
Cooperative Blockade of CK2 and ATM Kinases Drives Apoptosis in VHL-Deficient Renal Carcinoma Cells through ROS Overproduction


 , , , , , , , and
, , , , , , , and 
Abstract
Simple Summary
Abstract
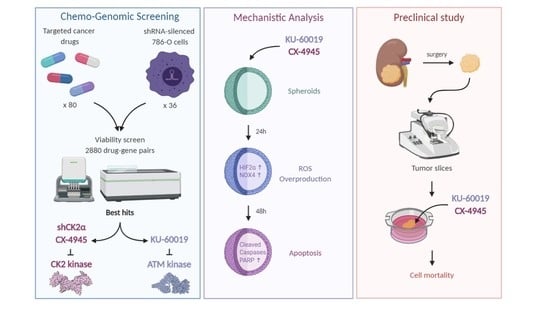
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Transduction in 786-O Cells
2.4. Fully Automated, High-Throughput Screening Assay
2.5. Data Processing and Hit Selection
2.6. Guide RNA Design and Cloning
2.7. Two-Dimension (2D) Viability Assay
2.8. Western Blot Analysis
2.9. Three-Dimension (3D) Assay
2.10. Bulk RNA Barcoding (BRB)-Seq Library Preparation and Sequencing
2.11. BRB-Seq Raw Data Preprocessing
2.12. Bioinformatics Analysis
2.13. 3D Cell Migration Assay
2.14. ROS and Cell Death Measurements
2.15. Mice Orthotopic Tumor Xenograft Models
2.16. Patients and Clinical Samples
2.17. Fresh Tissue Sectioning
2.18. Organotypic Tissue Cultures
2.19. Statistical Analysis
3. Results
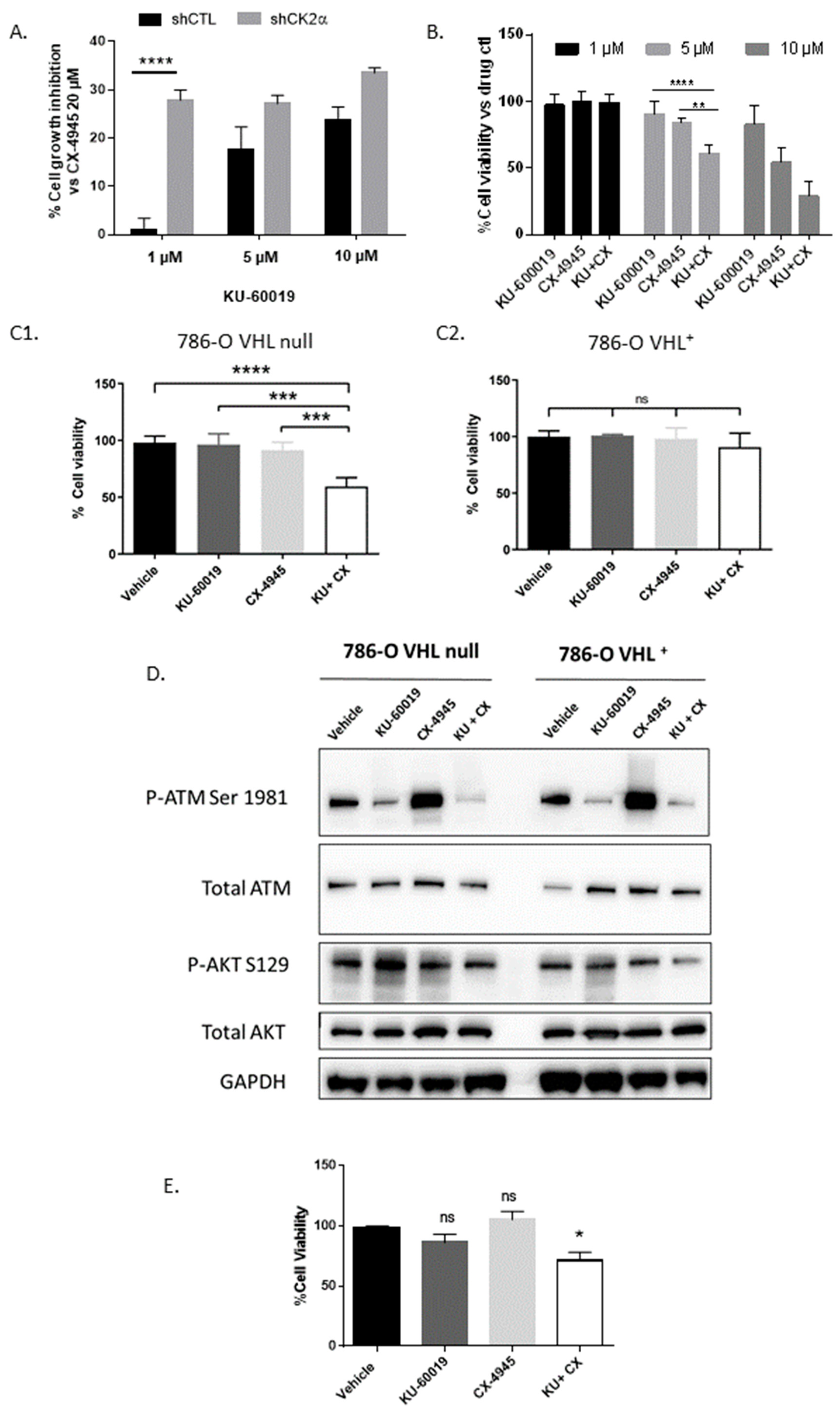
3.1. An Integrated RNAi and Drug Screen Identifies CK2 and ATM as Drug Targets in VHL-Deficient Renal Carcinoma Cells
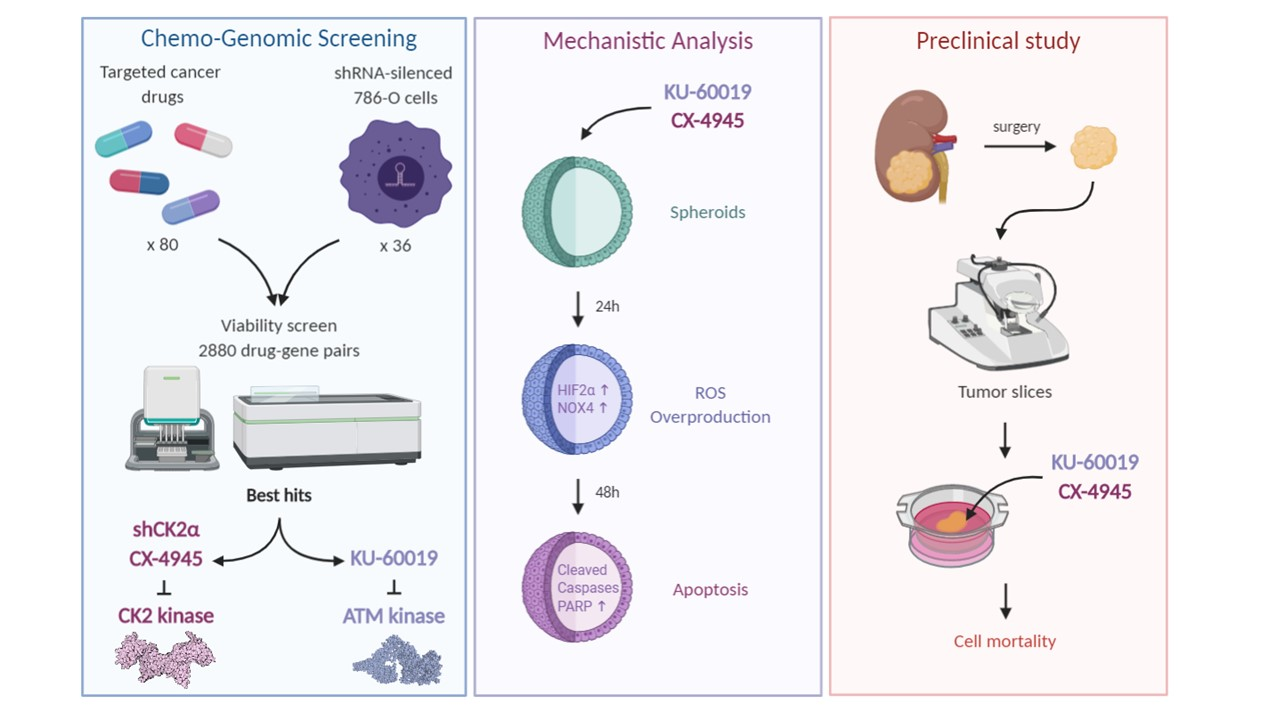
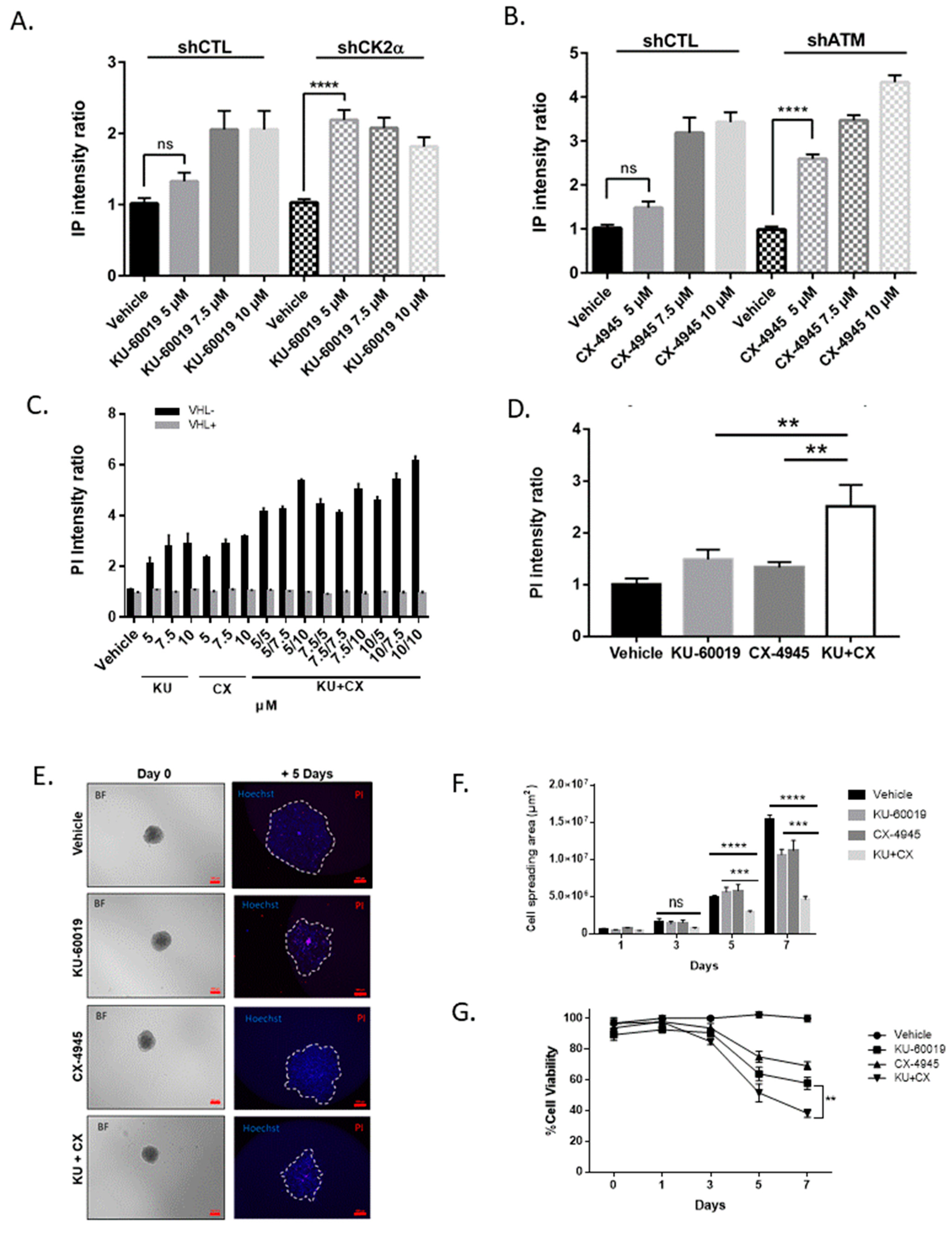
3.2. Combined Inhibition of CK2 and ATM Decreases Cell Migration and Promotes Apoptosis in Renal Multi-Cellular Tumor Spheroids
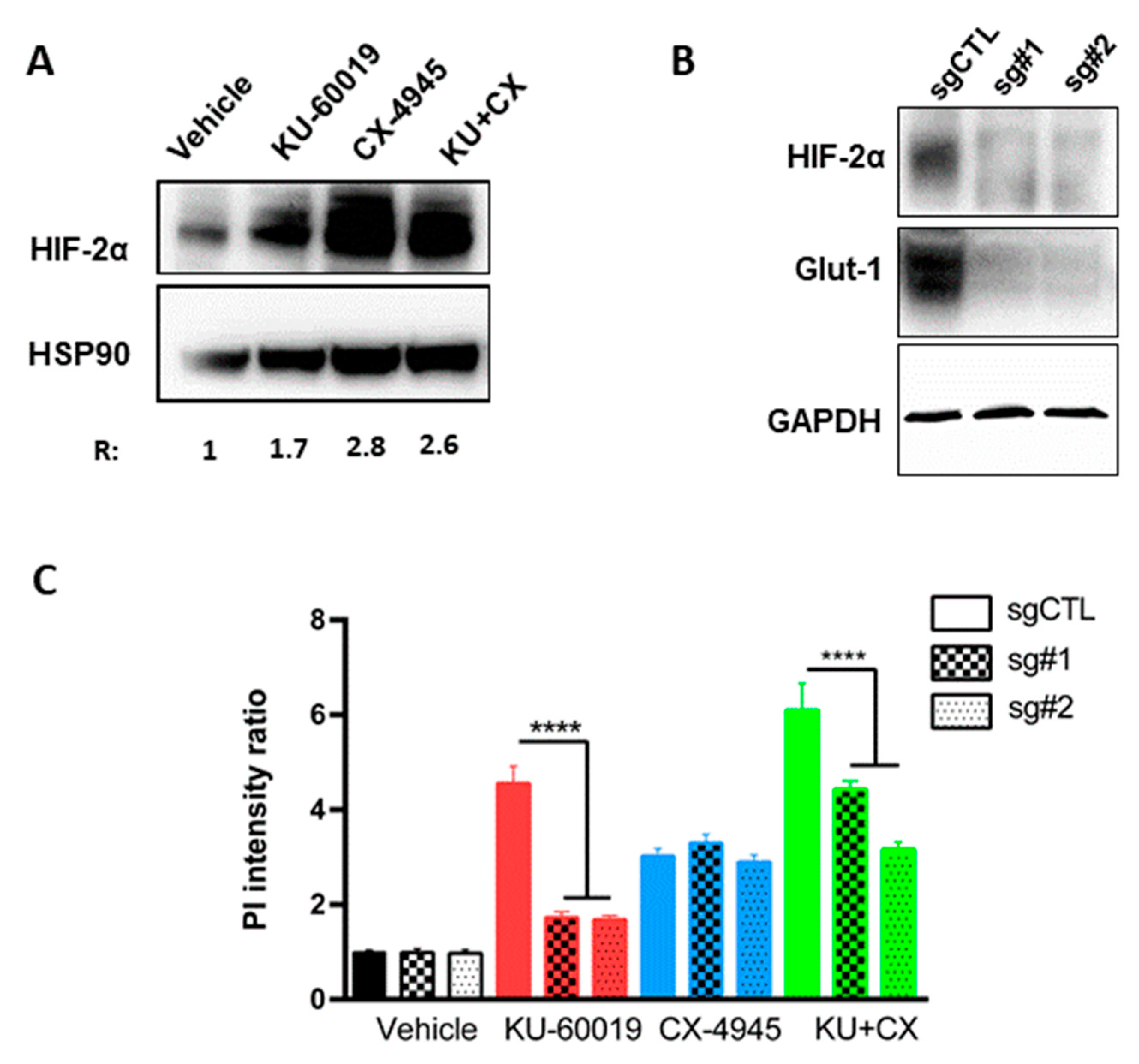
3.3. HIF-2α Expression Enhances Vulnerability to Combined CK2 and ATM Inhibition
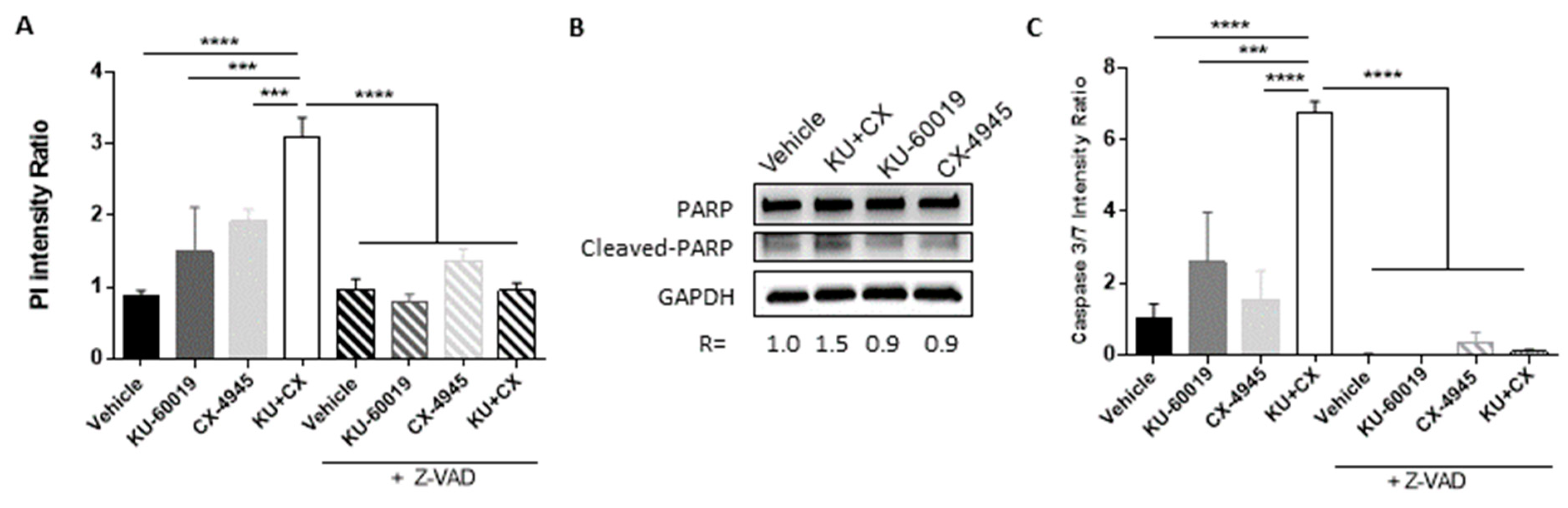
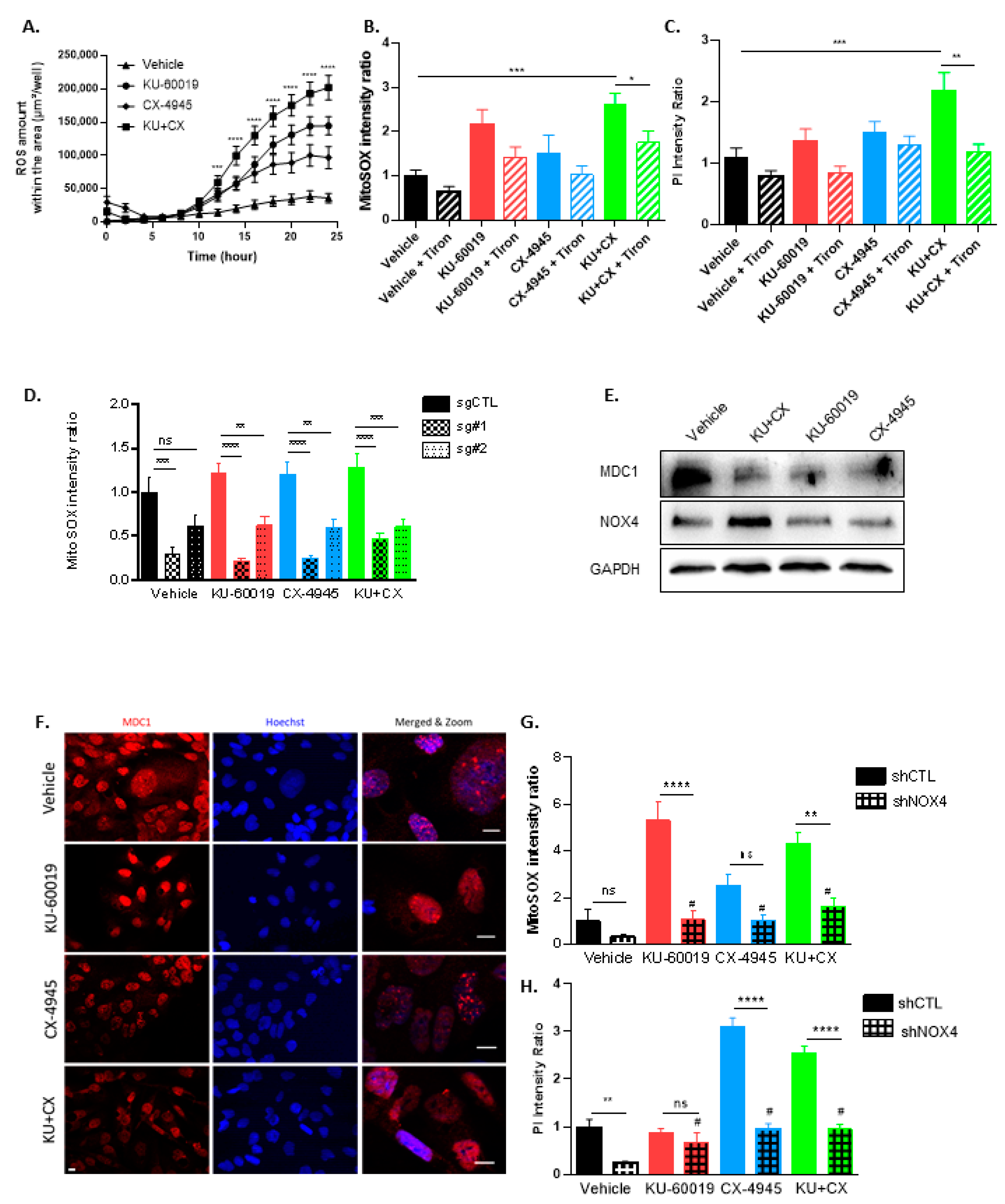
3.4. Cell Death in Response to Combined CK2 and ATM Inhibition Is Dependent on ROS Overproduction
3.5. KU-60019/CX-4945 Combination Leads to a Stronger Transcriptome Deregulation Than the Drugs Alone
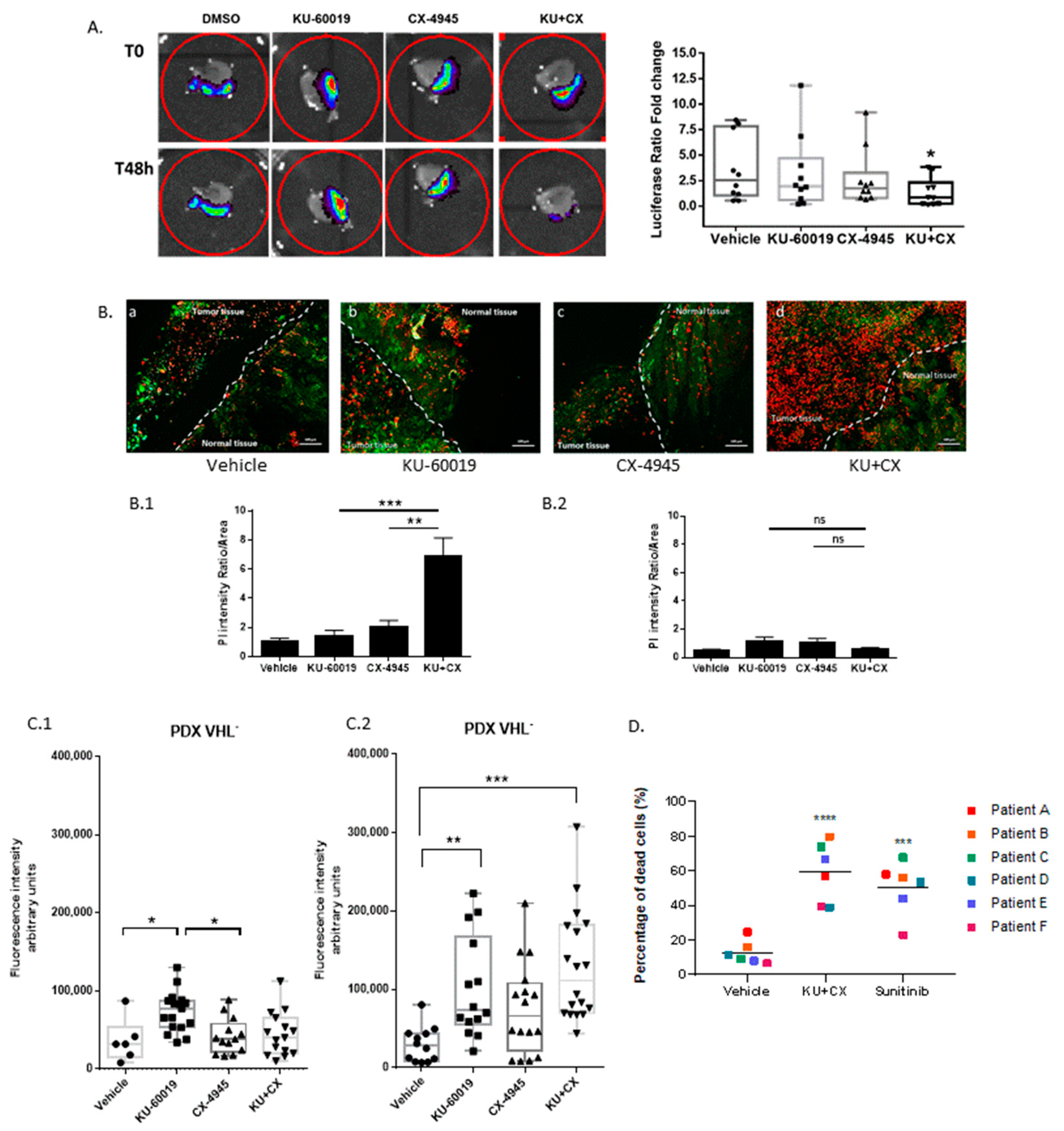
3.6. Cooperative Blockade of CK2 and ATM Induces Cell Death in Ex Vivo Human Renal Tumor Slice Cultures
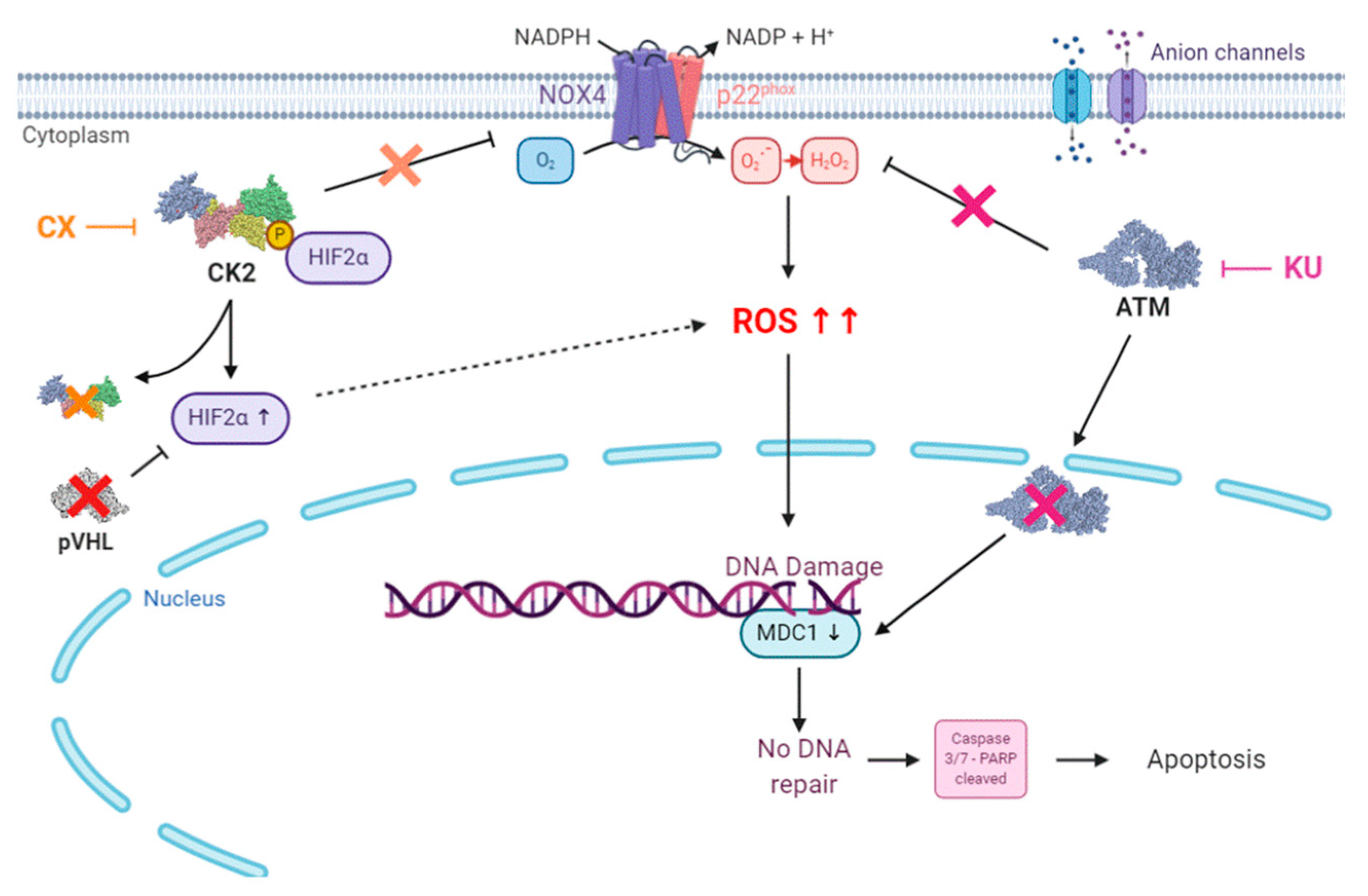
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McDermott, D.F.; Regan, M.M.; Clark, J.I.; Flaherty, L.E.; Weiss, G.R.; Logan, T.F.; Kirkwood, J.M.; Gordon, M.S.; Sosman, J.A.; Ernstoff, M.S.; et al. Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2005, 23, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.Y.; Harrison, D. WHO/ISUP classification, grading and pathological staging of renal cell carcinoma: Standards and controversies. World J. Urol. 2018, 36, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Figlin, R.; Sternberg, C.; Wood, C.G. Novel agents and approaches for advanced renal cell carcinoma. J. Urol. 2012, 188, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Santos, C.R.; Spencer-Dene, B.; Martinez, P.; Endesfelder, D.; Burrell, R.A.; Vetter, M.; Jiang, M.; Saunders, R.E.; Kelly, G.; et al. Genome-wide RNA interference analysis of renal carcinoma survival regulators identifies MCT4 as a Warburg effect metabolic target. J. Pathol. 2012, 227, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kim, W.Y.; Lechpammer, M.; Kaelin, W.G., Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003, 1, E83. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Crooks, D.R.; Linehan, W.M. Targeting HIF2alpha in Clear-Cell Renal Cell Carcinoma. Cancer Cell 2016, 30, 515–517. [Google Scholar] [CrossRef]
- Kondo, K.; Klco, J.; Nakamura, E.; Lechpammer, M.; Kaelin, W.G., Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002, 1, 237–246. [Google Scholar] [CrossRef]
- Roberts, A.M.; Ohh, M. Beyond the hypoxia-inducible factor-centric tumour suppressor model of von Hippel-Lindau. Curr. Opin. Oncol. 2008, 20, 83–89. [Google Scholar] [CrossRef]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer 2008, 8, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 2015, 125, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol 2010, 28, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomised phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef]
- Escudier, B.; Bellmunt, J.; Negrier, S.; Bajetta, E.; Melichar, B.; Bracarda, S.; Ravaud, A.; Golding, S.; Jethwa, S.; Sneller, V. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): Final analysis of overall survival. J. Clin. Oncol 2010, 28, 2144–2150. [Google Scholar] [CrossRef]
- Maj-Hes, A.; Medioni, J.; Scotte, F.; Schmidinger, M.; Kramer, G.; Combe, P.; Gornadha, Y.; Elaidi, R.; Oudard, S. Rechallenge with mTOR inhibitors in metastatic renal cell carcinoma patients who progressed on previous mTOR inhibitor therapy. Oncology 2013, 85, 8–13. [Google Scholar] [CrossRef]
- Posadas, E.M.; Limvorasak, S.; Figlin, R.A. Targeted therapies for renal cell carcinoma. Nat. Rev. Nephrol 2017, 13, 496–511. [Google Scholar] [CrossRef]
- Wheeler, D.A.; Takebe, N.; Hinoue, T.; Hoadley, K.A.; Cardenas, M.F.; Hamilton, A.M.; Laird, P.W.; Wang, L.; Johnson, A.; Dewal, N.; et al. Molecular Features of Cancers Exhibiting Exceptional Responses to Treatment. Cancer Cell 2021, 39, 38–53.e37. [Google Scholar] [CrossRef] [PubMed]
- Lehar, J.; Stockwell, B.R.; Giaever, G.; Nislow, C. Combination chemical genetics. Nat. Chem. Biol. 2008, 4, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Muellner, M.K.; Uras, I.Z.; Gapp, B.V.; Kerzendorfer, C.; Smida, M.; Lechtermann, H.; Craig-Mueller, N.; Colinge, J.; Duernberger, G.; Nijman, S.M. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat. Chem. Biol. 2011, 7, 787–793. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.C.; Burman, B.; Kruhlak, M.J.; Misteli, T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014, 9, 1703–1717. [Google Scholar] [CrossRef]
- Tang, S.; Yang, L.; Tang, X.; Liu, M. The role of oxidized ATM in the regulation of oxidative stress-induced energy metabolism reprogramming of CAFs. Cancer Lett 2014, 353, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.S.; Park, S.C. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Strzyz, P. Cell Senescence: A new role for ATM. Nat. Rev. Mol. Cell Biol. 2017, 18, 277. [Google Scholar] [CrossRef]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell. Mol. Life Sci. CMLS 2016, 73, 2829–2850. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Scott, A.; Murray, D. New insights into p53 signaling and cancer cell response to DNA damage: Implications for cancer therapy. J. Biomed. Biotechnol. 2012, 2012, 170325. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Luo, G.; Du, W.; Kong, T.; Liu, C.; Liu, Z. Recent Advances in Microfluidic Platforms Applied in Cancer Metastasis: Circulating Tumor Cells’ (CTCs) Isolation and Tumor-On-A-Chip. Small 2020, 16, e1903899. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta 2008, 1784, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: CK2: A key player in cancer biology. Cell. Mol. Life Sci. CMLS 2009, 66, 1858–1867. [Google Scholar] [CrossRef]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining CK2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar] [CrossRef]
- Rabalski, A.J.; Gyenis, L.; Litchfield, D.W. Molecular Pathways: Emergence of Protein Kinase CK2 (CSNK2) as a Potential Target to Inhibit Survival and DNA Damage Response and Repair Pathways in Cancer Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 2840–2847. [Google Scholar] [CrossRef]
- Chua, M.M.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef]
- Silva-Pavez, E.; Tapia, J.C. Protein Kinase CK2 in Cancer Energetics. Front. Oncol. 2020, 10, 893. [Google Scholar] [CrossRef]
- Roelants, C.; Giacosa, S.; Duchemin-Pelletier, E.; McLeer-Florin, A.; Tisseyre, C.; Aubert, C.; Champelovier, P.; Boutonnat, J.; Descotes, J.-L.; Rambeaud, J.-J. Dysregulated Expression of Protein Kinase CK2 in Renal Cancer; Advances in Biochemistry in Health; Khalil Ahmed, O.-G.I., Szyszka, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Vilardell, J.; Alcaraz, E.; Sarro, E.; Trilla, E.; Cuadros, T.; de Torres, I.; Plana, M.; Ramon, Y.C.S.; Pinna, L.A.; Ruzzene, M.; et al. Under-expression of CK2beta subunit in ccRCC represents a complementary biomarker of p-STAT3 Ser727 that correlates with patient survival. Oncotarget 2018, 9, 5736–5751. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-Chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2010, 54, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Alpern, D.; Gardeux, V.; Russeil, J.; Mangeat, B.; Meireles-Filho, A.C.A.; Breysse, R.; Hacker, D.; Deplancke, B. BRB-seq: Ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing. Genome Biol. 2019, 20, 71. [Google Scholar] [CrossRef] [PubMed]
- Draskau, M.K.; Lardenois, A.; Evrard, B.; Boberg, J.; Chalmel, F.; Svingen, T. Transcriptome analysis of fetal rat testis following intrauterine exposure to the azole fungicides triticonazole and flusilazole reveals subtle changes despite adverse endocrine effects. Chemosphere 2020, 264, 128468. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Gunther, W.; Pawlak, E.; Damasceno, R.; Arnold, H.; Terzis, A.J. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br. J. Cancer 2003, 88, 463–469. [Google Scholar] [CrossRef]
- Roelants, C.; Giacosa, S.; Pillet, C.; Bussat, R.; Champelovier, P.; Bastien, O.; Guyon, L.; Arnoux, V.; Cochet, C.; Filhol, O. Combined inhibition of PI3K and Src kinases demonstrates synergistic therapeutic efficacy in clear-cell renal carcinoma. Oncotarget 2018, 9, 30066–30078. [Google Scholar] [CrossRef]
- Roelants, C.; Pillet, C.; Franquet, Q.; Sarrazin, C.; Peilleron, N.; Giacosa, S.; Guyon, L.; Fontanell, A.; Fiard, G.; Long, J.A.; et al. Ex-Vivo Treatment of Tumor Tissue Slices as a Predictive Preclinical Method to Evaluate Targeted Therapies for Patients with Renal Carcinoma. Cancers 2020, 12, 232. [Google Scholar] [CrossRef]
- Astsaturov, I.; Ratushny, V.; Sukhanova, A.; Einarson, M.B.; Bagnyukova, T.; Zhou, Y.; Devarajan, K.; Silverman, J.S.; Tikhmyanova, N.; Skobeleva, N.; et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci. Signal. 2010, 3, ra67. [Google Scholar] [CrossRef] [PubMed]
- Mathews Griner, L.A.; Guha, R.; Shinn, P.; Young, R.M.; Keller, J.M.; Liu, D.; Goldlust, I.S.; Yasgar, A.; McKnight, C.; Boxer, M.B.; et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Kurose, A.; Tanaka, T.; Huang, X.; Halicka, H.D.; Traganos, F.; Dai, W.; Darzynkiewicz, Z. Assessment of ATM phosphorylation on Ser-1981 induced by DNA topoisomerase I and II inhibitors in relation to Ser-139-histone H2AX phosphorylation, cell cycle phase, and apoptosis. Cytom. A 2005, 68, 1–9. [Google Scholar] [CrossRef] [PubMed]
- So, S.; Davis, A.J.; Chen, D.J. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell Biol. 2009, 187, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; Ruzzene, M. Role of protein kinase CK2 in antitumor drug resistance. J. Exp. Clin. Cancer Res. CR 2019, 38, 287. [Google Scholar] [CrossRef]
- Kucejova, B.; Pena-Llopis, S.; Yamasaki, T.; Sivanand, S.; Tran, T.A.; Alexander, S.; Wolff, N.C.; Lotan, Y.; Xie, X.J.; Kabbani, W.; et al. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol. Cancer Res. MCR 2011, 9, 1255–1265. [Google Scholar] [CrossRef]
- Ashida, S.; Nishimori, I.; Tanimura, M.; Onishi, S.; Shuin, T. Effects of von Hippel-Lindau gene mutation and methylation status on expression of transmembrane carbonic anhydrases in renal cell carcinoma. J. Cancer Res. Clin. Oncol. 2002, 128, 561–568. [Google Scholar] [CrossRef]
- Martins, L.R.; Lucio, P.; Melao, A.; Antunes, I.; Cardoso, B.A.; Stansfield, R.; Bertilaccio, M.T.; Ghia, P.; Drygin, D.; Silva, M.G.; et al. Activity of the clinical-stage CK2-specific inhibitor CX-4945 against chronic lymphocytic leukemia. Leukemia 2014, 28, 179–182. [Google Scholar] [CrossRef]
- Marschke, R.F.; Borad, M.J.; McFarland, R.W.; Alvarez, R.H.; Lim, J.K.; Padgett, C.S.; von Hoff, D.D.; O’Brien, S.E.; Northfelt, D.W. Findings from the phase I clinical trials of CX-4945, an orally available inhibitor of CK2. ASCO Meet. Abstr. 2011, 29, 3087. [Google Scholar] [CrossRef]
- Giacosa, S.; Pillet, C.; Cochet, C.; Filhol, O.; Barette, C.; Soleilhac, E. A Synthetic Lethal Drug Combination for Treating Renal Cell Carcinoma. Google Patents WO2017050842A1, 13 September 2018. [Google Scholar]
- Dubessy, C.; Merlin, J.M.; Marchal, C.; Guillemin, F. Spheroids in radiobiology and photodynamic therapy. Crit. Rev. Oncol. Hematol. 2000, 36, 179–192. [Google Scholar] [CrossRef]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Chesnel, F.; Hascoet, P.; Gagne, J.P.; Couturier, A.; Jouan, F.; Poirier, G.G.; Le Goff, C.; Vigneau, C.; Danger, Y.; Verite, F.; et al. The von Hippel-Lindau tumour suppressor gene: Uncovering the expression of the pVHL172 isoform. Br. J. Cancer 2015, 113, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Beroukhim, R.; Schumacher, S.E.; Zhou, J.; Chang, M.; Signoretti, S.; Kaelin, W.G., Jr. Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov. 2011, 1, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef]
- Mottet, D.; Ruys, S.P.; Demazy, C.; Raes, M.; Michiels, C. Role for casein kinase 2 in the regulation of HIF-1 activity. Int. J. Cancer 2005, 117, 764–774. [Google Scholar] [CrossRef]
- Gradin, K.; Takasaki, C.; Fujii-Kuriyama, Y.; Sogawa, K. The transcriptional activation function of the HIF-like factor requires phosphorylation at a conserved threonine. J. Biol. Chem. 2002, 277, 23508–23514. [Google Scholar] [CrossRef]
- Lancaster, D.E.; McNeill, L.A.; McDonough, M.A.; Aplin, R.T.; Hewitson, K.S.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. Disruption of dimerization and substrate phosphorylation inhibit factor inhibiting hypoxia-inducible factor (FIH) activity. Biochem. J. 2004, 383, 429–437. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Watanabe, S.; Iimori, M.; Chan, D.V.; Hara, E.; Kitao, H.; Maehara, Y. MDC1 methylation mediated by lysine methyltransferases EHMT1 and EHMT2 regulates active ATM accumulation flanking DNA damage sites. Sci. Rep. 2018, 8, 10888. [Google Scholar] [CrossRef]
- Jungmichel, S.; Clapperton, J.A.; Lloyd, J.; Hari, F.J.; Spycher, C.; Pavic, L.; Li, J.; Haire, L.F.; Bonalli, M.; Larsen, D.H.; et al. The molecular basis of ATM-dependent dimerization of the Mdc1 DNA damage checkpoint mediator. Nucleic Acids Res. 2012, 40, 3913–3928. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Geiszt, M.; Kopp, J.B.; Varnai, P.; Leto, T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef] [PubMed]
- Gregg, J.L.; Turner, R.M., 2nd; Chang, G.; Joshi, D.; Zhan, Y.; Chen, L.; Maranchie, J.K. NADPH oxidase NOX4 supports renal tumorigenesis by promoting the expression and nuclear accumulation of HIF2alpha. Cancer Res. 2014, 74, 3501–3511. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Kiselyov, K.; Muallem, S. ROS and intracellular ion channels. Cell Calcium 2016, 60, 108–114. [Google Scholar] [CrossRef]
- Yde, C.W.; Olsen, B.B.; Meek, D.; Watanabe, N.; Guerra, B. The regulatory beta-subunit of protein kinase CK2 regulates cell-cycle progression at the onset of mitosis. Oncogene 2008, 27, 4986–4997. [Google Scholar] [CrossRef]
- Guerra, B.; Issinger, O.G. Role of Protein Kinase CK2 in Aberrant Lipid Metabolism in Cancer. Pharmaceuticals 2020, 13, 292. [Google Scholar] [CrossRef]
- Qaiser, F.; Trembley, J.H.; Kren, B.T.; Wu, J.J.; Naveed, A.K.; Ahmed, K. Protein kinase CK2 inhibition induces cell death via early impact on mitochondrial function. J. Cell. Biochem. 2014, 115, 2103–2115. [Google Scholar] [CrossRef]
- Montenarh, M.; Gotz, C. Protein kinase CK2 and ion channels (Review). Biomed. Rep. 2020, 13, 55. [Google Scholar] [CrossRef]
- Bleijs, M.; van de Wetering, M.; Clevers, H.; Drost, J. Xenograft and organoid model systems in cancer research. EMBO J. 2019, 38, e101654. [Google Scholar] [CrossRef] [PubMed]
- Schmeichel, K.L.; Bissell, M.J. Modeling tissue-specific signaling and organ function in three dimensions. J. Cell Sci. 2003, 116, 2377–2388. [Google Scholar] [CrossRef] [PubMed]
- Vaira, V.; Fedele, G.; Pyne, S.; Fasoli, E.; Zadra, G.; Bailey, D.; Snyder, E.; Faversani, A.; Coggi, G.; Flavin, R.; et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 8352–8356. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Pena-Llopis, S.; Zhao, H.; Kucejova, B.; Spence, P.; Pavia-Jimenez, A.; Yamasaki, T.; McBride, D.J.; Gillen, J.; Wolff, N.C.; et al. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci. Transl. Med. 2012, 4, 137ra175. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Atkins, M.B. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009, 10, 992–1000. [Google Scholar] [CrossRef]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef]
- Assrir, N.; Filhol, O.; Galisson, F.; Lipinski, M. HIRIP3 is a nuclear phosphoprotein interacting with and phosphorylated by the serine-threonine kinase CK2. Biol. Chem. 2007, 388, 391–398. [Google Scholar] [CrossRef]
- Kappes, F.; Damoc, C.; Knippers, R.; Przybylski, M.; Pinna, L.A.; Gruss, C. Phosphorylation by protein kinase CK2 changes the DNA binding properties of the human chromatin protein DEK. Mol. Cell Biol. 2004, 24, 6011–6020. [Google Scholar] [CrossRef]
- Wise, C.A.; Chiang, L.C.; Paznekas, W.A.; Sharma, M.; Musy, M.M.; Ashley, J.A.; Lovett, M.; Jabs, E.W. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc. Natl. Acad. Sci. USA 1997, 94, 3110–3115. [Google Scholar] [CrossRef]
- Ciccia, A.; Huang, J.W.; Izhar, L.; Sowa, M.E.; Harper, J.W.; Elledge, S.J. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2014, 111, 18631–18636. [Google Scholar] [CrossRef]
- Buljan, M.; Ciuffa, R.; van Drogen, A.; Vichalkovski, A.; Mehnert, M.; Rosenberger, G.; Lee, S.; Varjosalo, M.; Pernas, L.E.; Spegg, V.; et al. Kinase Interaction Network Expands Functional and Disease Roles of Human Kinases. Mol. Cell 2020, 79, 504–520.e9. [Google Scholar] [CrossRef] [PubMed]
- Pinna, L.A. Protein kinase CK2: A challenge to canons. J. Cell Sci. 2002, 115, 3873–3878. [Google Scholar] [CrossRef] [PubMed]
- Slaton, J.W.; Unger, G.M.; Sloper, D.T.; Davis, A.T.; Ahmed, K. Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol. Cancer Res. MCR 2004, 2, 712–721. [Google Scholar]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2016, 20, 319–340. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Bliesath, J.; Macalino, D.; Omori, M.; Huser, N.; Streiner, N.; Ho, C.B.; Anderes, K.; Proffitt, C.; O’Brien, S.E.; et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: Mechanistic rationale for drug combination therapy. Mol. Cancer Ther. 2012, 11, 994–1005. [Google Scholar] [CrossRef]
- Bliesath, J.; Huser, N.; Omori, M.; Bunag, D.; Proffitt, C.; Streiner, N.; Ho, C.; Siddiqui-Jain, A.; O’Brien, S.E.; Lim, J.K.; et al. Combined inhibition of EGFR and CK2 augments the attenuation of PI3K-Akt-mTOR signaling and the killing of cancer cells. Cancer Lett. 2012, 322, 113–118. [Google Scholar] [CrossRef]
- Parker, R.; Clifton-Bligh, R.; Molloy, M.P. Phosphoproteomics of MAPK inhibition in BRAF-mutated cells and a role for the lethal synergism of dual BRAF and CK2 inhibition. Mol. Cancer Ther. 2014, 13, 1894–1906. [Google Scholar] [CrossRef]
- Song, C.; Ge, Z.; Ding, Y.; Tan, B.H.; Desai, D.; Gowda, K.; Amin, S.; Gowda, R.; Robertson, G.P.; Yue, F.; et al. IKAROS and CK2 regulate expression of BCL-XL and chemosensitivity in high-risk B-cell acute lymphoblastic leukemia. Blood 2020, 136, 1520–1534. [Google Scholar] [CrossRef]
- Bian, Y.; Han, J.; Kannabiran, V.; Mohan, S.; Cheng, H.; Friedman, J.; Zhang, L.; VanWaes, C.; Chen, Z. MEK Inhibitor PD-0325901 Overcomes Resistance to CK2 Inhibitor CX-4945 and Exhibits Anti-Tumor Activity in Head and Neck Cancer. Int. J. Biol. Sci. 2015, 11, 411–422. [Google Scholar] [CrossRef]
- Khalil, H.S.; Tummala, H.; Hupp, T.R.; Zhelev, N. Pharmacological inhibition of ATM by KU55933 stimulates ATM transcription. Exp. Biol. Med. (Maywood) 2012, 237, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E. Banking on ATM as a new target in metabolic syndrome. Cell Metab. 2006, 4, 337–338. [Google Scholar] [CrossRef]
- Barascu, A.; Le Chalony, C.; Pennarun, G.; Genet, D.; Imam, N.; Lopez, B.; Bertrand, P. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J. 2012, 31, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Deshpande, R.; Paull, T.T. ATM activation in the presence of oxidative stress. Cell Cycle 2010, 9, 4805–4811. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Woo, D.K.; Green, P.D.; Santos, J.H.; D’Souza, A.D.; Walther, Z.; Martin, W.D.; Christian, B.E.; Chandel, N.S.; Shadel, G.S. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am. J. Pathol. 2012, 180, 24–31. [Google Scholar] [CrossRef]
- Liou, G.Y.; Doppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef]
- Teppo, H.R.; Soini, Y.; Karihtala, P. Reactive Oxygen Species-Mediated Mechanisms of Action of Targeted Cancer Therapy. Oxidative Med. Cell. Longev. 2017, 2017, 1485283. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Kirtonia, A.; Sethi, G.; Garg, M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell. Mol. Life Sci. CMLS 2020, 77, 4459–4483. [Google Scholar] [CrossRef] [PubMed]
- Gillissen, B.; Richter, A.; Richter, A.; Preissner, R.; Schulze-Osthoff, K.; Essmann, F.; Daniel, P.T. Bax/Bak-independent mitochondrial depolarization and reactive oxygen species induction by sorafenib overcome resistance to apoptosis in renal cell carcinoma. J. Biol. Chem. 2017, 292, 6478–6492. [Google Scholar] [CrossRef]
- Bokoch, G.M.; Diebold, B.; Kim, J.S.; Gianni, D. Emerging evidence for the importance of phosphorylation in the regulation of NADPH oxidases. Antioxid. Redox Signal. 2009, 11, 2429–2441. [Google Scholar] [CrossRef]
- Weyemi, U.; Redon, C.E.; Aziz, T.; Choudhuri, R.; Maeda, D.; Parekh, P.R.; Bonner, M.Y.; Arbiser, J.L.; Bonner, W.M. NADPH oxidase 4 is a critical mediator in Ataxia telangiectasia disease. Proc. Natl. Acad. Sci. USA 2015, 112, 2121–2126. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, Y.H.; Song, H.Z.; Ji, H.; Wang, X.L.; Wang, L.; Qian, J.; Ling, J.J.; Ping, F.F. 5d, a novel analogue of 3-n-butylphthalide, decreases NADPH oxidase activity through the positive regulation of CK2 after ischemia/reperfusion injury. Oncotarget 2016, 7, 39444–39457. [Google Scholar] [CrossRef][Green Version]
- Jung, S.N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: New developments in an old debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Maranchie, J.K.; Zhan, Y. Nox4 is critical for hypoxia-inducible factor 2-alpha transcriptional activity in von Hippel-Lindau-deficient renal cell carcinoma. Cancer Res. 2005, 65, 9190–9193. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. New horizons in hypoxia signaling pathways. Exp. Cell Res. 2017, 356, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Staller, P.; Sulitkova, J.; Lisztwan, J.; Moch, H.; Oakeley, E.J.; Krek, W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature 2003, 425, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Vanharanta, S.; Massague, J. Origins of metastatic traits. Cancer Cell 2013, 24, 410–421. [Google Scholar] [CrossRef]
- Wallace, E.M.; Rizzi, J.P.; Han, G.; Wehn, P.M.; Cao, Z.; Du, X.; Cheng, T.; Czerwinski, R.M.; Dixon, D.D.; Goggin, B.S.; et al. A Small-Molecule Antagonist of HIF2alpha Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016, 76, 5491–5500. [Google Scholar] [CrossRef]
- Bertout, J.A.; Majmundar, A.J.; Gordan, J.D.; Lam, J.C.; Ditsworth, D.; Keith, B.; Brown, E.J.; Nathanson, K.L.; Simon, M.C. HIF2alpha inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc. Natl. Acad. Sci. USA 2009, 106, 14391–14396. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Kaelin, W.G., Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef]
- Diao, S.; Zheng, Q.; Gao, J.; Yao, Y.; Ren, S.; Liu, Y.; Xu, Y. Trefoil factor 3 contributes to the malignancy of glioma via regulating HIF-1alpha. Oncotarget 2017, 8, 76770–76782. [Google Scholar] [CrossRef][Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacosa, S.; Pillet, C.; Séraudie, I.; Guyon, L.; Wallez, Y.; Roelants, C.; Battail, C.; Evrard, B.; Chalmel, F.; Barette, C.; et al. Cooperative Blockade of CK2 and ATM Kinases Drives Apoptosis in VHL-Deficient Renal Carcinoma Cells through ROS Overproduction. Cancers 2021, 13, 576. https://doi.org/10.3390/cancers13030576
Giacosa S, Pillet C, Séraudie I, Guyon L, Wallez Y, Roelants C, Battail C, Evrard B, Chalmel F, Barette C, et al. Cooperative Blockade of CK2 and ATM Kinases Drives Apoptosis in VHL-Deficient Renal Carcinoma Cells through ROS Overproduction. Cancers. 2021; 13(3):576. https://doi.org/10.3390/cancers13030576
Chicago/Turabian StyleGiacosa, Sofia, Catherine Pillet, Irinka Séraudie, Laurent Guyon, Yann Wallez, Caroline Roelants, Christophe Battail, Bertrand Evrard, Frédéric Chalmel, Caroline Barette, and et al. 2021. "Cooperative Blockade of CK2 and ATM Kinases Drives Apoptosis in VHL-Deficient Renal Carcinoma Cells through ROS Overproduction" Cancers 13, no. 3: 576. https://doi.org/10.3390/cancers13030576
APA StyleGiacosa, S., Pillet, C., Séraudie, I., Guyon, L., Wallez, Y., Roelants, C., Battail, C., Evrard, B., Chalmel, F., Barette, C., Soleilhac, E., Fauvarque, M.-O., Franquet, Q., Sarrazin, C., Peilleron, N., Fiard, G., Long, J.-A., Descotes, J.-L., Cochet, C., & Filhol, O. (2021). Cooperative Blockade of CK2 and ATM Kinases Drives Apoptosis in VHL-Deficient Renal Carcinoma Cells through ROS Overproduction. Cancers, 13(3), 576. https://doi.org/10.3390/cancers13030576