
Senolytics for Cancer Therapy: Is All that Glitters Really Gold?


Abstract
Simple Summary
Abstract
1. Introduction
2. Senolytic Therapies: Have We Hit Gold or Pyrite?
2.1. Established Success of Senolytic Therapy in the Mitigation of Aging-Associated Disease
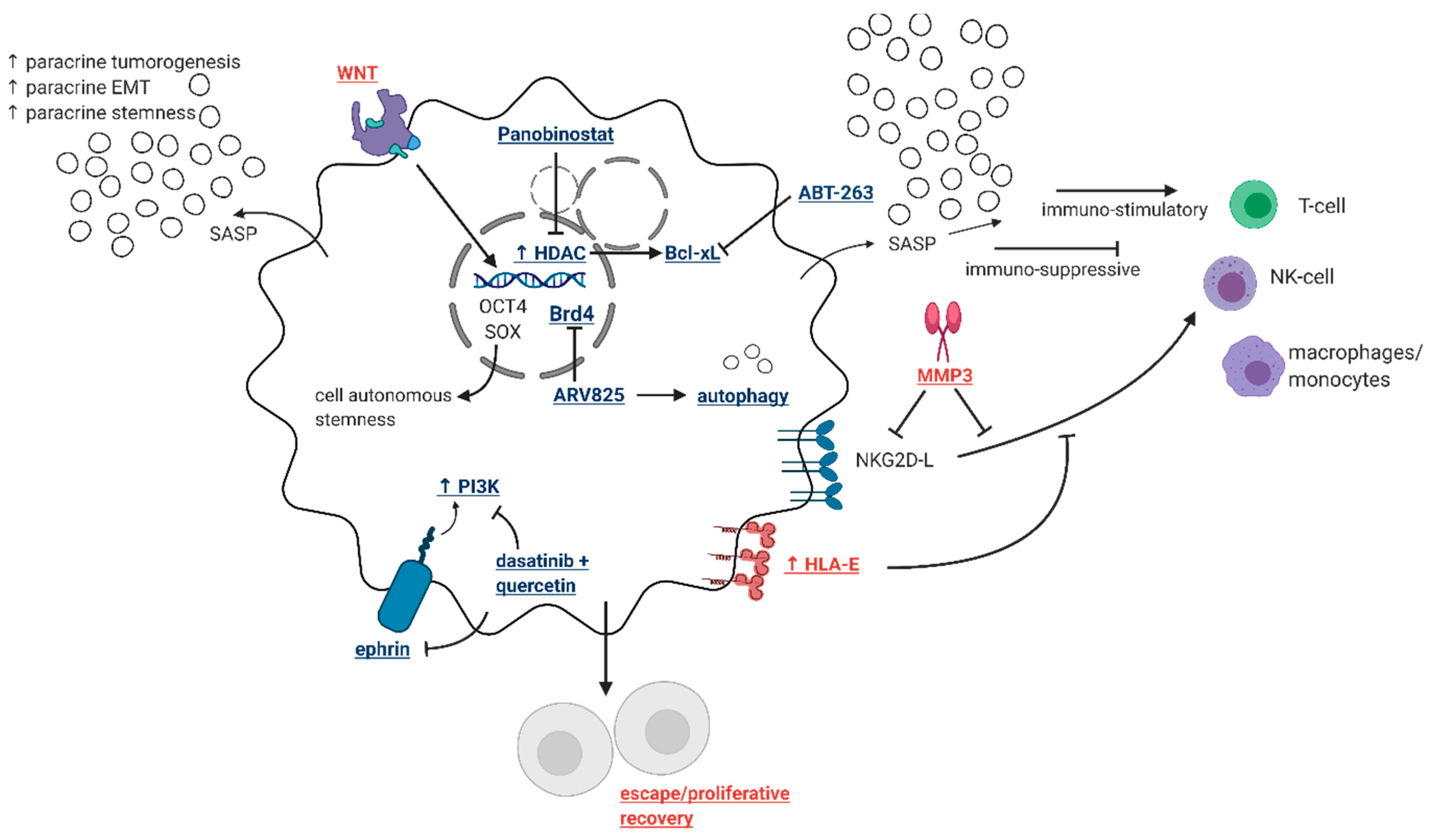
2.2. The Dual Faces of Therapy-Induced Senescence
2.2.1. Stability of the Senescent Growth Arrest
2.2.2. Impacts on the Immune System
2.2.3. TIS in Non-Malignant Cells
2.2.4. Evidence Supporting the Removal of Non-Malignant Therapy-Induced Senescent Cells
2.3. Early Evidence on Senolytics as Anti-Cancer Therapies
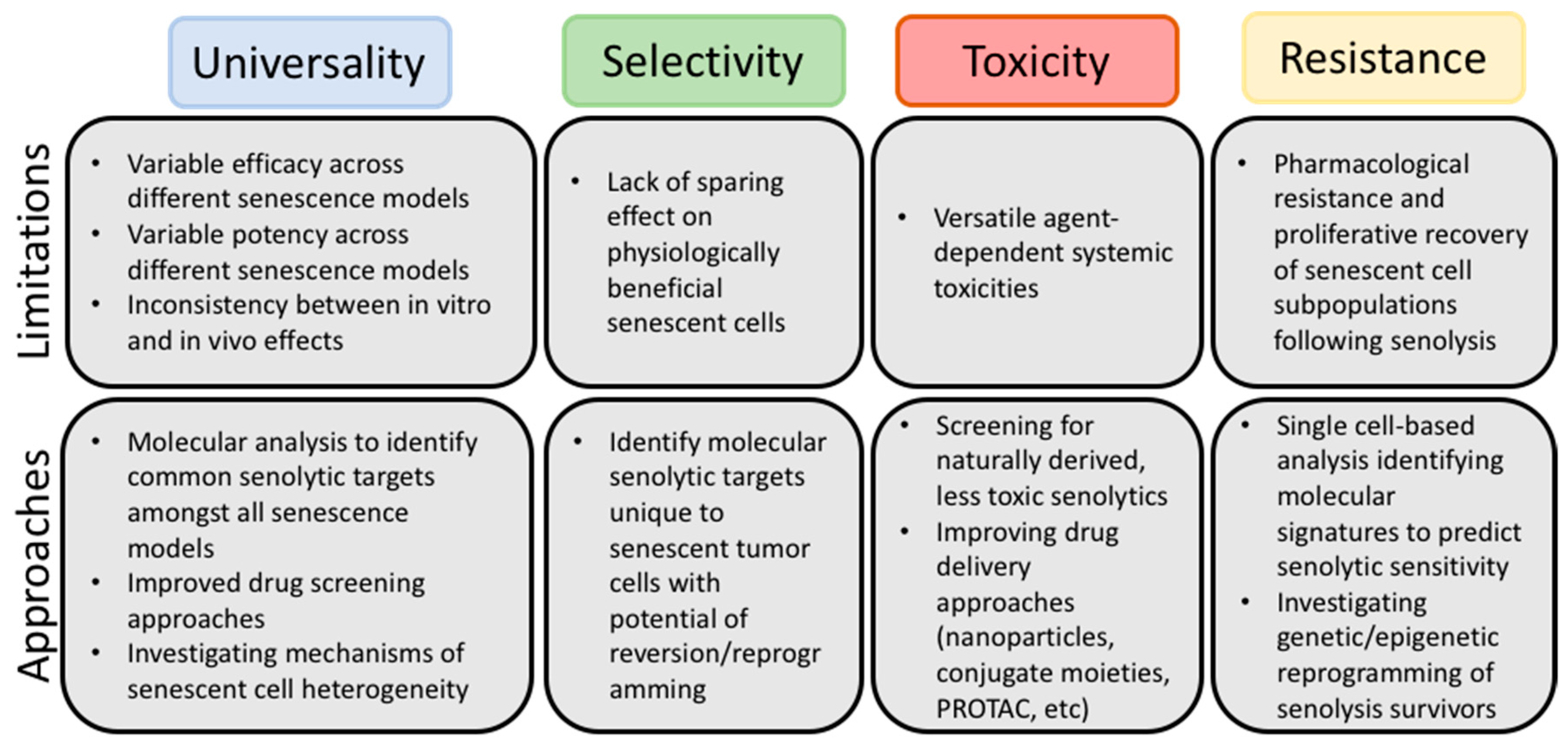
3. Possible Refinements of the Senolytic Strategy
3.1. Multi-Model Screenings
3.2. Single Cell-Omics
3.3. Improving Targeted Drug Delivery
3.4. Identification of Novel Drug Targets that Selectively Eliminate “Bad” Senescent Cells
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karlseder, J.; Smogorzewska, A.; De Lange, T. Senescence Induced by Altered Telomere State, Not Telomere Loss. Science 2002, 295, 2446–2449. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.F.M.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nat. Cell Biol. 2005, 436, 660–665. [Google Scholar] [CrossRef]
- Gewirtz, D.A.; Holt, S.E.; Elmore, L.W. Accelerated senescence: An emerging role in tumor cell response to chemotherapy and radiation. Biochem. Pharmacol. 2008, 76, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Lee, Y.-S.; Cho, K.A.; Park, S.C. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Res. Rev. 2018, 47, 176–182. [Google Scholar] [CrossRef]
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.-Y.; Andrew, E.P.; Fraser, P.; Reik, W. Global Reorganization of the Nuclear Landscape in Senescent Cells. Cell Rep. 2015, 10, 471–483. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- You, R.; Dai, J.; Zhang, P.; Barding, J.G.A.; Raftery, D. Dynamic Metabolic Response to Adriamycin-Induced Senescence in Breast Cancer Cells. Metabolites 2018, 8, 95. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial Dysfunction Accounts for the Stochastic Heterogeneity in Telomere-Dependent Senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; Zuffo, R.D.; Matti, V.; D’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; De Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.-M.; DeMaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Barascu, A.; Le Chalony, C.; Pennarun, G.; Genet, D.; Imam, N.; Lopez, B.; Bertrand, P. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J. 2012, 31, 1080–1094. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.-Y.; Campisi, J. Tumor Suppressor and Aging Biomarker p16INK4a Induces Cellular Senescence without the Associated Inflammatory Secretory Phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef]
- Chen, W.; Wang, X.; Wei, G.; Huang, Y.; Shi, Y.; Li, D.; Qiu, S.; Zhou, B.; Cao, J.; Chen, M.; et al. Single-Cell Transcriptome Analysis Reveals Six Subpopulations Reflecting Distinct Cellular Fates in Senescent Mouse Embryonic Fibroblasts. Front. Genet. 2020, 11, 867. [Google Scholar] [CrossRef]
- Tang, H.; Geng, A.; Zhang, T.; Wang, C.; Jiang, Y.; Mao, Z. Single senescent cell sequencing reveals heterogeneity in senescent cells induced by telomere erosion. Protein Cell 2018, 10, 370–375. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef]
- Mooi, W.; Peeper, D. Oncogene-Induced Cell Senescence—Halting on the Road to Cancer. N. Engl. J. Med. 2006, 355, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef]
- Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P.; Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA Damage Is Able to Induce Senescence in Tumor Cells in Vitro and in Vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor cell escape from therapy-induced senescence. Biochem. Pharmacol. 2019, 162, 202–212. [Google Scholar] [CrossRef]
- Saleh, T.; Carpenter, V.J.; Tyutyunyk-Massey, L.; Murray, G.; Leverson, J.D.; Souers, A.J.; Alotaibi, M.R.; Faber, A.C.; Reed, J.; Harada, H.; et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-X L –BAX interaction. Mol. Oncol. 2020, 14, 2504–2519. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, W.; Dong, H.; Li, Y.; Li, L.; Han, L.; Han, Z.; Wang, S.; Ma, D.; Wang, H. Cisplatin-induced senescence in ovarian cancer cells is mediated by GRP78. Oncol. Rep. 2014, 31, 2525–2534. [Google Scholar] [CrossRef]
- Qu, K.; Lin, T.; Wang, Z.; Liu, S.; Chang, H.; Xu, X.; Meng, F.; Zhou, L.; Wei, J.; Tai, M.; et al. Reactive oxygen species generation is essential for cisplatininduced accelerated senescence in hepatocellular carcinoma. Front. Med. 2014, 8, 227–235. [Google Scholar] [CrossRef]
- Sun, X.; Shi, B.; Zheng, H.; Min, L.; Yang, J.; Li, X.; Liao, X.; Huang, W.; Zhang, M.; Xu, S.; et al. Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M. Whole-exome sequencing reveals novel genetic variants associated with diverse phenotypes of melanoma cells. Mol. Carcinog. 2019, 58, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Haferkamp, S.; Borst, A.; Adam, C.; Becker, T.M.; Motschenbacher, S.; Windhövel, S.; Hufnagel, A.L.; Houben, R.; Meierjohann, S. Vemurafenib Induces Senescence Features in Melanoma Cells. J. Investig. Dermatol. 2013, 133, 1601–1609. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Kang, B.S.; Bae, Y.-S. Premature senescence in human breast cancer and colon cancer cells by tamoxifen-mediated reactive oxygen species generation. Life Sci. 2014, 97, 116–122. [Google Scholar] [CrossRef]
- Tuttle, R.; Miller, K.R.; Maiorano, J.N.; Termuhlen, P.M.; Gao, Y.; Berberich, S.J. Novel senescence associated gene, YPEL3, is repressed by estrogen in ER+ mammary tumor cells and required for tamoxifen-induced cellular senescence. Int. J. Cancer 2011, 130, 2291–2299. [Google Scholar] [CrossRef]
- Ewald, J.A.; Desotelle, J.A.; Church, D.R.; Yang, B.; Huang, W.; Laurila, T.A.; Jarrard, D.F. Androgen deprivation induces senescence characteristics in prostate cancer cells in vitro and in vivo. Prostate 2012, 73, 337–345. [Google Scholar] [CrossRef]
- Blute, M.L.; Damaschke, N.; Wagner, J.; Yang, B.; Gleave, M.; Fazli, L.; Shi, F.; Abel, E.J.; Downs, T.M.; Huang, W.; et al. Persistence of senescent prostate cancer cells following prolonged neoadjuvant androgen deprivation therapy. PLoS ONE 2017, 12, e0172048. [Google Scholar] [CrossRef]
- Barakat, D.J.; Zhang, J.; Barberi, T.; Denmeade, S.R.; Friedman, A.D.; Paz-Priel, I. CCAAT/Enhancer binding protein β controls androgen-deprivation-induced senescence in prostate cancer cells. Oncogene 2015, 34, 5912–5922. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12. [Google Scholar] [CrossRef]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.-Y.J.; Wu, D.Y. Escape from Therapy-Induced Accelerated Cellular Senescence in p53-Null Lung Cancer Cells and in Human Lung Cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef]
- Cotarelo, C.L.; Schad, A.; Kirkpatrick, C.J.; Sleeman, J.P.; Springer, E.; Schmidt, M.; Thaler, S. Detection of cellular senescence within human invasive breast carcinomas distinguishes different breast tumor subtypes. Oncotarget 2016, 7, 74846–74859. [Google Scholar] [CrossRef]
- Chang, B.-D.; Xuan, Y.; Broude, E.V.; Zhu, H.; Schott, B.; Fang, J.; Roninson, I.B. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 1999, 18, 4808–4818. [Google Scholar] [CrossRef] [PubMed]
- Cairney, C.J.; Bilsland, A.E.; Evans, T.J.; Roffey, J.; Bennett, D.C.; Narita, M.; Torrance, C.J.; Keith, W.N. Cancer cell senescence: A new frontier in drug development. Drug Discov. Today 2012, 17, 269–276. [Google Scholar] [CrossRef]
- Lee, S.; Lee, A.J.-S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef]
- Elmore, L.W.; Rehder, C.W.; Di, X.; McChesney, P.A.; Jackson-Cook, C.K.; Gewirtz, D.A.; Holt, S.E. Adriamycin-induced Senescence in Breast Tumor Cells Involves Functional p53 and Telomere Dysfunction. J. Biol. Chem. 2002, 277, 35509–35515. [Google Scholar] [CrossRef] [PubMed]
- Litwiniec, A.; Gackowska, L.; Helmin-Basa, A.; Żuryń, A.; Grzanka, A. Low-dose etoposide-treatment induces endoreplication and cell death accompanied by cytoskeletal alterations in A549 cells: Does the response involve senescence? The possible role of vimentin. Cancer Cell Int. 2013, 13, 9. [Google Scholar] [CrossRef]
- Puig, P.-E.; Guilly, M.-N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Alotaibi, M.; Sharma, K.; Saleh, T.; Povirk, L.F.; Hendrickson, E.A.; Gewirtz, D.A. Radiosensitization by PARP Inhibition in DNA Repair Proficient and Deficient Tumor Cells: Proliferative Recovery in Senescent Cells. Radiat. Res. 2016, 185, 229–245. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nat. Cell Biol. 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Jonchère, B.; Vétillard, A.; Toutain, B.; Lam, D.; Bernard, A.C.; Henry, C.; Trécesson, S.D.C.; Gamelin, E.; Juin, P.; Guette, C.; et al. Irinotecan treatment and senescence failure promote the emergence of more transformed and invasive cells that depend on anti-apoptotic Mcl-1. Oncotarget 2014, 6, 409–426. [Google Scholar] [CrossRef]
- Pluquet, O.; Abbadie, C.; Coqueret, O. Connecting cancer relapse with senescence. Cancer Lett. 2019, 463, 50–58. [Google Scholar] [CrossRef]
- DeMaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2016, 7, 165–176. [Google Scholar] [CrossRef]
- Duy, C.; Li, M.; Teater, M.; Meydan, C.; Garrett-Bakelman, F.E.; Lee, T.C.; Chin, C.R.; Durmaz, C.; Kawabata, K.C.; Dhimolea, E.; et al. Chemotherapy induces senescence-like resilient cells capable of initiating AML recurrence. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology 2013, 14, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.-M.; Awad, P.; Campisi, J.; Desprez, P.-Y. Epithelial-Mesenchymal Transition Induced by Senescent Fibroblasts. Cancer Microenviron. 2011, 5, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kurose, A.; Ogawa, A.; Ogasawara, K.; Traganos, F.; Darzynkiewicz, Z.; Sawai, T. Diversity of DNA damage response of astrocytes and glioblastoma cell lines with various p53 status to treatment with etoposide and temozolomide. Cancer Biol. Ther. 2009, 8, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.E.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; DeMaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; Von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; De Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nat. Cell Biol. 2019, 574, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Sieben, C.J.; Sturmlechner, I.; Van De Sluis, B.; Van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Nath, K.A.; O’Brien, D.R.; Croatt, A.J.; Grande, J.P.; Ackerman, A.W.; Nath, M.C.; Yamada, S.; Terzic, A.; Tchkonia, T.; Kirkland, J.L.; et al. The murine dialysis fistula model exhibits a senescence phenotype: Pathobiological mechanisms and therapeutic potential. Am. J. Physiol. Physiol. 2018, 315, F1493–F1499. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Sen, J.M.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Parikh, P.; Britt, R.D.; Manlove, L.J.; Wicher, S.A.; Roesler, A.; Ravix, J.; Teske, J.; Thompson, M.A.; Sieck, G.C.; Kirkland, J.L.; et al. Hyperoxia-induced Cellular Senescence in Fetal Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2019, 61, 51–60. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.; Tchkonia, T.; Krüger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1061–1077.e8. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; Van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Chen, R.; Gui, C.; Tao, T.; Ge, Y.; Zhao, X.; Qin, R.; Yao, W.; Gu, S.; Jiang, Y.; et al. Eliminating senescent chondrogenic progenitor cells enhances chondrogenesis under intermittent hydrostatic pressure for the treatment of OA. Stem Cell Res. Ther. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Chandra, A.; Lagnado, A.B.; Farr, J.N.; Monroe, D.G.; Park, S.; Hachfeld, C.; Tchkonia, T.; Kirkland, J.L.; Khosla, S.; Passos, J.F.; et al. Targeted Reduction of Senescent Cell Burden Alleviates Focal Radiotherapy-Related Bone Loss. J. Bone Miner. Res. 2020, 35, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Z.; Huang, Y.; Zhou, Y.; Sheng, X.; Jiang, Q.; Wang, Y.; Luo, P.; Luo, M.; Shi, C. Senolytics (DQ) Mitigates Radiation Ulcers by Removing Senescent Cells. Front. Oncol. 2020, 9, 1576. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Hickson, L.J.; Prata, L.G.L.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; Lebrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Kim, H.-N.; Chang, J.; Shao, L.; Han, L.; Iyer, S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L.; Zhou, D.; Almeida, M. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell 2017, 16, 693–703. [Google Scholar] [CrossRef]
- Pan, J.; Li, D.; Xu, Y.; Zhang, J.; Wang, Y.; Chen, M.; Lin, S.; Huang, L.; Chung, E.J.; Citrin, D.E.; et al. Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int. J. Radiat. Oncol. 2017, 99, 353–361. [Google Scholar] [CrossRef]
- Mikawa, R.; Suzuki, Y.; Baskoro, H.; Kanayama, K.; Sugimoto, K.; Sato, T.; Sugimoto, M. Elimination of p19ARF-expressing cells protects against pulmonary emphysema in mice. Aging Cell 2018, 17, e12827. [Google Scholar] [CrossRef]
- Xu, X.; Kim, J.J.; Li, Y.; Xie, J.; Shao, C.; Wei, J. Oxidative stress-induced miRNAs modulate AKT signaling and promote cellular senescence in uterine leiomyoma. J. Mol. Med. 2018, 96, 1095–1106. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nat. Cell Biol. 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Yabluchanskiy, A.; Tarantini, S.; Balasubramanian, P.; Kiss, T.; Csipo, T.; Fülöp, G.A.; Lipecz, A.; Ahire, C.; DelFavero, J.; Nyul-Toth, A.; et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation–induced impairment of neurovascular coupling responses protecting cognitive function in mice. Geroscience 2020, 42, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef] [PubMed]
- Dookun, E.; Walaszczyk, A.; Redgrave, R.; Palmowski, P.; Tual-Chalot, S.; Suwana, A.; Chapman, J.; Jirkovsky, E.; Sosa, L.D.; Gill, E.; et al. Clearance of senescent cells during cardiac ischemia–reperfusion injury improves recovery. Aging Cell 2020, 19, e13249. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Dai, Y.; Liu, A.; Li, X.; Wu, L.; Lu, L.; Bao, Y.; Jin, Q. Senolytic Agent Navitoclax Inhibits Angiotensin II-Induced Heart Failure in Mice. J. Cardiovasc. Pharmacol. 2020, 76, 452–460. [Google Scholar] [CrossRef]
- Van Der Feen, D.E.; Bossers, G.P.L.; Hagdorn, Q.A.J.; Moonen, J.-R.; Kurakula, K.; Szulcek, R.; Chappell, J.; Vallania, F.; Donato, M.; Kok, K.; et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci. Transl. Med. 2020, 12, 4974. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; Van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef]
- Sessions, G.A.; Copp, M.E.; Liu, J.; Sinkler, M.A.; D’Costa, S.; Diekman, B.O. Controlled induction and targeted elimination of p16 INK4a -expressing chondrocytes in cartilage explant culture. FASEB J. 2019, 33, 12364–12373. [Google Scholar] [CrossRef]
- Yang, H.; Chen, C.; Chen, H.; Duan, X.; Li, J.; Zhou, Y.; Zeng, W.; Yang, L. Navitoclax (ABT263) reduces inflammation and promotes chondrogenic phenotype by clearing senescent osteoarthritic chondrocytes in osteoarthritis. Aging 2020, 12, 12750–12770. [Google Scholar] [CrossRef]
- Chung, L.; Maestas, D.R.; Lebid, A.; Mageau, A.; Rosson, G.D.; Wu, X.; Wolf, M.T.; Tam, A.J.; Vanderzee, I.; Wang, X.; et al. Interleukin 17 and senescent cells regulate the foreign body response to synthetic material implants in mice and humans. Sci. Transl. Med. 2020, 12, 3799. [Google Scholar] [CrossRef]
- Sugihara, H.; Teramoto, N.; Nakamura, K.; Shiga, T.; Shirakawa, T.; Matsuo, M.; Ogasawara, M.; Nishino, I.; Matsuwaki, T.; Nishihara, M.; et al. Cellular senescence-mediated exacerbation of Duchenne muscular dystrophy. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef]
- Eradat, H.; Grosicki, S.; Catalono, J.; Cosolo, W.; Dyagil, I.; Kipps, T.J.; Zheng, B.; Yalamanchili, S.; Sahasranaman, S.; Hurst, D.; et al. Preliminary Results of a Phase II Open-Label, Randomized Study of the BH3 Mimetic Protein Navitoclax (ABT-263) with or without Rituximab for Treatment of Previously Untreated B-Cell Chronic Lymphocytic Leukemia. Blood 2012, 120, 190. [Google Scholar] [CrossRef]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients With Relapsed or Refractory Disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Anuar, N.N.M.; Hisam, N.S.N.; Liew, S.L.; Ugusman, A. Clinical Review: Navitoclax as a Pro-Apoptotic and Anti-Fibrotic Agent. Front. Pharmacol. 2020, 11, 1817. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 2016, 8, 2915–2926. [Google Scholar] [CrossRef]
- Cherif, H.; Bisson, D.G.; Jarzem, P.; Weber, M.; Ouellet, J.; Haglund, L. Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro. J. Clin. Med. 2019, 8, 433. [Google Scholar] [CrossRef]
- Li, W.; He, Y.; Zhang, R.; Zheng, G.; Zhou, D. The curcumin analog EF24 is a novel senolytic agent. Aging 2019, 11, 771–782. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef]
- Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 2006, 252, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef]
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D.A. Is Senescence Reversible? Curr. Drug Targets 2016, 17, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Elmore, L.W.; Di, X.; Dumur, C.; Holt, S.E.; Gewirtz, D.A. Evasion of a Single-Step, Chemotherapy-Induced Senescence in Breast Cancer Cells: Implications for Treatment Response. Clin. Cancer Res. 2005, 11, 2637–2643. [Google Scholar] [CrossRef]
- Huang, D.; Leslie, K.A.; Guest, D.; Yeshcheulova, O.; Roy, I.J.; Piva, M.; Moriceau, G.; Zangle, T.A.; Lo, R.S.; Teitell, M.A.; et al. High-Speed Live-Cell Interferometry: A New Method for Quantifying Tumor Drug Resistance and Heterogeneity. Anal. Chem. 2018, 90, 3299–3306. [Google Scholar] [CrossRef]
- Guillon, J.; Petit, C.; Moreau, M.; Toutain, B.; Henry, C.; Roché, H.; Bonichon-Lamichhane, N.; Salmon, J.P.; Lemonnier, J.; Campone, M.; et al. Regulation of senescence escape by TSP1 and CD47 following chemotherapy treatment. Cell Death Dis. 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, 4445. [Google Scholar] [CrossRef] [PubMed]
- Bojko, A.; Czarnecka-Herok, J.; Charzyńska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef]
- Prata, L.G.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef] [PubMed]
- Brighton, P.J.; Maruyama, Y.; Fishwick, K.; Vrljicak, P.; Tewary, S.; Fujihara, R.; Muter, J.; Lucas, E.S.; Yamada, T.; Woods, L.; et al. Clearance of senescent decidual cells by uterine natural killer cells in cycling human endometrium. eLife 2017, 6, e31274. [Google Scholar] [CrossRef]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-Cell-Autonomous Tumor Suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Ruscetti, M.; Morris, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.-J.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441.e21. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, L.; Ferlito, L.; Di Mauro, S.; Imbesi, R.M.; Scalia, G. Immunosenescence and cancer: A review. Arch. Gerontol. Geriatr. 2001, 32, 77–93. [Google Scholar] [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Dou, J.; Mo, W.; Liu, S.; Han, B.; Huang, Y.; Zhang, Y.; Varvares, M.A.; et al. TLR 8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol. Med. 2014, 6, 1294–1311. [Google Scholar] [CrossRef]
- Ramello, M.C.; Tosello, J.; Canale, F.; Mena, H.A.; Negrotto, S.; Gastman, B.; Gruppi, A.; Acosta-Rodriguez, E.; Montes, C.L. Tumor-induced senescent T cells promote the secretion of pro-inflammatory cytokines and angiogenic factors by human monocytes/macrophages through a mechanism that involves Tim-3 and CD40L. Cell Death Dis. 2014, 5, e1507. [Google Scholar] [CrossRef]
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-Induced Senescent T Cells with Suppressor Function: A Potential Form of Tumor Immune Evasion. Cancer Res. 2008, 68, 870–879. [Google Scholar] [CrossRef]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.-H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef] [PubMed]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing Chemotherapy Efficacy in Pten -Deficient Prostate Tumors by Activating the Senescence-Associated Antitumor Immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, D.P.; Yannone, S.M.; Daemen, A.; Sun, Y.; Vakar-Lopez, F.; Kawahara, M.; Freund, A.M.; Rodier, F.; Wu, J.D.; Desprez, P.-Y.; et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight 2019, 4, e124716. [Google Scholar] [CrossRef]
- Yao, Z.; Murali, B.; Ren, Q.; Luo, X.; Faget, D.V.; Cole, T.; Ricci, B.; Thotala, D.; Monahan, J.; Van Deursen, J.M.; et al. Therapy-Induced Senescence Drives Bone Loss. Cancer Res. 2020, 80, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Tyutynuk-Massey, L.; Cudjoe, E.K.J.; Idowu, M.O.; Landry, J.W.; Gewirtz, D.A. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front. Oncol. 2018, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Acklin, S.; Zhang, M.; Du, W.; Zhao, X.; Plotkin, M.; Chang, J.; Campisi, J.; Zhou, D.; Xia, F. Depletion of senescent-like neuronal cells alleviates cisplatin-induced peripheral neuropathy in mice. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. Targeting normal and cancer senescent cells as a strategy of senotherapy. Ageing Res. Rev. 2019, 55, 100941. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A Senescence Program Controlled by p53 and p16INK4a Contributes to the Outcome of Cancer Therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef]
- Haugstetter, A.M.; Loddenkemper, C.; Lenze, D.; Gröne, J.; Standfuss, C.; Petersen, I.; Dörken, B.; Schmitt, C.A. Cellular senescence predicts treatment outcome in metastasised colorectal cancer. Br. J. Cancer 2010, 103, 505–509. [Google Scholar] [CrossRef]
- Schönlein, M.; Fan, D.N.-Y.; Ihlow, J.; Schwarzer, R.; Vick, B.; Spiekermann, K.; Burmeister, T.; Westermann, J.; Jeremias, I.; Schmitt, C. Therapy-Induced Senescence Is a Predictor of Treatment Outcome in Acute Myeloid Leukemia. Blood 2017, 130, 1393. [Google Scholar] [CrossRef]
- Wang, L.; De Oliveira, R.L.; Wang, C.; Neto, J.M.F.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willemsen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep. 2017, 21, 773–783. [Google Scholar] [CrossRef]
- Shahbandi, A.; Rao, S.G.; Anderson, A.Y.; Frey, W.D.; Olayiwola, J.O.; Ungerleider, N.A.; Jackson, J.G. BH3 mimetics selectively eliminate chemotherapy-induced senescent cells and improve response in TP53 wild-type breast cancer. Cell Death Differ. 2020, 27, 3097–3116. [Google Scholar] [CrossRef]
- Gayle, S.S.; Sahni, J.M.; Webb, B.M.; Weber-Bonk, K.L.; Shively, M.S.; Spina, R.; Bar, E.E.; Summers, M.K.; Keri, R.A. Targeting BCL-xL improves the efficacy of bromodomain and extra-terminal protein inhibitors in triple-negative breast cancer by eliciting the death of senescent cells. J. Biol. Chem. 2019, 294, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, N.; Vancayseele, A.; Gilbert, S.; Antenor-Habazac, L.; Olivier, M.; Brahem, Z.A.A.; Saad, F.; Delouya, G.; Rodier, F. DNA Damage- But Not Enzalutamide-Induced Senescence in Prostate Cancer Promotes Senolytic Bcl-xL Inhibitor Sensitivity. Cells 2020, 9, 1593. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, J.; Cardin, G.B.; Malaquin, N.; Boisvert, J.-S.; Rodier, F.; Wong, P. Senolytic Targeting of Bcl-2 Anti-Apoptotic Family Increases Cell Death in Irradiated Sarcoma Cells. Cancers 2021, 13, 386. [Google Scholar] [CrossRef] [PubMed]
- Pungsrinont, T.; Sutter, M.F.; Ertingshausen, M.C.C.M.; Lakshmana, G.; Kokal, M.; Khan, A.S.; Baniahmad, A. Senolytic compounds control a distinct fate of androgen receptor agonist- and antagonist-induced cellular senescent LNCaP prostate cancer cells. Cell Biosci. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Suvarna, V.; Singh, V.; Murahari, M. Current overview on the clinical update of Bcl-2 anti-apoptotic inhibitors for cancer therapy. Eur. J. Pharmacol. 2019, 862, 172655. [Google Scholar] [CrossRef]
- Sharma, A.K.; Roberts, R.L.; Benson, R.D.J.; Pierce, J.L.; Yu, K.; Hamrick, M.W.; McGee-Lawrence, M.E. The Senolytic Drug Navitoclax (ABT-263) Causes Trabecular Bone Loss and Impaired Osteoprogenitor Function in Aged Mice. Front. Cell Dev. Biol. 2020, 8, 354. [Google Scholar] [CrossRef]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic Cocktail Dasatinib+Quercetin (D+Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front. Oncol. 2018, 8, 459. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900. [Google Scholar] [CrossRef] [PubMed]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Workman, P. Hsp90 Molecular Chaperone Inhibitors: Are We There Yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Qin, C.; Bai, L.; Wang, S. Small-molecule PROTAC degraders of the Bromodomain and Extra Terminal (BET) proteins—A review. Drug Discov. Today Technol. 2019, 31, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Beck, J.; Turnquist, C.; Horikawa, I.; Harris, C.C. Targeting cellular senescence in cancer and aging: Roles of p53 and its isoforms. Carcinogenesis 2020, 41, 1017–1029. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.-M.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nat. Cell Biol. 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- DeMaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Van Baarle, D.; Tsegaye, A.; Miedema, F.; Akbar, A. Significance of senescence for virus-specific memory T cell responses: Rapid ageing during chronic stimulation of the immune system. Immunol. Lett. 2005, 97, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, S.; Liu, X.; Wang, Y.; Chang, J.; Zhang, X.; Mackintosh, S.G.; Tackett, A.J.; He, Y.; Lv, D.; et al. Oxidation resistance 1 is a novel senolytic target. Aging Cell 2018, 17, e12780. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.-J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nat. Cell Biol. 2020, 583, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Cojocari, D. Hematologic Tumor Cell Resistance to the BCL-2 Inhibitor Venetoclax: A Product of Its Microenvironment? Front. Oncol. 2018, 8, 458. [Google Scholar] [CrossRef]
- González-Gualda, E.; Pàez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R.; González-López, C.; Ou, H.; Mirón-Barroso, S.; Zhang, Z.; Lérida-Viso, A.; et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef]
- Pisco, A.O.; Schaum, N.; McGeever, A.; Karkanias, J.; Neff, N.F.; Darmanis, S.; Wyss-Coray, T.; Quake, S.R. The Tabula Muris Consortium A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nat. Cell Biol. 2020, 583, 590–595. [Google Scholar] [CrossRef]
- Galiana, I.; Lozano-Torres, B.; Sancho, M.; Alfonso, M.; Bernardos, A.; Bisbal, V.; Serrano, M.; Martínez-Máñez, R.; Orzáez, M. Preclinical antitumor efficacy of senescence-inducing chemotherapy combined with a nanoSenolytic. J. Control. Release 2020, 323, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018, 10, e9355. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhang, X.; Chang, J.; Kim, H.; Zhang, P.; Wang, Y.; Khan, S.; Liu, X.; Zhang, X.; Lv, D.-W.; et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2012, 132, 1505–1515. [Google Scholar] [CrossRef]
- Ogden, A.; Rida, P.C.; Knudsen, B.S.; Kucuk, O.; Aneja, R. Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Lett. 2015, 367, 89–92. [Google Scholar] [CrossRef]
- Saleh, T.; Carpenter, V.J.; Bloukh, S.; Gewirtz, D.A. Targeting tumor cell senescence and polyploidy as potential therapeutic strategies. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Senolytic | Mechanism | Model | Reference(s) |
|---|---|---|---|
| Dasatinib + Quercetin | Dasatinib: tyrosine kinase inhibitor Quercetin: flavanol that inhibits various kinases, including PI3K | Irradiated preadipocytes, HUVEC cells, and MEFS; chronic atherosclerotic vascular disease; radiation- or bleomycin-induced lung fibrosis; aging-associated hepatic steatosis; arteriovenous fisulation in chronic kidney disease; Alzheimer’s disease; hyperoxia-induced airway dysfunction; obesity-associated anxiety; obesity-induced metabolic dysfunction; osteoarthritis; radiation-induced bone degeneration; radiation-induced ulceration; doxorubicin-treated HepG2 and Huh-7 cells | [60,68,69,70,71,72,73,74,75,76,77,78,79,80,81,150] |
| Navitoclax (ABT-263) | Inhibits BCL-2, BCL-XL, and BCL-W | Radiation-treated HUVEC, IMR90, and MEF cells; Irradiated bone marrow stem cells; Aging associated bone loss; radiation-induced and bleomycin-induced lung fibrosis; lung emphysema; utrine leiomyoma; tau-dependent neurogeneration; radiation-induced neurodegeneration; myocardial infarction; heart failure; pulmonary hypertension; insulin resistance; osteoarthritis; syntehetic implant-mediated fibrosis; Duchenne muscular dystrophy; doxorubicin-treated and MDA-MB-231 cells; doxorubicin-treated SKBR7, Cal 51, 4226, HCC712, MDA-MB-175, MCF-7, HCC1428, ZR75-30, T47D, U2OS, and MPE600 cells; etoposide-treated and irradiated A549 cells, PARPi-treated OV1369(R2), OV90, OV4453, and OV1946 cells; PARPi-treated and irradiated LNCaP and PC3 cancer cells; irradiated STS93, STS109, and STS117 cells. | [27,50,62,84,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,143,145] |
| Panobinostat | HDAC inhibitor | Cisplatin and taxol treated A549, H460, H1355, FaDu, UMSCC47, and UMSCC1 cells | [153] |
| 17-DMAG | HSP-90 inhibitor | Oxidative stress induced senescent MEFs and MSCs; etoposide-treated IMR90 cells; telomere-shortening induced senescent WI38 cells | [61] |
| Piperlongumine | Multi-faceted; potentially via inhibition of oxidation resistance 1 protein | Irradiated, RAS-overexpressing, and replication-induced senescent WI-38 cells; PARPi-treated OV1369(R2), OV90, OV4453, and OV1946 cells; enzalutamide-treated LNCaP cancer cells | [50,107,145,164] |
| Curcumin/EF-24 | Unidentified; proteasomal degradation of MCL-1 and BCL-XL | Senescent intervertebral disc cells; irradiated and replication-induced senescent WI-38, IMR-90, HUVEC, HREC, and preadipocyte cells | [108,109] |
| Fisetin | Unidentified | Oxidative-stressed induced MEFs; etoposide-treated or irradiated IMR90 cells; irradiated HUVECs; irradiated primary human preadipocytes; progeroid mice; PARPi-treated OV1369(R2), OV90, OV4453, and OV1946 cells | [50,105,106] |
| Cardiac glycosides | Inhibit Na+/K+ ATPase pump; increase expression of NOXA | Bleomycin-, gemcitabine-, doxorubicin-, etoposide-, and palbociclib-treated A549 cells; palbociclib-treated SK-MEL-103; RAS-overexpressing and H202-treated senescent primary BJ cells; senescent HaCat, H1299, U373-MG, H1755, and MCF-7 cells; osteoarthritic chondrocytes; breast cancer PDXs; lung fibrosis; RAS-overexpressing, replicative-induced, and etoposide-, doxorubicin-, and palbociclib-treated IMR90 cells; primary bronchial epithelial cells; | [151,152] |
| Chimeric antigen receptor (CAR) T cells | T cells engineered to target specific cell membrane proteins such as urokinase-type plasminogen activator receptor to redirect specificity to senescent cells | MEKi/CDK4/6i-treated KP cells; hepatic NRASG12V-expressing NSG mice; murine liver fibrosis | [165] |
| Bromodomain and extra-terminal (BET) family protein inhibitors/degraders | Prevent DNA repair and increase autophagy via degradation of the BET protein BRD4 | RAS-overexpressing IMR90 cells; replication-induced, RAS-overexpressing, and doxorubicin-treated TIG-3 cells; obesity-induced murine hepatocellular carcinoma; doxorubicin-treated HCT116 cells; | [154] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. Senolytics for Cancer Therapy: Is All that Glitters Really Gold? Cancers 2021, 13, 723. https://doi.org/10.3390/cancers13040723
Carpenter VJ, Saleh T, Gewirtz DA. Senolytics for Cancer Therapy: Is All that Glitters Really Gold? Cancers. 2021; 13(4):723. https://doi.org/10.3390/cancers13040723
Chicago/Turabian StyleCarpenter, Valerie J., Tareq Saleh, and David A. Gewirtz. 2021. "Senolytics for Cancer Therapy: Is All that Glitters Really Gold?" Cancers 13, no. 4: 723. https://doi.org/10.3390/cancers13040723
APA StyleCarpenter, V. J., Saleh, T., & Gewirtz, D. A. (2021). Senolytics for Cancer Therapy: Is All that Glitters Really Gold? Cancers, 13(4), 723. https://doi.org/10.3390/cancers13040723