
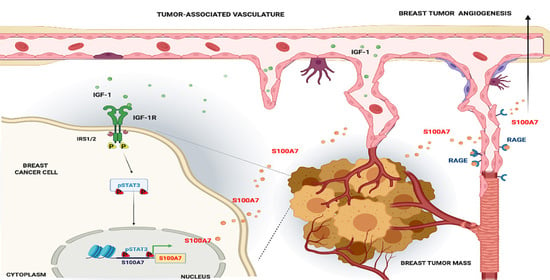
Activation of the S100A7/RAGE Pathway by IGF-1 Contributes to Angiogenesis in Breast Cancer


 ,
,  ,
,  ,
,  and
and 
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. S100A7 Correlates with Negative Prognostic Features in Breast Cancer Patients
2.2. IGF-1/IGF-1R Induces the Expression of S100A7
2.3. IGF-1 Triggers Angiocrine Actions through the Involvement of S100A7/RAGE Signaling
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Publicly Available Molecular Datasets
4.3. Survival Analysis
4.4. Cell Cultures
4.5. Lentiviral Gene Transduction
4.6. Gene Silencing and Plasmids
4.7. Gene Reporter Assays
4.8. Gene Expression Studies
4.9. Western Blot Analysis
4.10. Chromatin Immunoprecipitation (ChIP) Assay
4.11. Conditioned Medium
4.12. ELISA
4.13. Tube Formation Assay
4.14. Sulphorhodamine B (SRB) Assay
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Esposito, V.; Ambrosio, M.R.; Giuliano, M.; Cabaro, S.; Miele, C.; Beguinot, F.; Formisano, P. Mammary Adipose Tissue Control of Breast Cancer Progression: Impact of Obesity and Diabetes. Front. Oncol. 2020, 10, 1554. [Google Scholar] [CrossRef]
- Andò, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, Leptin and Breast Cancer: Epidemiological Evidence and Proposed Mechanisms. Cancers 2019, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Gracheck, P.J.; Vona-Davis, L. The Interactions of Obesity, Inflammation and Insulin Resistance in Breast Cancer. Cancers 2015, 7, 2147–2168. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.F.; Msaouel, P.; Koutsilieris, M. The Role of the Insulin-like Growth Factor-1 System in Breast Cancer. Mol. Cancer 2015, 14, 1–14. [Google Scholar] [CrossRef]
- Vella, V.; De Francesco, E.M.; Lappano, R.; Muoio, M.G.; Manzella, L.; Maggiolini, M.; Belfiore, A. Microenvironmental determinants of BC metastasis: Focus on the crucial interplay between estrogen and insulin/IGF signaling. Front. Cell Dev. Biol. 2020, 8, 1458. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sims, A.H.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P.; Clarke, R.B. GPER Mediates the Angiocrine Actions Induced by IGF1 through the HIF-1α/VEGF Pathway in the Breast Tumor Microenvironment. Breast Cancer Res. 2017, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Matà, R.; Palladino, C.; Nicolosi, M.L.; Presti, A.R.L.; Malaguarnera, R.; Ragusa, M.; Sciortino, D.; Morrione, A.; Maggiolini, M.; Vella, V.; et al. IGF-I Induces Upregulation of DDR1 Collagen Receptor in Breast Cancer Cells by Suppressing MIR-199a-5p through the PI3K/AKT Pathway. Oncotarget 2016, 7, 7683–7700. [Google Scholar] [CrossRef]
- Emberley, E.D.; Murphy, L.C.; Watson, P.H. S100A7 and the Progression of Breast Cancer. Breast Cancer Res. 2004, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.E.; Jang, Y.K. The Histone Demethylase LSD1 Is Required for Estrogen-Dependent S100A7 Gene Expression in Human Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2012, 427, 336–342. [Google Scholar] [CrossRef]
- Skliris, G.P.; Lewis, A.; Emberley, E.; Peng, B.; Weebadda, W.K.; Kemp, A.; Davie, J.R.; Shiu, R.P.C.; Watson, P.H.; Murphy, L.C. Estrogen Receptor-β Regulates Psoriasin (S100A7) in Human Breast Cancer. Breast Cancer Res. Treat. 2007, 104, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Paruchuri, V.; Prasad, A.; McHugh, K.; Bhat, H.K.; Polyak, K.; Ganju, R.K. S100A7-Downregulation Inhibits Epidermal Growth Factor-Induced Signaling in Breast Cancer Cells and Blocks Osteoclast Formation. PLoS ONE 2008, 3, e1741. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.W.; Wani, N.A.; Ahirwar, D.K.; Powell, C.A.; Ravi, J.; Elbaz, M.; Zhao, H.; Padilla, L.; Zhang, X.; Shilo, K.; et al. RAGE Mediates S100A7-Induced Breast Cancer Growth and Metastasis by Modulating the Tumor Microenvironment. Cancer Res. 2015, 75, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, L.; Wang, X.; Cao, Z.; Yang, Q.; Zhang, K. S100A7 Enhances Invasion of Human Breast Cancer MDA-MB-468 Cells through Activation of Nuclear Factor-ΚB Signaling. World J. Surg. Oncol. 2013, 11, 1–18. [Google Scholar] [CrossRef]
- Nasser, M.W.; Qamri, Z.; Deol, Y.S.; Ravi, J.; Powell, C.A.; Trikha, P.; Schwendener, R.A.; Bai, X.-F.; Shilo, K.; Zou, X.; et al. S100A7 Enhances Mammary Tumorigenesis through Upregulation of Inflammatory Pathways. Cancer Res. 2012, 72, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Emberley, E.D.; Niu, Y.; Curtis, L.; Troup, S.; Mandal, S.K.; Myers, J.N.; Gibson, S.B.; Murphy, L.C.; Watson, P.H. The S100A7-c-Jun Activation Domain Binding Protein 1 Pathway Enhances Prosurvival Pathways in Breast Cancer. Cancer Res. 2005, 65, 5696–5702. [Google Scholar] [CrossRef]
- Sakurai, M.; Miki, Y.; Takagi, K.; Suzuki, T.; Ishida, T.; Ohuchi, N.; Sasano, H. Interaction with Adipocyte Stromal Cells Induces Breast Cancer Malignancy via S100A7 Upregulation in Breast Cancer Microenvironment. Breast Cancer Res. 2017, 19, 1–12. [Google Scholar] [CrossRef]
- Lapeire, L.; Hendrix, A.; Lambein, K.; Van Bockstal, M.; Braems, G.; Van Den Broecke, R.; Limame, R.; Mestdagh, P.; Vandesompele, J.; Vanhove, C.; et al. Cancer-Associated Adipose Tissue Promotes Breast Cancer Progression by Paracrine Oncostatin M and Jak/STAT3 Signaling. Cancer Res. 2014, 74, 6806–6819. [Google Scholar] [CrossRef]
- Deol, Y.S.; Nasser, M.W.; Yu, L.; Zou, X.; Ganju, R.K. Tumor-Suppressive Effects of Psoriasin (S100A7) Are Mediated through the β-Catenin/T Cell Factor 4 Protein Pathway in Estrogen Receptor-Positive Breast Cancer Cells. J. Biol. Chem. 2011, 286, 44845–44854. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and Expression of a Cell Surface Receptor for Advanced Glycosylation End Products of Proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Feng, Z.; Zhu, L.; Wu, J. RAGE Signalling in Obesity and Diabetes: Focus on the Adipose Tissue Macrophage. Adipocyte 2020, 9, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Al-Haddad, S.; Zhang, Z.; Leygue, E.; Snell, L.; Huang, A.; Niu, Y.; Hiller-Hitchcock, T.; Hole, K.; Murphy, L.C.; Watson, P.H. Psoriasin (S100A7) Expression and Invasive Breast Cancer. Am. J. Pathol. 1999, 155, 2057–2066. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The Genomic and Transcriptomic Architecture of 2,000 Breast Tumours Reveals Novel Subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like Growth Factor 1 Induces Hypoxia-Inducible Factor 1-Mediated Vascular Endothelial Growth Factor Expression, Which Is Dependent on MAP Kinase and Phosphatidylinositol 3-Kinase Signaling in Colon Cancer Cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef]
- Zhang, W.; Zong, C.S.; Hermanto, U.; Lopez-Bergami, P.; Ronai, Z.; Wang, L.-H. RACK1 Recruits STAT3 Specifically to Insulin and Insulin-Like Growth Factor 1 Receptors for Activation, Which Is Important for Regulating Anchorage-Independent Growth. Mol. Cell. Biol. 2006, 26, 413–424. [Google Scholar] [CrossRef]
- Zong, C.S.; Chan, J.; Levy, D.E.; Horvath, C.; Sadowski, H.B.; Wang, L.-H. Mechanism of STAT3 Activation by Insulin-like Growth Factor I Receptor. J. Biol. Chem. 2000, 275, 15099–15105. [Google Scholar] [CrossRef]
- Fan, J.-J.; Hsu, W.-H.; Lee, K.-H.; Chen, K.-C.; Lin, C.-W.; Lee, Y.-L.; Ko, T.-P.; Lee, L.-T.; Lee, M.-T.; Chang, M.-S.; et al. Dietary Flavonoids Luteolin and Quercetin Inhibit Migration and Invasion of Squamous Carcinoma through Reduction of Src/Stat3/S100A7 Signaling. Antioxidants 2019, 8, 557. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Watson, P.H. S100A7 (Psoriasin) Is Induced by the Proinflammatory Cytokines Oncostatin-M and Interleukin-6 in Human Breast Cancer. Oncogene 2010, 29, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Vegfors, J.; Ekman, A.-K.; Stoll, S.W.; Eding, C.B.; Enerbäck, C. Psoriasin (S100A7) Promotes Stress-Induced Angiogenesis. Br. J. Dermatol. 2016, 175, 1263–1273. [Google Scholar] [CrossRef]
- Padilla, L.; Dakhel, S.; Adan, J.; Masa, M.; Martinez, J.M.; Roque, L.; Coll, T.; Hervas, R.; Calvis, C.; Llinas, L.; et al. S100A7: From Mechanism to Cancer Therapy. Oncogene 2017, 36, 6749–6761. [Google Scholar] [CrossRef]
- Chung, A.S.; Ferrara, N. Developmental and Pathological Angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A Multimodal RAGE-Specific Inhibitor Reduces Amyloid β–Mediated Brain Disorder in a Mouse Model of Alzheimer Disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Staton, C.A.; Reed, M.W.R.; Brown, N.J. A Critical Analysis of Current in Vitro and in Vivo Angiogenesis Assays: Current in Vitro and in Vivo Angiogenesis Assays. Int. J. Exp. Pathol. 2009, 90, 195–221. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Nohata, N.; Lappano, R.; Cirillo, F.; Talia, M.; Scordamaglia, D.; Gutkind, J.S.; Maggiolini, M. IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells 2020, 9, 1010. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Nicolosi, M.L.; Sacco, A.; Morcavallo, A.; Vella, V.; Voci, C.; Spatuzza, M.; Xu, S.-Q.; Iozzo, R.V.; Vigneri, R.; et al. Novel Cross-Talk between IGF-IR and DDR1 Regulates IGF-IR Trafficking, Signaling and Biological Responses. Oncotarget 2015, 6, 16084–16105. [Google Scholar] [CrossRef] [PubMed]
- Pisano, A.; Santolla, M.F.; De Francesco, E.M.; De Marco, P.; Rigiracciolo, D.C.; Perri, M.G.; Vivacqua, A.; Abonante, S.; Cappello, A.R.; Dolce, V.; et al. GPER, IGF-IR, and EGFR Transduction Signaling Are Involved in Stimulatory Effects of Zinc in Breast Cancer Cells and Cancer-Associated Fibroblasts. Mol. Carcinog. 2017, 56, 580–593. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 Proteins in Cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Allgöwer, C.; Kretz, A.-L.; von Karstedt, S.; Wittau, M.; Henne-Bruns, D.; Lemke, J. Friend or Foe: S100 Proteins in Cancer. Cancers 2020, 12, 2037. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.; Mascia, F.; Dharamsi, A.; Howard, O.M.Z.; Cataisson, C.; Bliskovski, V.; Winston, J.; Feigenbaum, L.; Lichti, U.; Ruzicka, T.; et al. Gene from a Psoriasis Susceptibility Locus Primes the Skin for Inflammation. Sci. Transl. Med. 2010, 2, 61ra90. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, F.; Skarmoutsou, E.; Granata, M.; Trovato, C.; Rossi, G.A.; Mazzarino, M.C. S100A7: A RAMPing up AMP Molecule in Psoriasis. Cytokine Growth Factor Rev. 2016, 32, 97–104. [Google Scholar] [CrossRef]
- Krop, I.; März, A.; Carlsson, H.; Li, X.; Bloushtain-Qimron, N.; Hu, M.; Gelman, R.; Sabel, M.S.; Schnitt, S.; Ramaswamy, S.; et al. A Putative Role for Psoriasin in Breast Tumor Progression. Cancer Res. 2005, 65, 11326–11334. [Google Scholar] [CrossRef]
- Mayama, A.; Takagi, K.; Suzuki, H.; Sato, A.; Onodera, Y.; Miki, Y.; Sakurai, M.; Watanabe, T.; Sakamoto, K.; Yoshida, R.; et al. OLFM4, LY6D and S100A7 as Potent Markers for Distant Metastasis in Estrogen Receptor-Positive Breast Carcinoma. Cancer Sci. 2018, 109, 3350–3359. [Google Scholar] [CrossRef]
- Jiang, W.G.; Watkins, G.; Douglas-Jones, A.; Mansel, R.E. Psoriasin Is Aberrantly Expressed in Human Breast Cancer and Is Related to Clinical Outcomes. Int. J. Oncol. 2004, 25, 81–85. [Google Scholar] [CrossRef]
- Cancemi, P.; Buttacavoli, M.; Di Cara, G.; Albanese, N.N.; Bivona, S.; Pucci-Minafra, I.; Feo, S. A Multiomics Analysis of S100 Protein Family in Breast Cancer. Oncotarget 2018, 9, 29064–29081. [Google Scholar] [CrossRef] [PubMed]
- Moog-Lutz, C.; Bouillet, P.; Régnier, C.H.; Tomasetto, C.; Mattei, M.-G.; Chenard, M.-P.; Anglard, P.; Rio, M.-C.; Basset, P. Comparative Expression of Thepsoriasin (S100A7) AndS100C Genes in Breast Carcinoma and Co-Localization to Human Chromosome 1q21-Q22. Int. J. Cancer 1995, 63, 297–303. [Google Scholar] [CrossRef]
- Goh, J.Y.; Feng, M.; Wang, W.; Oguz, G.; Yatim, S.M.J.M.; Lee, P.L.; Bao, Y.; Lim, T.H.; Wang, P.; Tam, W.L.; et al. Chromosome 1q21.3 Amplification Is a Trackable Biomarker and Actionable Target for Breast Cancer Recurrence. Nat. Med. 2017, 23, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Sutton, K.M.; Hayat, S.; Chau, N.-M.; Cook, S.; Pouyssegur, J.; Ahmed, A.; Perusinghe, N.; Floch, R.L.; Yang, J.; Ashcroft, M. Selective Inhibition of MEK1/2 Reveals a Differential Requirement for ERK1/2 Signalling in the Regulation of HIF-1 in Response to Hypoxia and IGF-1. Oncogene 2007, 26, 3920–3929. [Google Scholar] [CrossRef]
- Lee, H.; Ryu, W.-I.; Kim, H.J.; Bae, H.C.; Ryu, H.J.; Shin, J.J.; Song, K.-H.; Kim, T.W.; Son, S.W. TSLP Down-Regulates S100A7 and ß-Defensin 2 Via the JAK2/STAT3-Dependent Mechanism. J. Investig. Dermatol. 2016, 136, 2427–2435. [Google Scholar] [CrossRef] [PubMed]
- Schaaij-Visser, T.B.M.; de Wit, M.; Lam, S.W.; Jiménez, C.R. The Cancer Secretome, Current Status and Opportunities in the Lung, Breast and Colorectal Cancer Context. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2013, 1834, 2242–2258. [Google Scholar] [CrossRef]
- Song, F.; Hurtado del Pozo, C.; Rosario, R.; Zou, Y.S.; Ananthakrishnan, R.; Xu, X.; Patel, P.R.; Benoit, V.M.; Yan, S.F.; Li, H.; et al. RAGE Regulates the Metabolic and Inflammatory Response to High-Fat Feeding in Mice. Diabetes 2014, 63, 1948–1965. [Google Scholar] [CrossRef]
- Pearce, D.A.; Nirmal, A.J.; Freeman, T.C.; Sims, A.H. Continuous Biomarker Assessment by Exhaustive Survival Analysis. bioRxiv 2018, 208660. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1α/GPER Signaling Mediates the Expression of VEGF Induced by Hypoxia in Breast Cancer Associated Fibroblasts (CAFs). Breast Cancer Res. 2013, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Orellana, E.; Kasinski, A. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio-Protocol 2016, 6, e1984. [Google Scholar] [CrossRef]
- Rojas, A.; Morales, M.; Gonzalez, I.; Araya, P. Inhibition of RAGE Axis Signaling: A Pharmacological Challenge. Curr. Durg Targets 2019, 20, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Hardaway, A.L.; Podgorski, I. IL-1β, RAGE and FABP4: Targeting the Dynamic Trio in Metabolic Inflammation and Related Pathologies. Future Med. Chem. 2013, 5, 1089–1108. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muoio, M.G.; Talia, M.; Lappano, R.; Sims, A.H.; Vella, V.; Cirillo, F.; Manzella, L.; Giuliano, M.; Maggiolini, M.; Belfiore, A.; et al. Activation of the S100A7/RAGE Pathway by IGF-1 Contributes to Angiogenesis in Breast Cancer. Cancers 2021, 13, 621. https://doi.org/10.3390/cancers13040621
Muoio MG, Talia M, Lappano R, Sims AH, Vella V, Cirillo F, Manzella L, Giuliano M, Maggiolini M, Belfiore A, et al. Activation of the S100A7/RAGE Pathway by IGF-1 Contributes to Angiogenesis in Breast Cancer. Cancers. 2021; 13(4):621. https://doi.org/10.3390/cancers13040621
Chicago/Turabian StyleMuoio, Maria Grazia, Marianna Talia, Rosamaria Lappano, Andrew H. Sims, Veronica Vella, Francesca Cirillo, Livia Manzella, Marika Giuliano, Marcello Maggiolini, Antonino Belfiore, and et al. 2021. "Activation of the S100A7/RAGE Pathway by IGF-1 Contributes to Angiogenesis in Breast Cancer" Cancers 13, no. 4: 621. https://doi.org/10.3390/cancers13040621
APA StyleMuoio, M. G., Talia, M., Lappano, R., Sims, A. H., Vella, V., Cirillo, F., Manzella, L., Giuliano, M., Maggiolini, M., Belfiore, A., & De Francesco, E. M. (2021). Activation of the S100A7/RAGE Pathway by IGF-1 Contributes to Angiogenesis in Breast Cancer. Cancers, 13(4), 621. https://doi.org/10.3390/cancers13040621