
The Potential of T Cell Factor 1 in Sustaining CD8+ T Lymphocyte-Directed Anti-Tumor Immunity



Abstract
Simple Summary
Abstract
1. Introduction
2. T Cell Factor 1
3. CD8+ T Lymphocytes: Heterogeneity & Differentiation
3.1. CD8+ Effector T (TEFF) Cells
3.2. CD8+ Memory T Cells
4. CD8+ T Lymphocyte Responses in Anti-Tumor Immunity
4.1. Effector CD8+ T Cell-Mediated Immunity against Tumors
4.2. Memory CD8+ T Cell-Mediated Immunity against Tumors
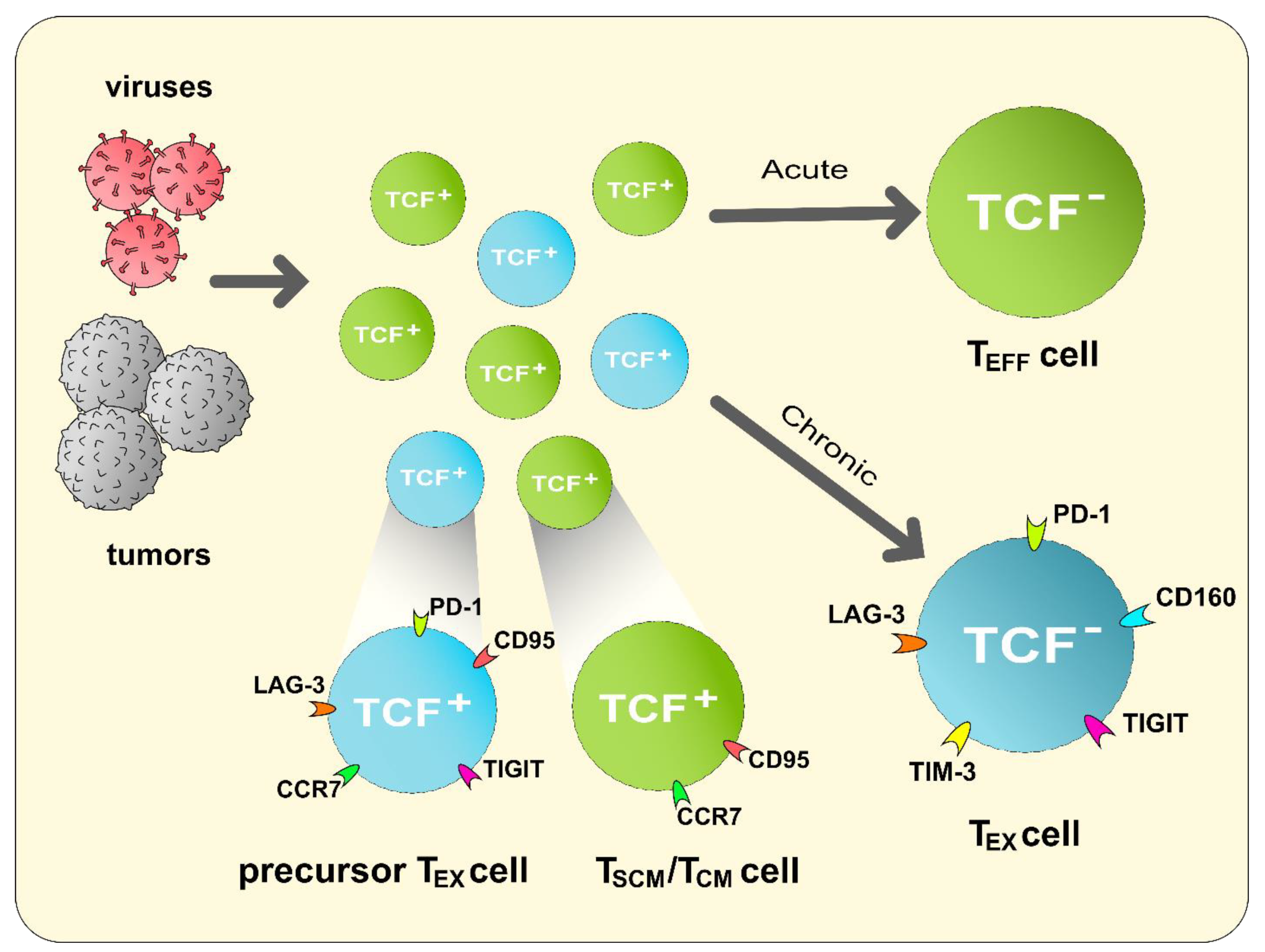
5. Exhaustion of CD8+ T Lymphocytes
5.1. Mechanisms of CD8+ T Cell Exhaustion
5.2. Inhibitory Immune Checkpoints
5.3. CD8+ TEX Cells in the TME
6. ICB as a Method of T Cell Reinvigoration
6.1. Resistance to ICB Therapy
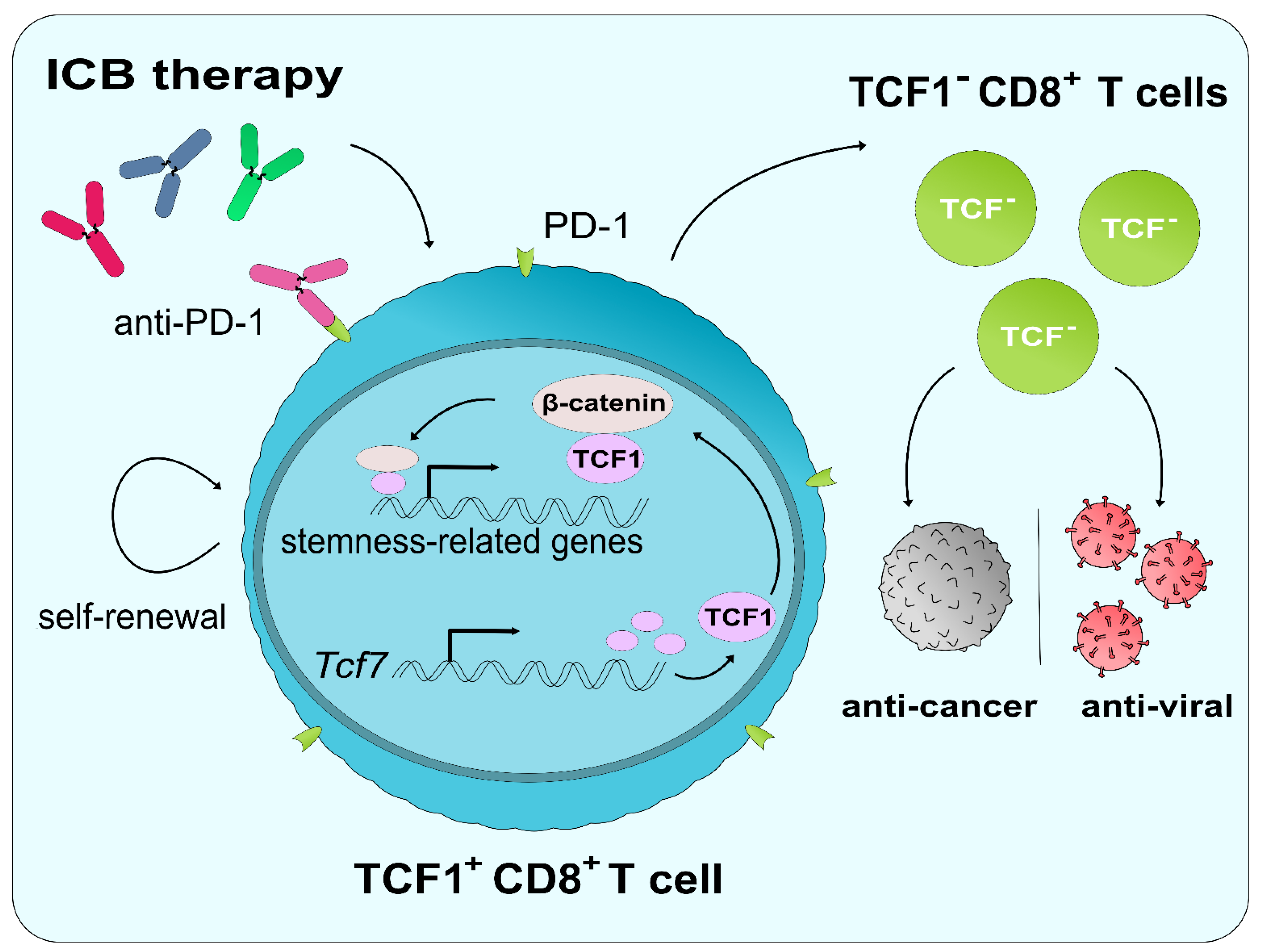
6.2. The Role of TCF1 in ICB and CD8+ T Cell Reinvigoration
7. Role of TCF1 in Tregs
8. Conclusions: Challenges and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APC | antigen-presenting cell |
| CAR | chimeric antigen receptor |
| CTL | cytotoxic T lymphocytes |
| CTLA-4 | cytotoxic-T-lymphocyte-associated protein 4 |
| DC | dendritic cell |
| FDA | (U.S.) Food and Drug Administration |
| ICB | immune checkpoint blockade therapy |
| IFN | interferon |
| iPSC | induced pluripotent stem cell |
| MHC | major histocompatibility complex |
| MPEC | memory precursor effector cell |
| LEF | lymphocyte enhancer factor |
| MDSC | myeloid-derived suppressor cell |
| PD-1 | programmed cell death 1 |
| PD-L | programmed cell death ligand |
| SLEC | short-lived effector cell |
| T9 CAR-T | interleukin-9-secreting chimeric antigen receptor-T cell |
| TAM | tumor-associated macrophage |
| TCF1 | T cell factor 1 |
| TCM | central memory T cell |
| TCR | T cell receptor |
| TEFF | effector T cell |
| TEM | effector memory T cell |
| TEX | exhausted T cell |
| TIGIT | T cell immunoreceptor with Ig and ITIM domain |
| TIL | tumor-infiltrating lymphocyte |
| TME | tumor microenvironment |
| Treg | regulatory T cell |
| TRM | tissue-resident memory T cell |
| TSCM | stem-cell memory T cell |
| WRE | Wnt-response element |
References
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Popat, V.; Gerber, D.E. Hyperprogressive disease: A distinct effect of immunotherapy? J. Thorac. Dis. 2019, 11, S262–S265. [Google Scholar] [CrossRef] [PubMed]
- Meder, L.; Schuldt, P.; Thelen, M.; Schmitt, A.; Dietlein, F.; Klein, S.; Borchmann, S.; Wennhold, K.; Vlasic, I.; Oberbeck, S.; et al. Combined VEGF and PD-L1 Blockade Displays Synergistic Treatment Effects in an Autochthonous Mouse Model of Small Cell Lung Cancer. Cancer Res. 2018, 78, 4270–4281. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [PubMed]
- Escobar, G.; Mangani, D.; Anderson, A.C. T cell factor 1: A master regulator of the T cell response in disease. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Welten, S.P.M.; Yermanos, A.; Baumann, N.S.; Wagen, F.; Oetiker, N.; Sandu, I.; Pedrioli, A.; Oduro, J.D.; Reddy, S.T.; Cicin-Sain, L.; et al. Tcf1+ cells are required to maintain the inflationary T cell pool upon MCMV infection. Nat. Commun. 2020, 11, 2295. [Google Scholar] [CrossRef]
- Kurtulus, S.; Madi, A.; Escobar, G.; Klapholz, M.; Nyman, J.; Christian, E.; Pawlak, M.; Dionne, D.; Xia, J.; Rozenblatt-Rosen, O.; et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1. Immunity 2019, 50, 181–194.e186. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+ PD-1+ CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e110. [Google Scholar] [CrossRef]
- Wang, K.; Fu, W. Transcriptional regulation of Treg homeostasis and functional specification. Cell Mol. Life Sci. 2020, 77, 4269–4287. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Rha, M.S.; Jeong, H.W.; Ko, J.H.; Choi, S.J.; Seo, I.H.; Lee, J.S.; Sa, M.; Kim, A.R.; Joo, E.J.; Ahn, J.Y.; et al. PD-1-Expressing SARS-CoV-2-Specific CD8+ T Cells Are Not Exhausted, but Functional in Patients with COVID-19. Immunity 2020, 54, 44–52.e3. [Google Scholar] [CrossRef] [PubMed]
- Van Beest, M.; Dooijes, D.; van De Wetering, M.; Kjaerulff, S.; Bonvin, A.; Nielsen, O.; Clevers, H. Sequence-Specific high mobility group box factors recognize 10-12-base pair minor groove motifs. J. Biol. Chem. 2000, 275, 27266–27273. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destrée, O.; Clevers, H. XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Boudousquié, C.; Danilo, M.; Pousse, L.; Jeevan-Raj, B.; Angelov, G.S.; Chennupati, V.; Zehn, D.; Held, W. Differences in the transduction of canonical Wnt signals demarcate effector and memory CD8 T cells with distinct recall proliferation capacity. J. Immunol. 2014, 193, 2784–2791. [Google Scholar] [CrossRef]
- Raghu, D.; Xue, H.H.; Mielke, L.A. Control of Lymphocyte Fate, Infection, and Tumor Immunity by TCF-1. Trends Immunol. 2019, 40, 1149–1162. [Google Scholar] [CrossRef]
- Danilo, M.; Chennupati, V.; Silva, J.G.; Siegert, S.; Held, W. Suppression of Tcf1 by Inflammatory Cytokines Facilitates Effector CD8 T Cell Differentiation. Cell Rep. 2018, 22, 2107–2117. [Google Scholar] [CrossRef]
- Zhao, M.; Kiernan, C.H.; Stairiker, C.J.; Hope, J.L.; Leon, L.G.; van Meurs, M.; Brouwers-Haspels, I.; Boers, R.; Boers, J.; Gribnau, J.; et al. Rapid in vitro generation of bona fide exhausted CD8+ T cells is accompanied by Tcf7 promotor methylation. PLoS Pathog. 2020, 16, e1008555. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Van Genderen, C.; Okamura, R.M.; Fariñas, I.; Quo, R.G.; Parslow, T.G.; Bruhn, L.; Grosschedl, R. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 1994, 8, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, S.; Izon, D.; Hofhuis, F.; Robanus-Maandag, E.; te Riele, H.; van de Wetering, M.; Oosterwegel, M.; Wilson, A.; MacDonald, H.R.; Clevers, H. An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature 1995, 374, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Okamura, R.M.; Sigvardsson, M.; Galceran, J.; Verbeek, S.; Clevers, H.; Grosschedl, R. Redundant regulation of T cell differentiation and TCRα gene expression by the transcription factors LEF-1 and TCF-1. Immunity 1998, 8, 11–20. [Google Scholar] [CrossRef]
- Hoppler, S.; Kavanagh, C.L. Wnt signalling: Variety at the core. J. Cell Sci. 2007, 120, 385–393. [Google Scholar] [CrossRef]
- Held, W.; Clevers, H.; Grosschedl, R. Redundant functions of TCF-1 and LEF-1 during T and NK cell development, but unique role of TCF-1 for Ly49 NK cell receptor acquisition. Eur. J. Immunol. 2003, 33, 1393–1398. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.; Tan, A.; Song, Y.; Lee, H.; Ying, Q.L.; Jho, E.H. The Distinct Role of Tcfs and Lef1 in the Self-Renewal or Differentiation of Mouse Embryonic Stem Cells. Int. J. Stem Cells 2020, 13, 192–201. [Google Scholar] [CrossRef]
- Ucar, D.; Márquez, E.J.; Chung, C.H.; Marches, R.; Rossi, R.J.; Uyar, A.; Wu, T.C.; George, J.; Stitzel, M.L.; Palucka, A.K.; et al. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. J. Exp. Med. 2017, 214, 3123–3144. [Google Scholar] [CrossRef]
- Tserel, L.; Kolde, R.; Limbach, M.; Tretyakov, K.; Kasela, S.; Kisand, K.; Saare, M.; Vilo, J.; Metspalu, A.; Milani, L.; et al. Age-related profiling of DNA methylation in CD8+ T cells reveals changes in immune response and transcriptional regulator genes. Sci. Rep. 2015, 5, 13107. [Google Scholar] [CrossRef]
- Abdelsamed, H.A.; Moustaki, A.; Fan, Y.; Dogra, P.; Ghoneim, H.E.; Zebley, C.C.; Triplett, B.M.; Sekaly, R.P.; Youngblood, B. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J. Exp. Med. 2017, 214, 1593–1606. [Google Scholar] [CrossRef]
- Roser, M.; Ritchie, H. Cancer. Available online: https://ourworldindata.org/cancer (accessed on 31 December 2020).
- Kim, C.; Jin, J.; Weyand, C.M.; Goronzy, J.J. The Transcription Factor TCF1 in T Cell Differentiation and Aging. Int. J. Mol. Sci. 2020, 21, 6497. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.; Sarkar, S.; Gourley, T.S.; Rouse, B.T.; Ahmed, R. Differentiation of memory B and T cells. Curr. Opin. Immunol. 2006, 18, 255–264. [Google Scholar] [CrossRef]
- Kaech, S.M.; Ahmed, R. Memory CD8+ T cell differentiation: Initial antigen encounter triggers a developmental program in naïve cells. Nat. Immunol. 2001, 2, 415–422. [Google Scholar] [CrossRef]
- Opferman, J.T.; Ober, B.T.; Ashton-Rickardt, P.G. Linear differentiation of cytotoxic effectors into memory T lymphocytes. Science 1999, 283, 1745–1748. [Google Scholar] [CrossRef]
- Van Stipdonk, M.J.; Lemmens, E.E.; Schoenberger, S.P. Naïve CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat. Immunol. 2001, 2, 423–429. [Google Scholar] [CrossRef]
- Van Stipdonk, M.J.; Hardenberg, G.; Bijker, M.S.; Lemmens, E.E.; Droin, N.M.; Green, D.R.; Schoenberger, S.P. Dynamic programming of CD8+ T lymphocyte responses. Nat. Immunol. 2003, 4, 361–365. [Google Scholar] [CrossRef]
- Wong, P.; Pamer, E.G. Cutting edge: Antigen-independent CD8 T cell proliferation. J. Immunol. 2001, 166, 5864–5868. [Google Scholar] [CrossRef]
- Yuzefpolskiy, Y.; Baumann, F.M.; Kalia, V.; Sarkar, S. Early CD8 T-cell memory precursors and terminal effectors exhibit equipotent in vivo degranulation. Cell Mol. Immunol. 2015, 12, 400–408. [Google Scholar] [CrossRef]
- Obar, J.J.; Molloy, M.J.; Jellison, E.R.; Stoklasek, T.A.; Zhang, W.; Usherwood, E.J.; Lefrançois, L. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc. Natl. Acad. Sci. USA 2010, 107, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Mosaheb, M.M.; Flavell, R.A. Opposing effects of TGF-βand IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity 2009, 31, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Wiesel, M.; Crouse, J.; Bedenikovic, G.; Sutherland, A.; Joller, N.; Oxenius, A. Type-I IFN drives the differentiation of short-lived effector CD8+ T cells in vivo. Eur. J. Immunol. 2012, 42, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, M.; Kurachi, J.; Suenaga, F.; Tsukui, T.; Abe, J.; Ueha, S.; Tomura, M.; Sugihara, K.; Takamura, S.; Kakimi, K.; et al. Chemokine receptor CXCR3 facilitates CD8+ T cell differentiation into short-lived effector cells leading to memory degeneration. J. Exp. Med. 2011, 208, 1605–1620. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Teichgräber, V.; Becker, T.C.; Masopust, D.; Kaech, S.M.; Antia, R.; von Andrian, U.H.; Ahmed, R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003, 4, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef]
- Hikono, H.; Kohlmeier, J.E.; Takamura, S.; Wittmer, S.T.; Roberts, A.D.; Woodland, D.L. Activation phenotype, rather than central- or effector-memory phenotype, predicts the recall efficacy of memory CD8+ T cells. J. Exp. Med. 2007, 204, 1625–1636. [Google Scholar] [CrossRef]
- Olson, J.A.; McDonald-Hyman, C.; Jameson, S.C.; Hamilton, S.E. Effector-like CD8⁺ T cells in the memory population mediate potent protective immunity. Immunity 2013, 38, 1250–1260. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Wolint, P.; Schwarz, K.; Jäger, P.; Oxenius, A. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. J. Immunol. 2005, 175, 4686–4696. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Förster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.K.; Paukovics, G.; Cashin, K.; Borm, K.; Ellett, A.; Roche, M.; Jakobsen, M.R.; Churchill, M.J.; Gorry, P.R. Quantifying susceptibility of CD4+ stem memory T-cells to infection by laboratory adapted and clinical HIV-1 strains. Viruses 2014, 6, 709–726. [Google Scholar] [CrossRef] [PubMed]
- Newell, E.W.; Davis, M.M. Beyond model antigens: High-Dimensional methods for the analysis of antigen-specific T cells. Nat. Biotechnol. 2014, 32, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Sant, A.J.; McMichael, A. Revealing the role of CD4+ T cells in viral immunity. J. Exp. Med. 2012, 209, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Conley, J.M.; Gallagher, M.P.; Berg, L.J. T Cells and Gene Regulation: The Switching on and Turning Up of Genes after T Cell Receptor Stimulation in CD8 T Cells. Front. Immunol. 2016, 7, 76. [Google Scholar] [CrossRef]
- Lanzavecchia, A.; Sallusto, F. Regulation of T cell immunity by dendritic cells. Cell 2001, 106, 263–266. [Google Scholar] [CrossRef]
- Mehlhop-Williams, E.R.; Bevan, M.J. Memory CD8+ T cells exhibit increased antigen threshold requirements for recall proliferation. J. Exp. Med. 2014, 211, 345–356. [Google Scholar] [CrossRef]
- Unsoeld, H.; Krautwald, S.; Voehringer, D.; Kunzendorf, U.; Pircher, H. Cutting edge: CCR7+ and CCR7- memory T cells do not differ in immediate effector cell function. J. Immunol. 2002, 169, 638–641. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 2006, 211, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Menares, E.; Gálvez-Cancino, F.; Cáceres-Morgado, P.; Ghorani, E.; López, E.; Díaz, X.; Saavedra-Almarza, J.; Figueroa, D.A.; Roa, E.; Quezada, S.A.; et al. Tissue-Resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019, 10, 4401. [Google Scholar] [CrossRef] [PubMed]
- Gallimore, A.; Glithero, A.; Godkin, A.; Tissot, A.C.; Plückthun, A.; Elliott, T.; Hengartner, H.; Zinkernagel, R. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J. Exp. Med. 1998, 187, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef]
- Fuller, M.J.; Zajac, A.J. Ablation of CD8 and CD4 T cell responses by high viral loads. J. Immunol. 2003, 170, 477–486. [Google Scholar] [CrossRef]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef]
- Shin, H.; Wherry, E.J. CD8 T cell dysfunction during chronic viral infection. Curr. Opin. Immunol. 2007, 19, 408–415. [Google Scholar] [CrossRef]
- Shin, H.; Blackburn, S.D.; Blattman, J.N.; Wherry, E.J. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J. Exp. Med. 2007, 204, 941–949. [Google Scholar] [CrossRef]
- Wherry, E.J.; Barber, D.L.; Kaech, S.M.; Blattman, J.N.; Ahmed, R. Antigen-Independent memory CD8 T cells do not develop during chronic viral infection. Proc. Natl. Acad. Sci. USA 2004, 101, 16004–16009. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 9th ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 397–416. [Google Scholar]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-Associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef]
- Fang, M.; Yuan, J.; Peng, C.; Li, Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 2014, 35, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Schmielau, J.; Nalesnik, M.A.; Finn, O.J. Suppressed T-cell receptor ζ chain expression and cytokine production in pancreatic cancer patients. Clin. Cancer Res. 2001, 7, 933s–939s. [Google Scholar]
- Nagaraj, S.; Schrum, A.G.; Cho, H.I.; Celis, E.; Gabrilovich, D.I. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J. Immunol. 2010, 184, 3106–3116. [Google Scholar] [CrossRef]
- Duraiswamy, J.; Freeman, G.J.; Coukos, G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013, 73, 6900–6912. [Google Scholar] [CrossRef]
- Loke, P.; Allison, J.P. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5336–5341. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, M.; De Placido, S.; Ascierto, P.A. Recent success and limitations of immune checkpoint inhibitors for cancer: A lesson from melanoma. Virchows Arch. 2019, 474, 421–432. [Google Scholar] [CrossRef]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef]
- Hudson, W.H.; Gensheimer, J.; Hashimoto, M.; Wieland, A.; Valanparambil, R.M.; Li, P.; Lin, J.X.; Konieczny, B.T.; Im, S.J.; Freeman, G.J.; et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ stem-line CD8+ T Cells during Chronic Infection. Immunity 2019, 51, 1043–1058.e1044. [Google Scholar] [CrossRef]
- Principe, N.; Kidman, J.; Goh, S.; Tilsed, C.M.; Fisher, S.A.; Fear, V.S.; Forbes, C.A.; Zemek, R.M.; Chopra, A.; Watson, M.; et al. Tumor infiltrating Effector Memory Antigen-Specific CD8+ T Cells Predict Response to Immune Checkpoint Therapy. Front. Immunol. 2020, 11, 584423. [Google Scholar] [CrossRef]
- Jeannet, G.; Boudousquié, C.; Gardiol, N.; Kang, J.; Huelsken, J.; Held, W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. USA 2010, 107, 9777–9782. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, S.; Zhao, D.M.; Harty, J.T.; Badovinac, V.P.; Xue, H.H. Differentiation and persistence of memory CD8+ T cells depend on T cell factor 1. Immunity 2010, 33, 229–240. [Google Scholar] [CrossRef]
- Wu, J.; Madi, A.; Mieg, A.; Hotz-Wagenblatt, A.; Weisshaar, N.; Ma, S.; Mohr, K.; Schlimbach, T.; Hering, M.; Borgers, H.; et al. T Cell Factor 1 Suppresses CD103+ Lung Tissue-Resident Memory T Cell Development. Cell Rep. 2020, 31, 107484. [Google Scholar] [CrossRef]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Baharom, F.; Ramirez-Valdez, R.A.; Tobin, K.K.S.; Yamane, H.; Dutertre, C.A.; Khalilnezhad, A.; Reynoso, G.V.; Coble, V.L.; Lynn, G.M.; Mulè, M.P.; et al. Intravenous nanoparticle vaccination generates stem-like TCF1. Nat. Immunol. 2021, 22, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Van der Veeken, J.; Glasner, A.; Zhong, Y.; Hu, W.; Wang, Z.M.; Bou-Puerto, R.; Charbonnier, L.M.; Chatila, T.A.; Leslie, C.S.; Rudensky, A.Y. The Transcription Factor Foxp3 Shapes Regulatory T Cell Identity by Tuning the Activity of trans-Acting Intermediaries. Immunity 2020, 53, 971–984.e975. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.H.; Wang, K.; Wan, S.; Liang, Y.; Yuan, X.; Dong, Y.; Cho, S.; Xu, W.; Jepsen, K.; Feng, G.S.; et al. TCF1 and LEF1 Control Treg Competitive Survival and Tfr Development to Prevent Autoimmune Diseases. Cell Rep. 2019, 27, 3629–3645.e3626. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.M.; Höllbacher, B.; Campbell, D.J. Cutting Edge: Dynamic Expression of Id3 Defines the Stepwise Differentiation of Tissue-Resident Regulatory T Cells. J. Immunol. 2019, 202, 31–36. [Google Scholar] [CrossRef]
- Peligero-Cruz, C.; Givony, T.; Sebé-Pedrós, A.; Dobeš, J.; Kadouri, N.; Nevo, S.; Roncato, F.; Alon, R.; Goldfarb, Y.; Abramson, J. IL18 signaling promotes homing of mature Tregs into the thymus. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Gai, K.; Li, X.; Shao, P.; Zeng, Z.; Zhao, X.; Chen, X.; Paradee, W.J.; Meyerholz, D.K.; Peng, W.; et al. Tcf1 and Lef1 are required for the immunosuppressive function of regulatory T cells. J. Exp. Med. 2019, 216, 847–866. [Google Scholar] [CrossRef]
- Van Loosdregt, J.; Fleskens, V.; Tiemessen, M.M.; Mokry, M.; van Boxtel, R.; Meerding, J.; Pals, C.E.; Kurek, D.; Baert, M.R.; Delemarre, E.M.; et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity 2013, 39, 298–310. [Google Scholar] [CrossRef]
- Keerthivasan, S.; Aghajani, K.; Dose, M.; Molinero, L.; Khan, M.W.; Venkateswaran, V.; Weber, C.; Emmanuel, A.O.; Sun, T.; Bentrem, D.J.; et al. Β-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci. Transl. Med. 2014, 6, 225ra28. [Google Scholar] [CrossRef]
- Chu, T.; Berner, J.; Zehn, D. Two parallel worlds of memory T cells. Nat. Immunol. 2020, 21, 1484–1485. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e828. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, D.T.; Gabriel, S.S.; Chisanga, D.; Gloury, R.; Gubser, P.M.; Vasanthakumar, A.; Shi, W.; Kallies, A. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol. 2020, 21, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.; Zehn, D. Charting the Roadmap of T Cell Exhaustion. Immunity 2020, 52, 724–726. [Google Scholar] [CrossRef]
- Liu, L.; Bi, E.; Ma, X.; Xiong, W.; Qian, J.; Ye, L.; Su, P.; Wang, Q.; Xiao, L.; Yang, M.; et al. Enhanced CAR-T activity against established tumors by polarizing human T cells to secrete interleukin-9. Nat. Commun. 2020, 11, 5902. [Google Scholar] [CrossRef]
- Balkhi, M.Y.; Wittmann, G.; Xiong, F.; Junghans, R.P. YY1 Upregulates Checkpoint Receptors and Downregulates Type I Cytokines in Exhausted, Chronically Stimulated Human T Cells. iScience 2018, 2, 105–122. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Crawford, A.; Shin, H.; Polley, A.; Freeman, G.J.; Wherry, E.J. Tissue-Specific differences in PD-1 and PD-L1 expression during chronic viral infection: Implications for CD8 T-cell exhaustion. J. Virol. 2010, 84, 2078–2089. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, S.; Baek, J.-H. The Potential of T Cell Factor 1 in Sustaining CD8+ T Lymphocyte-Directed Anti-Tumor Immunity. Cancers 2021, 13, 515. https://doi.org/10.3390/cancers13030515
Jung S, Baek J-H. The Potential of T Cell Factor 1 in Sustaining CD8+ T Lymphocyte-Directed Anti-Tumor Immunity. Cancers. 2021; 13(3):515. https://doi.org/10.3390/cancers13030515
Chicago/Turabian StyleJung, Sungmin, and Jea-Hyun Baek. 2021. "The Potential of T Cell Factor 1 in Sustaining CD8+ T Lymphocyte-Directed Anti-Tumor Immunity" Cancers 13, no. 3: 515. https://doi.org/10.3390/cancers13030515
APA StyleJung, S., & Baek, J.-H. (2021). The Potential of T Cell Factor 1 in Sustaining CD8+ T Lymphocyte-Directed Anti-Tumor Immunity. Cancers, 13(3), 515. https://doi.org/10.3390/cancers13030515