
Testicular Germ Cell Tumors Acquire Cisplatin Resistance by Rebalancing the Usage of DNA Repair Pathways

 , ,
, ,  ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Drugs
2.3. Micronuclei (MN) Assay
2.4. Colony Forming Assay
2.5. Cell Cycle Analysis and Quantification of Phospho-H2AX (Ser139) Signal by Flow Cytometry
2.6. Immunodetection of DNA Damage Repair Foci
2.7. DNA Repair Assays
2.8. Crystal Violet Assay
2.9. Generation of Stable Cell Lines
2.10. Immunoblotting and Biochemical Fractionation
2.11. Determination of Synergy
2.12. In Silico Analysis of Progression-Free Survival
2.13. Statistical Analysis
3. Results
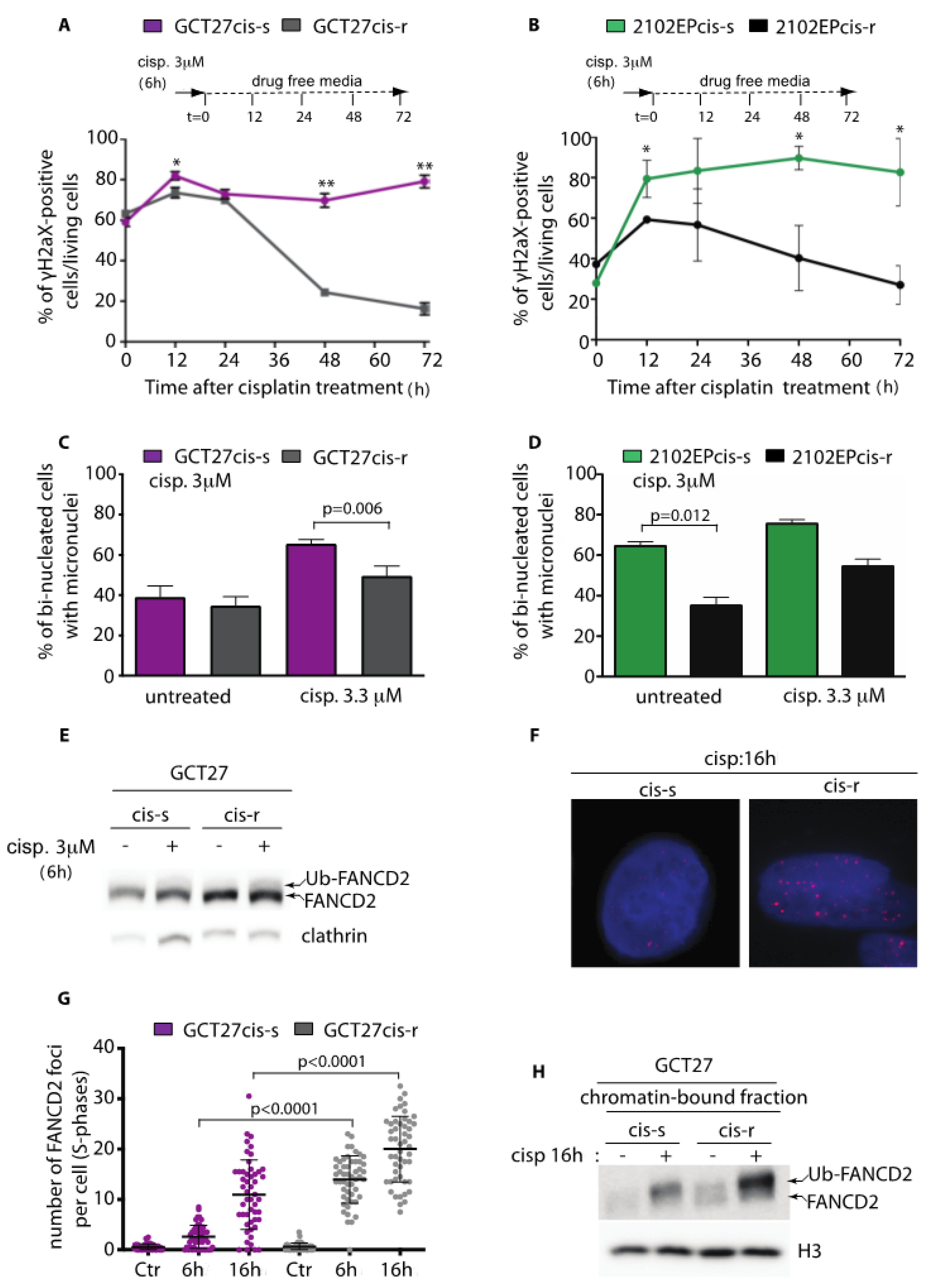
3.1. Testicular GCT Cell Lines with Acquired Resistance to Cisplatin Show Improved DNA Damage Repair Proficiency and Decreased Genome Instability
3.2. GCT27cis-r Cells Activate the FA-Pathway with Higher Efficiency than GCT27cis-s Cells
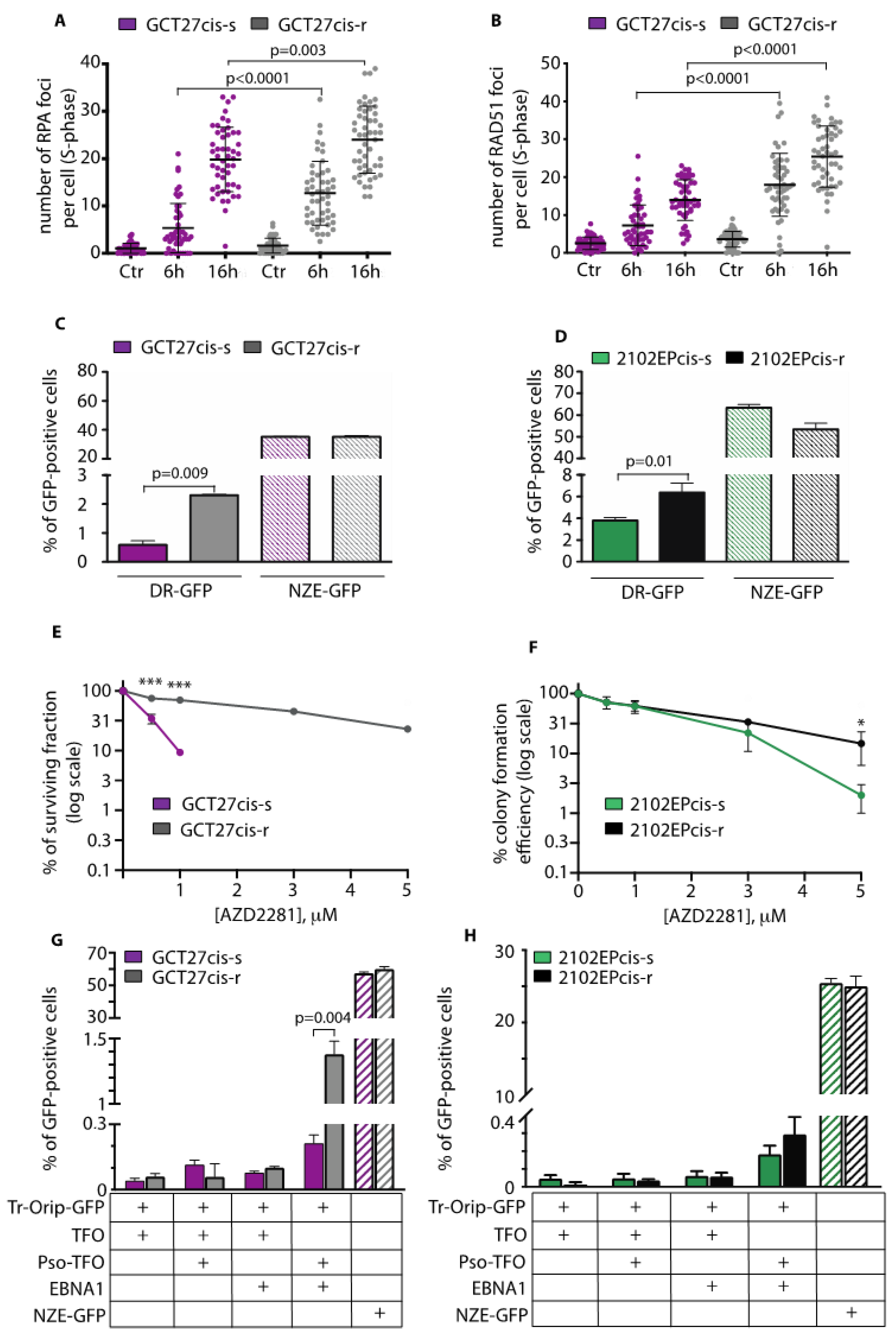
3.3. GCT27cis-r and 2102EPcis-r Cells Show an Increased Proficiency of Repair of DSBs by HR
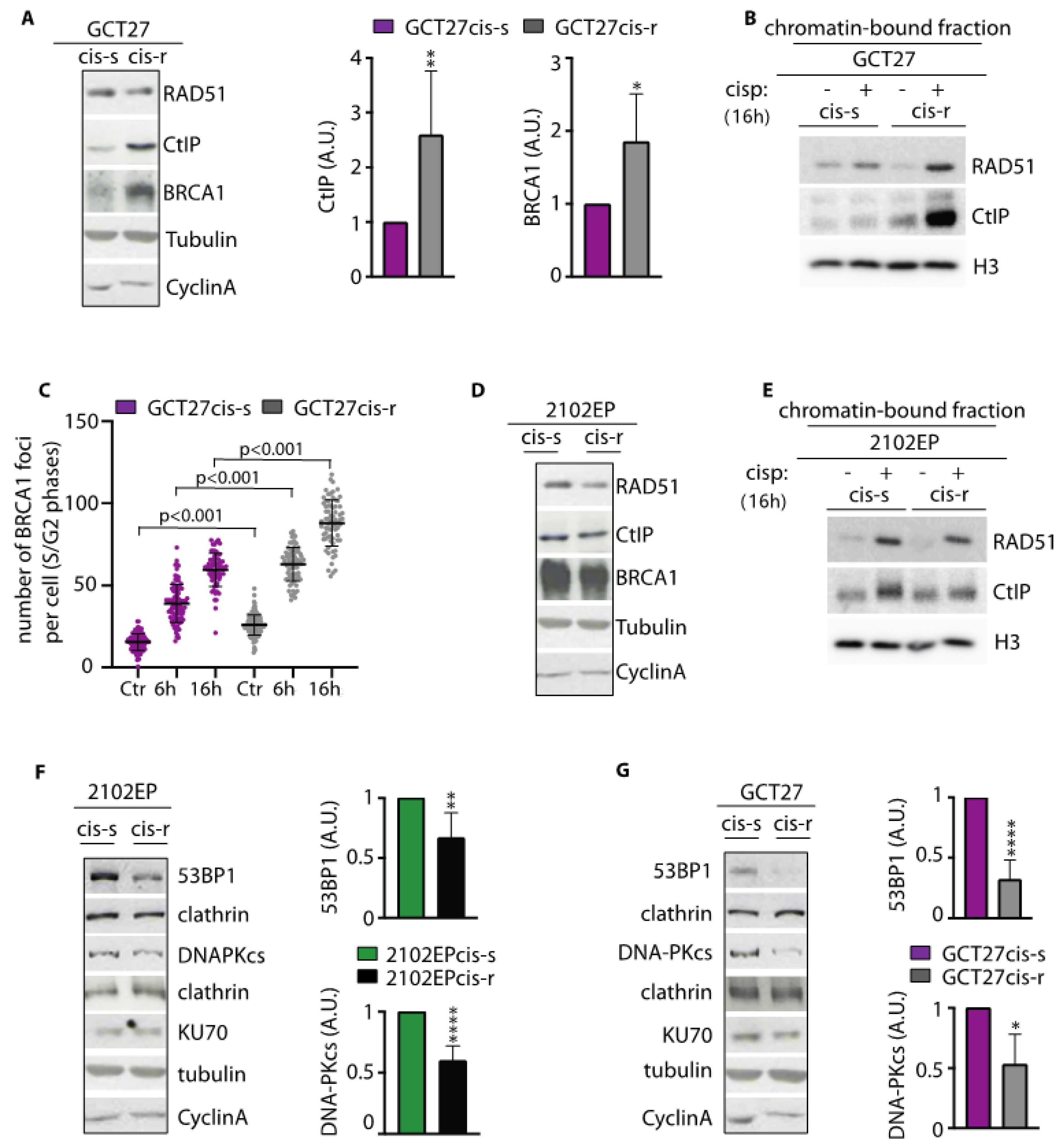
3.4. Analysis of the Expression and Binding onto Chromatin of HR-Repair Factors
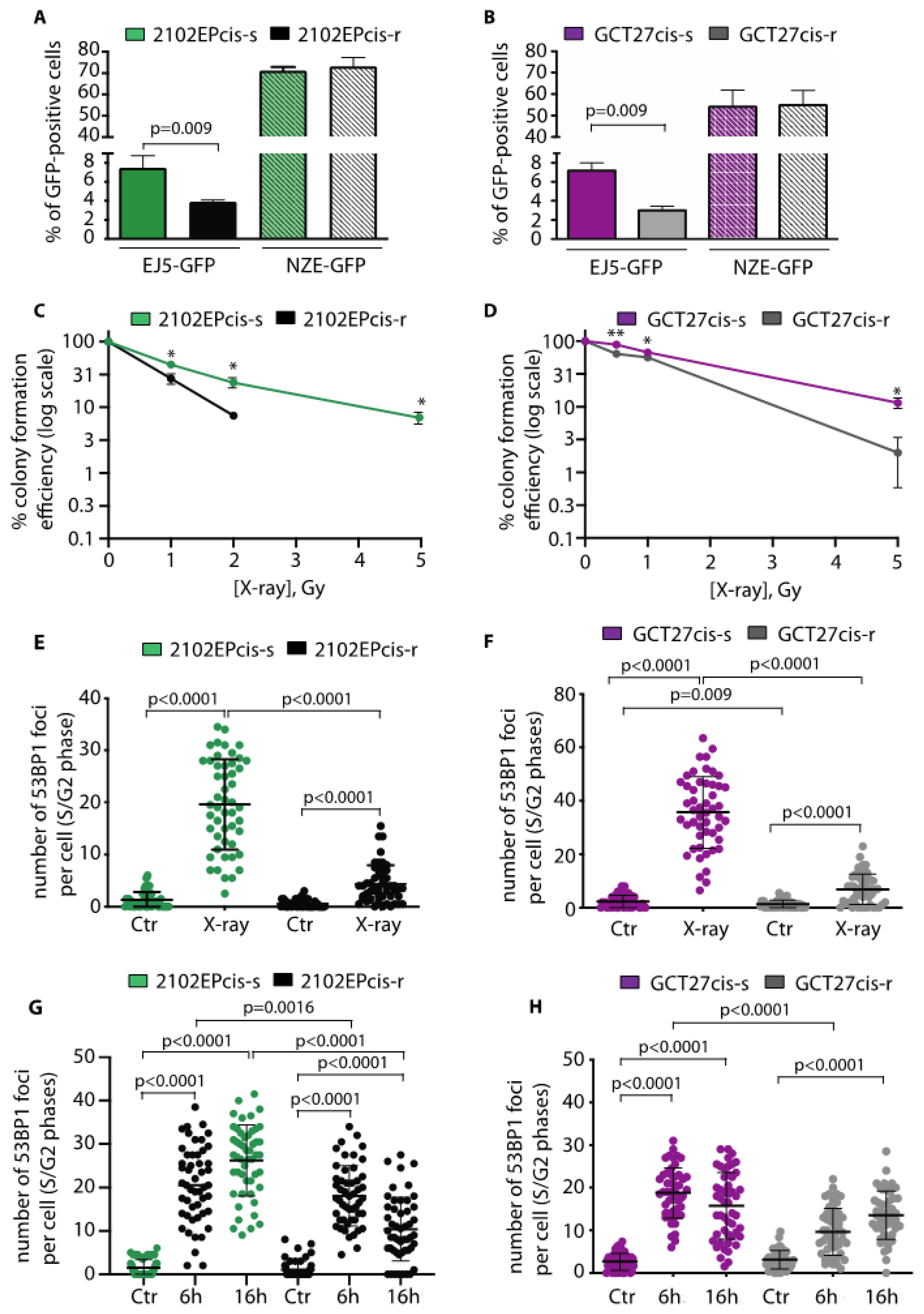
3.5. In cis-r Cell Lines 53BP1 and DNA-PKcs Expressions Are Dampened and NHEJ Is Attenuated
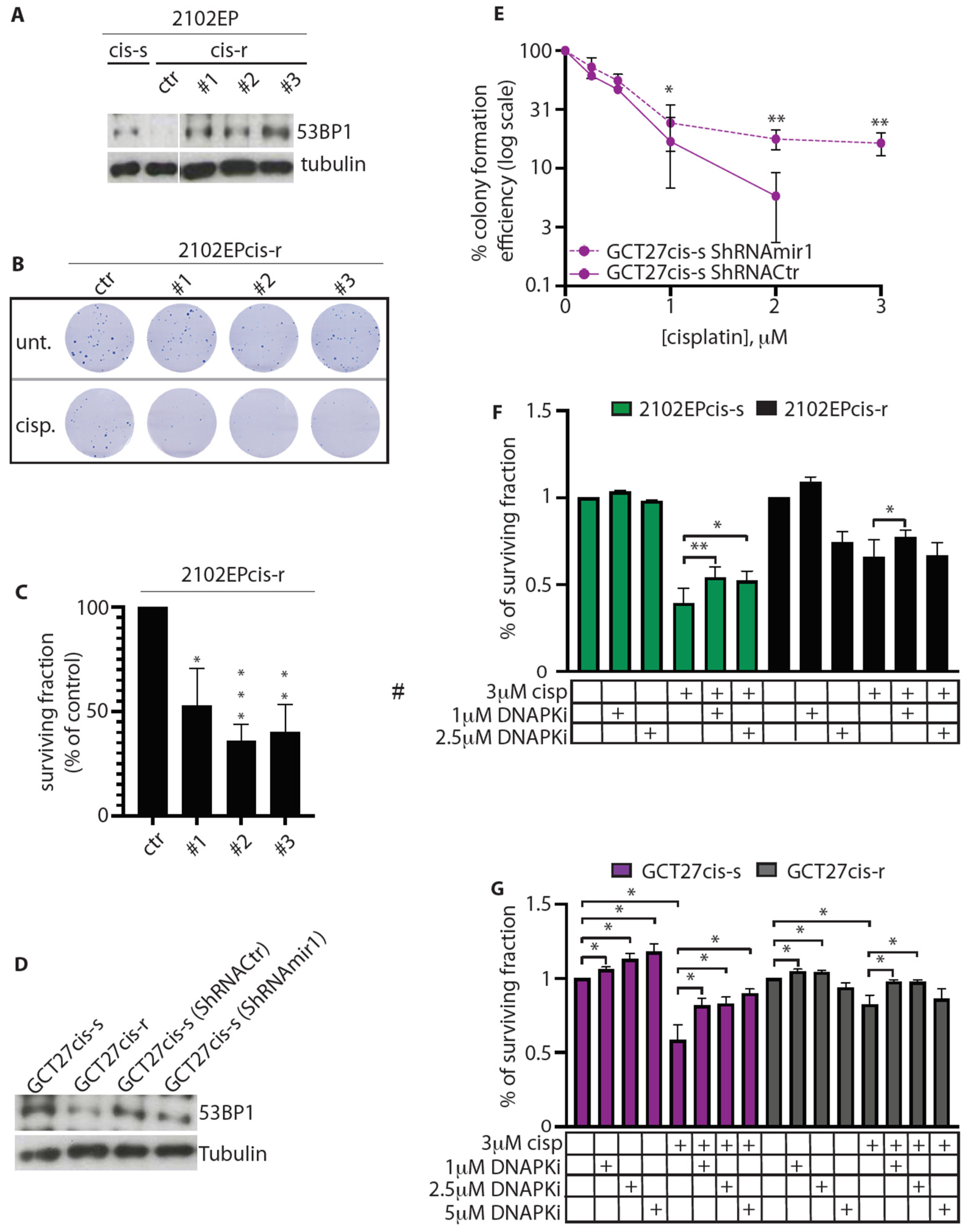
3.6. Modulation of 53BP1 Protein Expression Level Alters 2102EP and GCT27 Cells Sensitivity to Cisplatin
3.7. Inhibition of DNA-PKcs Activity Reduces the Cytotoxic Effect of Cisplatin
3.8. Expression of TP53BP1 and PRKDC in Non-Seminomas Patients
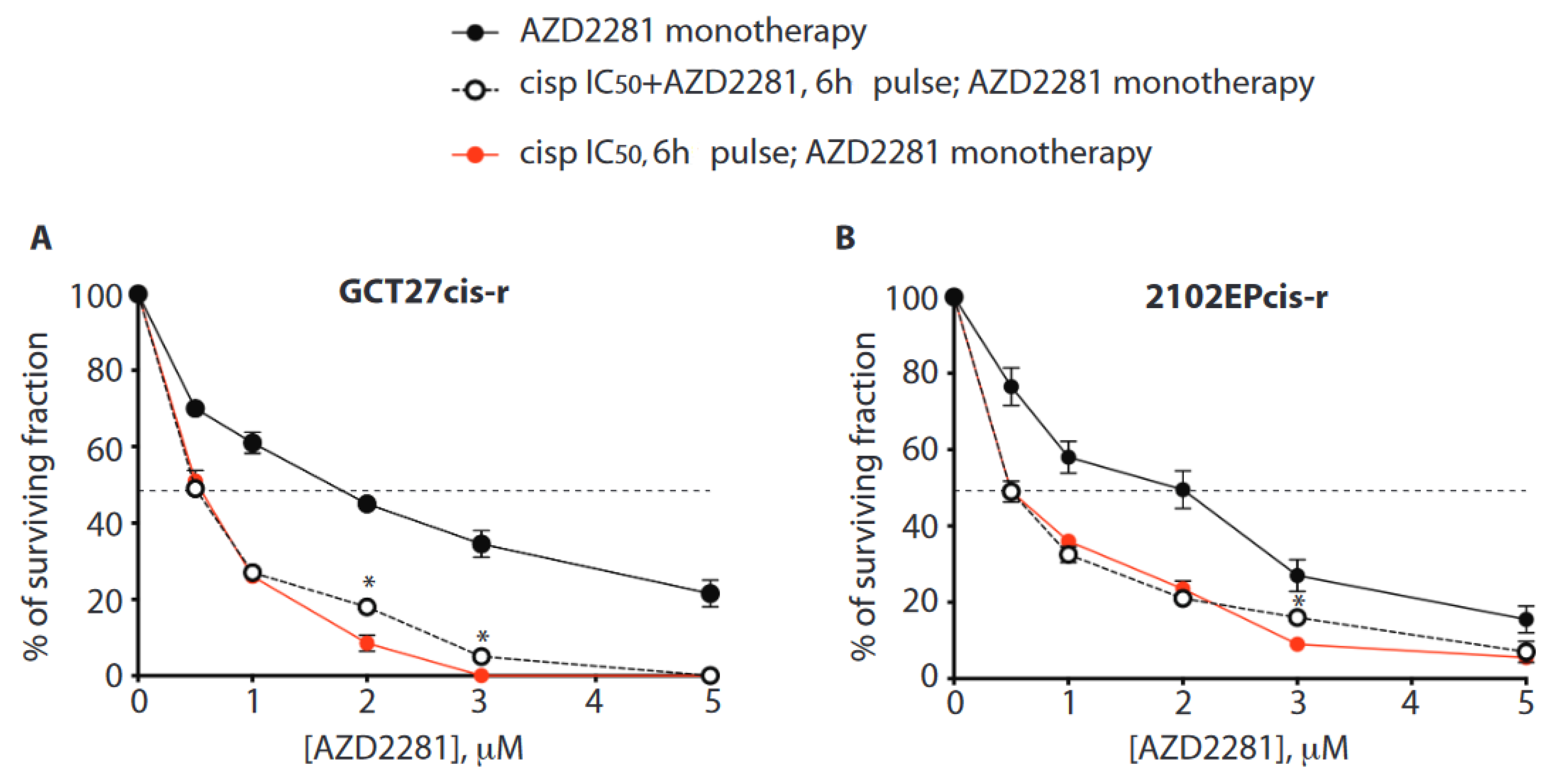
3.9. In Cis-r Cells PARP Inhibition Has an Additive/Synergistic Interaction with Cisplatin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McHugh, D.J.; Feldman, D.R. Conventional-Dose versus High-Dose Chemotherapy for Relapsed Germ Cell Tumors. Adv. Urol. 2018, 2018, 7272541. [Google Scholar] [CrossRef] [PubMed]
- Ghazarian, A.A.; Kelly, S.P.; Altekruse, S.F.; Rosenberg, P.S.; McGlynn, K.A. Future of testicular germ cell tumor incidence in the United States: Forecast through 2026. Cancer 2017, 123, 2320–2328. [Google Scholar] [CrossRef] [PubMed]
- Znaor, A.; Lortet-Tieulent, J.; Jemal, A.; Bray, F. International variations and trends in testicular cancer incidence and mortality. Eur. Urol. 2014, 65, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Delahunt, B.; Magi-Galluzzi, C.; Algaba, F.; Egevad, L.; Ulbright, T.M.; Tickoo, S.K.; Srigley, J.R.; Epstein, J.I.; Berney, D.M.; et al. The World Health Organization 2016 classification of testicular germ cell tumours: A review and update from the International Society of Urological Pathology Testis Consultation Panel. Histopathology 2017, 70, 335–346. [Google Scholar] [CrossRef]
- Moul, J.W.; McCarthy, W.F.; Fernandez, E.B.; Sesterhenn, I.A. Percentage of embryonal carcinoma and of vascular invasion predicts pathological stage in clinical stage I nonseminomatous testicular cancer. Cancer Res. 1994, 54, 362–364. [Google Scholar]
- Heidenreich, A.; Sesterhenn, I.A.; Mostofi, F.K.; Moul, J.W. Prognostic risk factors that identify patients with clinical stage I nonseminomatous germ cell tumors at low risk and high risk for metastasis. Cancer 1998, 83, 1002–1011. [Google Scholar] [CrossRef]
- Pont, J.; Holtl, W.; Kosak, D.; Machacek, E.; Kienzer, H.; Julcher, H.; Honetz, N. Risk-adapted treatment choice in stage I nonseminomatous testicular germ cell cancer by regarding vascular invasion in the primary tumor: A prospective trial. J. Clin. Oncol. 1990, 8, 16–20. [Google Scholar] [CrossRef]
- Ondrus, D.; Matoska, J.; Belan, V.; Kausitz, J.; Goncalves, F.; Hornak, M. Prognostic factors in clinical stage I nonseminomatous germ cell testicular tumors: Rationale for different risk-adapted treatment. Eur. Urol. 1998, 33, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Spierings, D.C.; de Vries, E.G.; Vellenga, E.; de Jong, S. The attractive Achilles heel of germ cell tumours: An inherent sensitivity to apoptosis-inducing stimuli. J. Pathol. 2003, 200, 137–148. [Google Scholar] [CrossRef]
- Kartalou, M.; Essigmann, J.M. Recognition of cisplatin adducts by cellular proteins. Mutat. Res. 2001, 478, 1–21. [Google Scholar] [CrossRef]
- Cavallo, F.; Graziani, G.; Antinozzi, C.; Feldman, D.R.; Houldsworth, J.; Bosl, G.J.; Chaganti, R.S.; Moynahan, M.E.; Jasin, M.; Barchi, M. Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to Cisplatin and poly (adp-ribose) polymerase inhibition. PLoS ONE 2012, 7, e51563. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, F.; Feldman, D.R.; Barchi, M. Revisiting DNA damage repair, p53-mediated apoptosis and cisplatin sensitivity in germ cell tumors. Int. J. Dev. Biol. 2013, 57, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [PubMed]
- De Silva, I.U.; McHugh, P.J.; Clingen, P.H.; Hartley, J.A. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell Biol. 2000, 20, 7980–7990. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef]
- Jensen, R.; Glazer, P.M. Cell-interdependent cisplatin killing by Ku/DNA-dependent protein kinase signaling transduced through gap junctions. Proc. Natl. Acad. Sci. USA 2004, 101, 6134–6139. [Google Scholar] [CrossRef]
- Thongthip, S.; Conti, B.A.; Lach, F.P.; Smogorzewska, A. Suppression of non-homologous end joining does not rescue DNA repair defects in Fanconi anemia patient cells. Cell Cycle 2020, 19, 2553–2561. [Google Scholar] [CrossRef]
- Pera, M.F.; Friedlos, F.; Mills, J.; Roberts, J.J. Inherent sensitivity of cultured human embryonal carcinoma cells to adducts of cis-diamminedichloroplatinum(II) on DNA. Cancer Res. 1987, 47, 6810–6813. [Google Scholar]
- Kelland, L.R.; Mistry, P.; Abel, G.; Freidlos, F.; Loh, S.Y.; Roberts, J.J.; Harrap, K.R. Establishment and characterization of an in vitro model of acquired resistance to cisplatin in a human testicular nonseminomatous germ cell line. Cancer Res. 1992, 52, 1710–1716. [Google Scholar] [PubMed]
- Juliachs, M.; Munoz, C.; Moutinho, C.A.; Vidal, A.; Condom, E.; Esteller, M.; Graupera, M.; Casanovas, O.; Germa, J.R.; Villanueva, A.; et al. The PDGFRbeta-AKT pathway contributes to CDDP-acquired resistance in testicular germ cell tumors. Clin. Cancer Res. 2014, 20, 658–667. [Google Scholar] [CrossRef]
- Andrews, P.W.; Goodfellow, P.N.; Shevinsky, L.H.; Bronson, D.L.; Knowles, B.B. Cell-surface antigens of a clonal human embryonal carcinoma cell line: Morphological and antigenic differentiation in culture. Int. J. Cancer 1982, 29, 523–531. [Google Scholar] [CrossRef]
- Oechsle, K.; Honecker, F.; Cheng, T.; Mayer, F.; Czaykowski, P.; Winquist, E.; Wood, L.; Fenner, M.; Glaesener, S.; Hartmann, J.T.; et al. Preclinical and clinical activity of sunitinib in patients with cisplatin-refractory or multiply relapsed germ cell tumors: A Canadian Urologic Oncology Group/German Testicular Cancer Study Group cooperative study. Ann. Oncol. 2011, 22, 2654–2660. [Google Scholar] [CrossRef]
- Cavallo, F.; Caggiano, C.; Jasin, M.; Barchi, M. Assessing Homologous Recombination and Interstrand Cross-Link Repair in Embryonal Carcinoma Testicular Germ Cell Tumor Cell Lines. Methods Mol. Biol. 2021, 2195, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef]
- Urien, S.; Brain, E.; Bugat, R.; Pivot, X.; Lochon, I.; Van, M.L.; Vauzelle, F.; Lokiec, F. Pharmacokinetics of platinum after oral or intravenous cisplatin: A phase 1 study in 32 adult patients. Cancer Chemother. Pharmacol. 2005, 55, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Ohnishi, T. Does gammaH2AX foci formation depend on the presence of DNA double strand breaks? Cancer Lett. 2005, 229, 171–179. [Google Scholar] [CrossRef]
- Räschle, M.; Knipscheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Schärer, O.D.; Walter, J.C. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef]
- Vollebergh, M.A.; Jonkers, J.; Linn, S.C. Genomic instability in breast and ovarian cancers: Translation into clinical predictive biomarkers. Cell. Mol. Life Sci. 2012, 69, 223–245. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Zimmer, J.; Tarsounas, M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016, 35, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Cavallo, F.; Perrouault, L.; Giovannangeli, C.; Moynahan, M.E.; Barchi, M.; Brunet, E.; Jasin, M. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nat. Struct. Mol. Biol. 2011, 18, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Ismail, A.; Katsuki, Y.; Freire, R.; Shibata, A.; Jeggo, P.A. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 2013, 41, 10298–10311. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Lobrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef]
- Dong, J.; Ren, Y.; Zhang, T.; Wang, Z.; Ling, C.C.; Li, G.C.; He, F.; Wang, C.; Wen, B. Inactivation of DNA-PK by knockdown DNA-PKcs or NU7441 impairs non-homologous end-joining of radiation-induced double strand break repair. Oncol. Rep. 2018, 39, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Van Echten, J.; van der Vloedt, W.S.; van de Pol, M.; Dam, A.; te Meerman, G.J.; Schraffordt Koops, H.; Sleijfer, D.T.; Oosterhuis, J.W.; de Jong, B. Comparison of the chromosomal pattern of primary testicular nonseminomas and residual mature teratomas after chemotherapy. Cancer Genet. Cytogenet. 1997, 99, 59–67. [Google Scholar] [CrossRef]
- Van Echten, J.; Sleijfer, D.T.; Wiersema, J.; Schraffordt Koops, H.; Oosterhuis, J.W.; de Jong, B. Cytogenetics of primary testicular nonseminoma, residual mature teratoma, and growing teratoma lesion in individual patients. Cancer Genet. Cytogenet. 1997, 96, 1–6. [Google Scholar] [CrossRef]
- Friedlander, M.; Matulonis, U.; Gourley, C.; du Bois, A.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Long-term efficacy, tolerability and overall survival in patients with platinum-sensitive, recurrent high-grade serous ovarian cancer treated with maintenance olaparib capsules following response to chemotherapy. Br. J. Cancer 2018, 119, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Murphy, C.G.; Doubrovina, E.; Jasin, M.; Moynahan, M.E. PARP Inhibitors in Clinical Use Induce Genomic Instability in Normal Human Cells. PLoS ONE 2016, 11, e0159341. [Google Scholar] [CrossRef]
- Koul, S.; Houldsworth, J.; Mansukhani, M.M.; Donadio, A.; McKiernan, J.M.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.; Murty, V.V. Characteristic promoter hypermethylation signatures in male germ cell tumors. Mol. Cancer 2002, 1, 8. [Google Scholar] [CrossRef]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef]
- Nacson, J.; Krais, J.J.; Bernhardy, A.J.; Clausen, E.; Feng, W.; Wang, Y.; Nicolas, E.; Cai, K.Q.; Tricarico, R.; Hua, X.; et al. BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell Rep. 2018, 25, 1384. [Google Scholar] [CrossRef] [PubMed]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.B.; Lukas, J.; Lukas, C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Lesport, E.; Ferster, A.; Biver, A.; Roch, B.; Vasquez, N.; Jabado, N.; Vives, F.L.; Revy, P.; Soulier, J.; de Villartay, J.P. Reduced recruitment of 53BP1 during interstrand crosslink repair is associated with genetically inherited attenuation of mitomycin C sensitivity in a family with Fanconi anemia. Oncotarget 2018, 9, 3779–3793. [Google Scholar] [CrossRef]
- De Giorgi, U.; Schepisi, G.; Gurioli, G.; Pisano, C.; Basso, U.; Lolli, C.; Petracci, E.; Casadei, C.; Cecere, S.C.; Attademo, L.; et al. Olaparib as salvage treatment for advanced germ cell tumors after chemotherapy failure: Results of the open-label, single-arm, IGG-02 phase II trial. J. Clin. Oncol. 2020, 38, 5058. [Google Scholar] [CrossRef]
- Urien, S.; Lokiec, F. Population pharmacokinetics of total and unbound plasma cisplatin in adult patients. Br. J. Clin. Pharmacol. 2004, 57, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, M.; Fujihara, H.; Fujimori, H.; Kawaguchi, K.; Yamada, H.; Nakayama, R.; Yamamoto, N.; Kishi, Y.; Hamada, Y.; Masutani, M. Synergetic Effects of PARP Inhibitor AZD2281 and Cisplatin in Oral Squamous Cell Carcinoma In Vitro and In Vivo. Int. J. Mol. Sci. 2016, 17, 272. [Google Scholar] [CrossRef]
- Hegan, D.C.; Lu, Y.; Stachelek, G.C.; Crosby, M.E.; Bindra, R.S.; Glazer, P.M. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc. Natl. Acad. Sci. USA 2010, 107, 2201–2206. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Source | Code | Working Solution |
|---|---|---|---|
| RAD51 (H-92) | Santa Cruz | Sc-8349 | 1:500 |
| BRCA1 (C-20) | Santa Cruz | Sc-642 | 1:250 |
| CtIP | Bethyl Laboratories | A300-488A-T | 1:1000 |
| KU70 | Novus Biologicals | NB 100-1915 | 1:2000 |
| DNAPKcs | Thermofisher | MA5-13244 | 1:200 |
| TP53BP1 | OriGene | TA309918 | 1:1000 |
| FANCD2 | Novus Biologicals | NB 100-182 | 1:1000 |
| TUBULIN | Sigma-Aldrich | T4026 | 1:15,000 |
| CLATHRIN | BD Bioscience | 610500 | 1:1000 |
| CYCLIN A (H-432) | Santa Cruz | Sc-751 | 1:1000 |
| 6 h cisp 3 µM | GCT27 cis-s IC50 | GCT27 cis-r IC50 | 2101EP cis-s IC50 | 2101EP cis-r IC50 |
| 0.31 ± 0.03 | 1.27 ± 0.11 | 0.29 ± 0.05 | 1.05 ± 0.22 | |
| RF | 4 | 3.6 | ||
| IC90 | IC90 | IC90 | IC90 | |
| 0.84 ± 0.02 | 4.91 ± 0.16 | 0.86 ± 0.08 | 5.07 ± 1.12 | |
| RF | 5.8 | 5.9 |
| IC90 Value for AZD2281 | ||
|---|---|---|
| GCT27 cis-r IC90 (µM) | 2102EP cis-r IC90 (µM) | |
| AZD2281 mono | 15.7 ± 1.7 | 9.4 ± 1.2 |
| 6 h cisp IC50 + AZD2281 mono | 1.7 ± 0.007 | 4 ± 0.6 |
| 6 h cisp IC50/AZD2281 + AZD2281 mono | 2.4 ± 0.07 | 4.6 ±0.3 |
| GCT27 cis-r | 2102EP cis-r | ||
|---|---|---|---|
| [AZD2281] | CI | CI | |
| 6 h cisp IC50 + | 1 µM | 0.67 | 1.1 |
| AZD2281 | 2 µM | 0.28 | 0.93 |
| mono | 3 µM | 0.04 | 0.46 |
| 5 µM | 0.05 | 0.35 | |
| [AZD2281] | CI | CI | |
| 6 h cisp | 1 µM | 0.7 | 1 |
| IC50/AZD2281 | 2 µM | 0.58 | 0.85 |
| + AZD2281 | 3 µM | 0.19 | 0.79 |
| mono | 5 µM | 0.05 | 0.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caggiano, C.; Cavallo, F.; Giannattasio, T.; Cappelletti, G.; Rossi, P.; Grimaldi, P.; Feldman, D.R.; Jasin, M.; Barchi, M. Testicular Germ Cell Tumors Acquire Cisplatin Resistance by Rebalancing the Usage of DNA Repair Pathways. Cancers 2021, 13, 787. https://doi.org/10.3390/cancers13040787
Caggiano C, Cavallo F, Giannattasio T, Cappelletti G, Rossi P, Grimaldi P, Feldman DR, Jasin M, Barchi M. Testicular Germ Cell Tumors Acquire Cisplatin Resistance by Rebalancing the Usage of DNA Repair Pathways. Cancers. 2021; 13(4):787. https://doi.org/10.3390/cancers13040787
Chicago/Turabian StyleCaggiano, Cinzia, Francesca Cavallo, Teresa Giannattasio, Gioia Cappelletti, Pellegrino Rossi, Paola Grimaldi, Darren R. Feldman, Maria Jasin, and Marco Barchi. 2021. "Testicular Germ Cell Tumors Acquire Cisplatin Resistance by Rebalancing the Usage of DNA Repair Pathways" Cancers 13, no. 4: 787. https://doi.org/10.3390/cancers13040787
APA StyleCaggiano, C., Cavallo, F., Giannattasio, T., Cappelletti, G., Rossi, P., Grimaldi, P., Feldman, D. R., Jasin, M., & Barchi, M. (2021). Testicular Germ Cell Tumors Acquire Cisplatin Resistance by Rebalancing the Usage of DNA Repair Pathways. Cancers, 13(4), 787. https://doi.org/10.3390/cancers13040787