K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014, 514, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Luskin, M.R.; Murakami, M.A.; Manalis, S.R.; Weinstock, D.M. Targeting minimal residual disease: A path to cure? Nat. Rev. Cancer 2018, 18, 255–263. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Shiokawa, D.; Sakai, H.; Ohata, H.; Miyazaki, T.; Kanda, Y.; Sekine, S.; Narushima, D.; Hosokawa, M.; Kato, M.; Suzuki, Y.; et al. Slow-Cycling Cancer Stem Cells Regulate Progression and Chemoresistance in Colon Cancer. Cancer Res. 2020, 80, 4451–4464. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, T.S.R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Shaked, Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer 2019, 19, 667–685. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walter, R.J.; Sonnentag, S.J.; Orian-Rousseau, V.; Munoz-Sagredo, L. Plasticity in Colorectal Cancer: Why Cancer Cells Differentiate. Cancers 2021, 13, 918. https://doi.org/10.3390/cancers13040918
Walter RJ, Sonnentag SJ, Orian-Rousseau V, Munoz-Sagredo L. Plasticity in Colorectal Cancer: Why Cancer Cells Differentiate. Cancers. 2021; 13(4):918. https://doi.org/10.3390/cancers13040918
Chicago/Turabian StyleWalter, Romina Judith, Steffen Joachim Sonnentag, Véronique Orian-Rousseau, and Leonel Munoz-Sagredo. 2021. "Plasticity in Colorectal Cancer: Why Cancer Cells Differentiate" Cancers 13, no. 4: 918. https://doi.org/10.3390/cancers13040918
APA StyleWalter, R. J., Sonnentag, S. J., Orian-Rousseau, V., & Munoz-Sagredo, L. (2021). Plasticity in Colorectal Cancer: Why Cancer Cells Differentiate. Cancers, 13(4), 918. https://doi.org/10.3390/cancers13040918