
Side-by-Side Comparison of Five Chelators for 89Zr-Labeling of Biomolecules: Investigation of Chemical/Radiochemical Properties and Complex Stability

 , ,
, ,  ,
, 

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. General
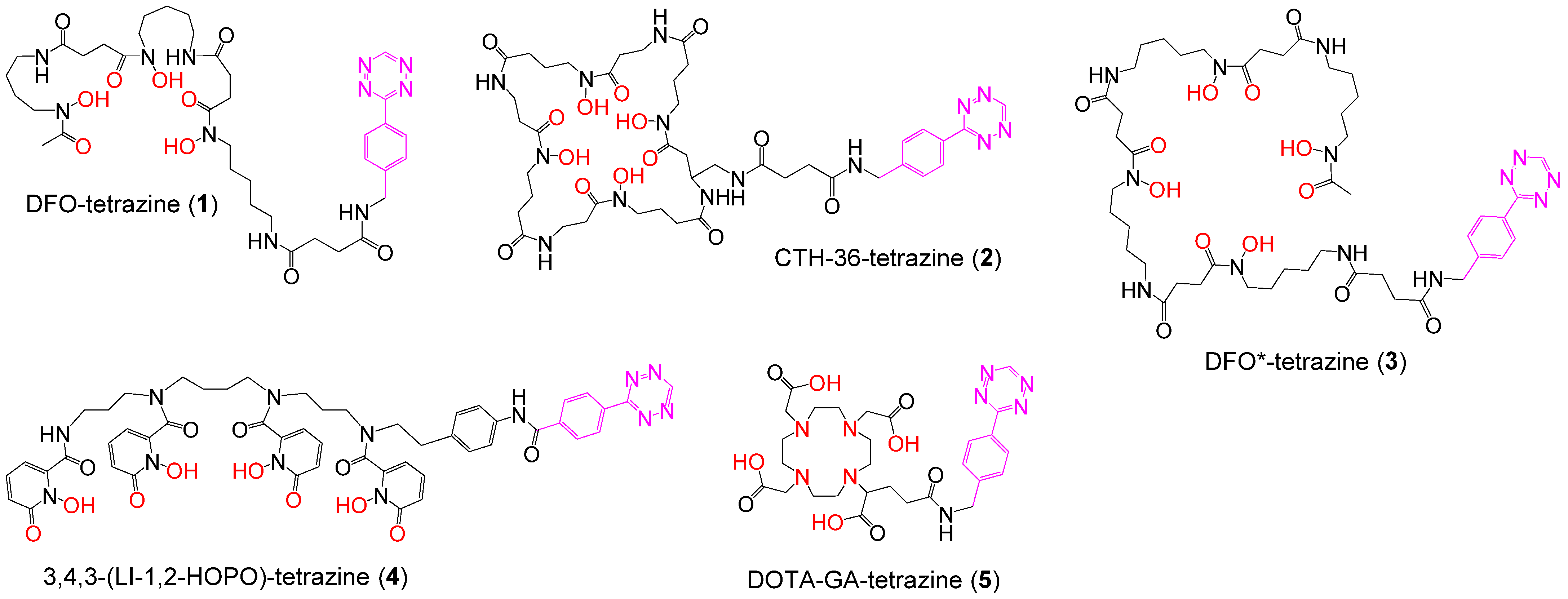
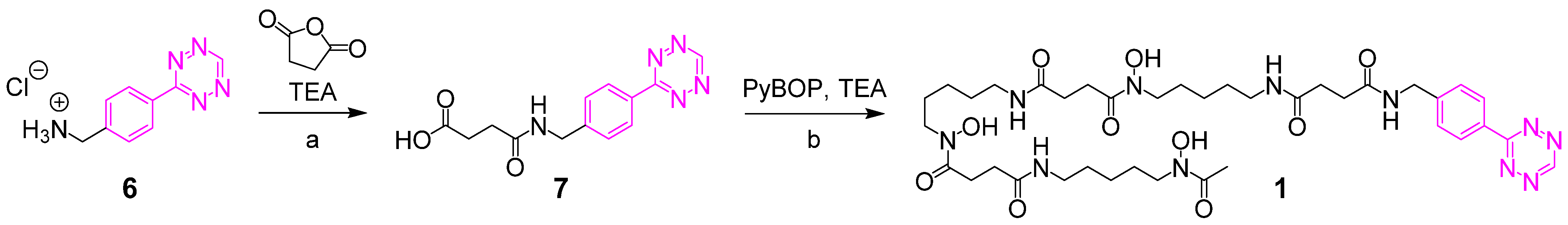
2.2. Syntheses of Chelator Tetrazines
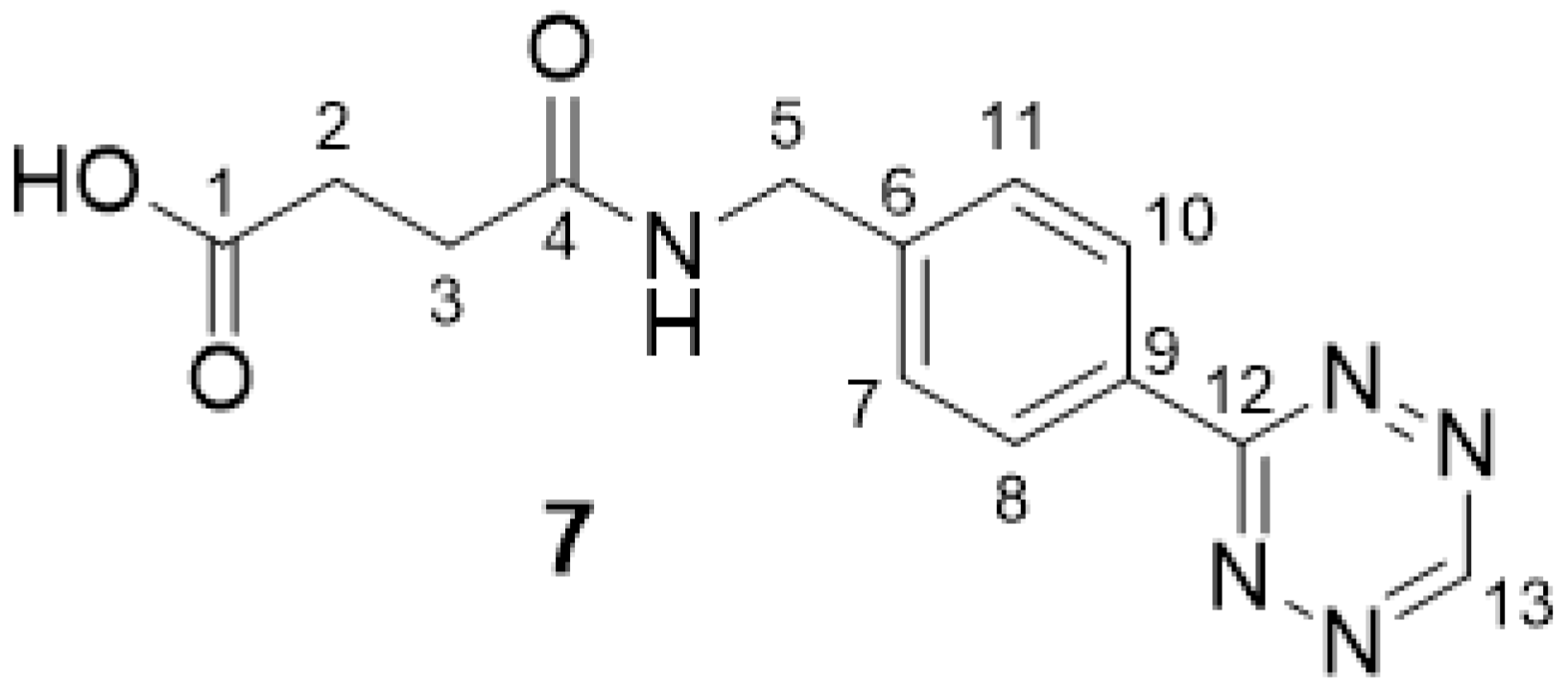
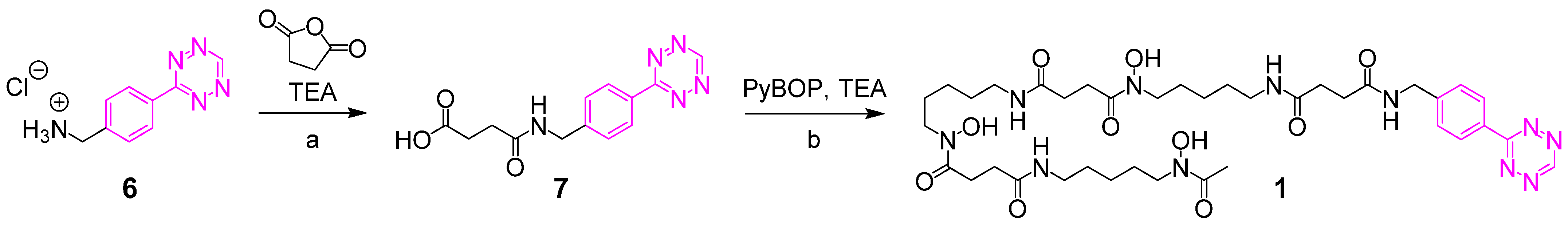
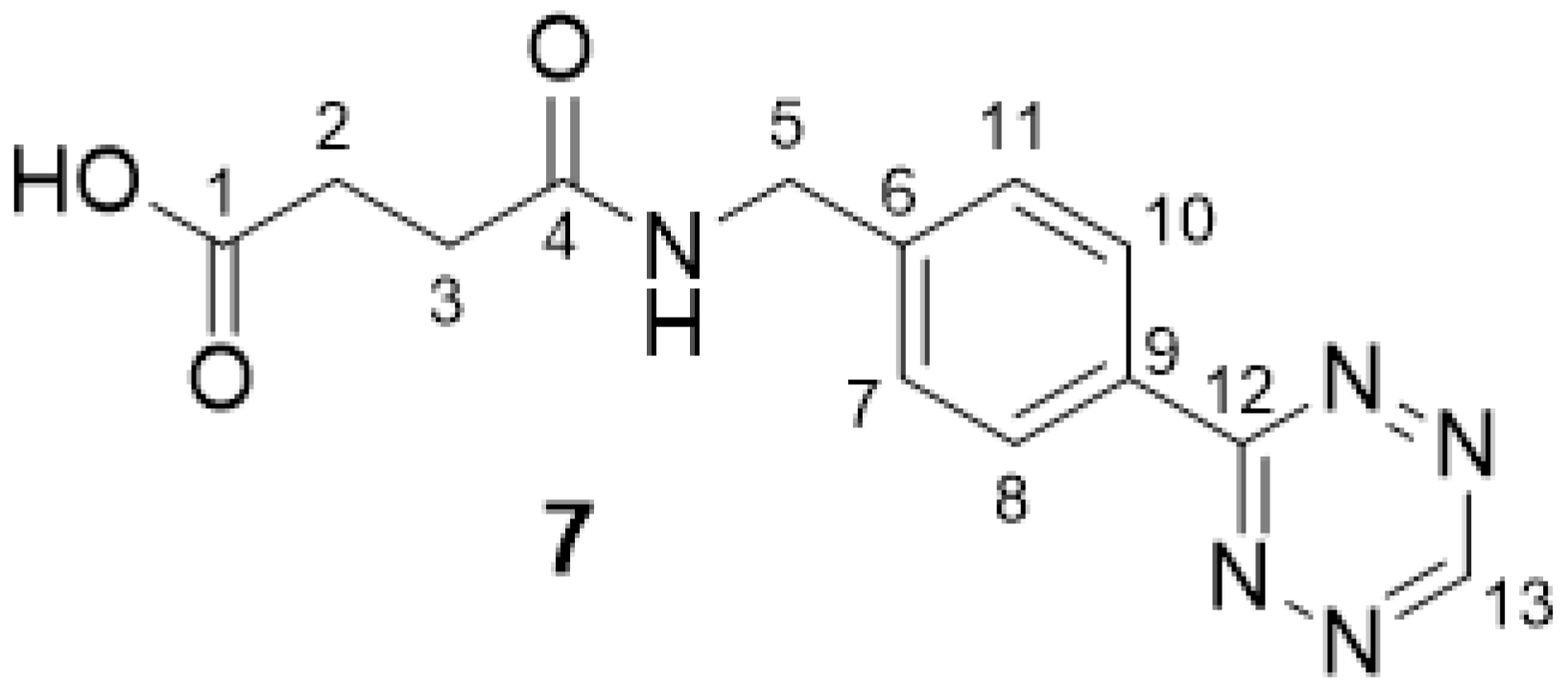
2.2.1. 4-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)amino)-4-oxobutanoic Acid 7
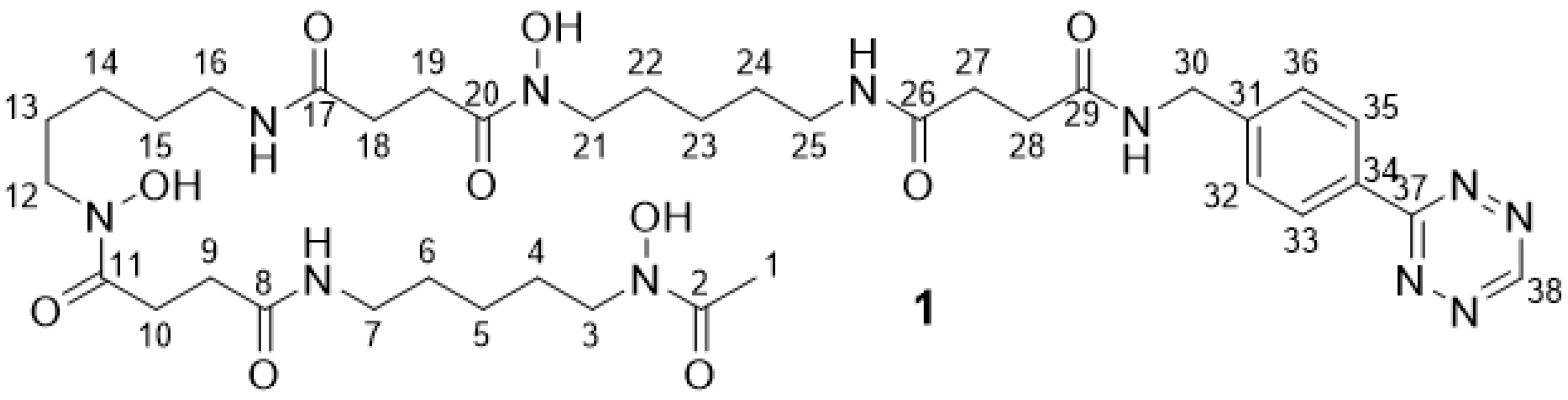
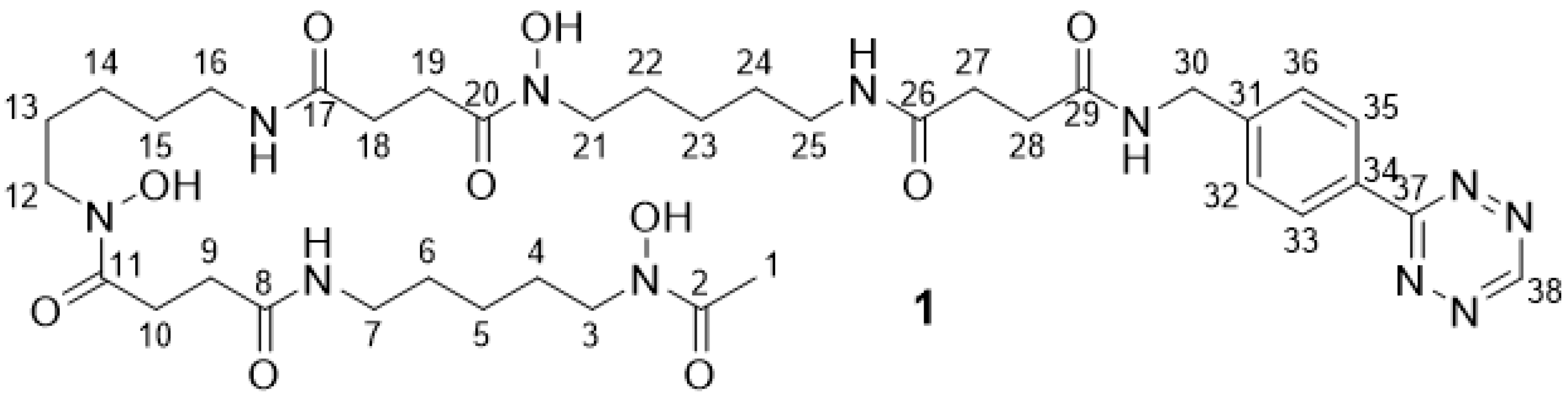
2.2.2. DFO Tetrazine 1
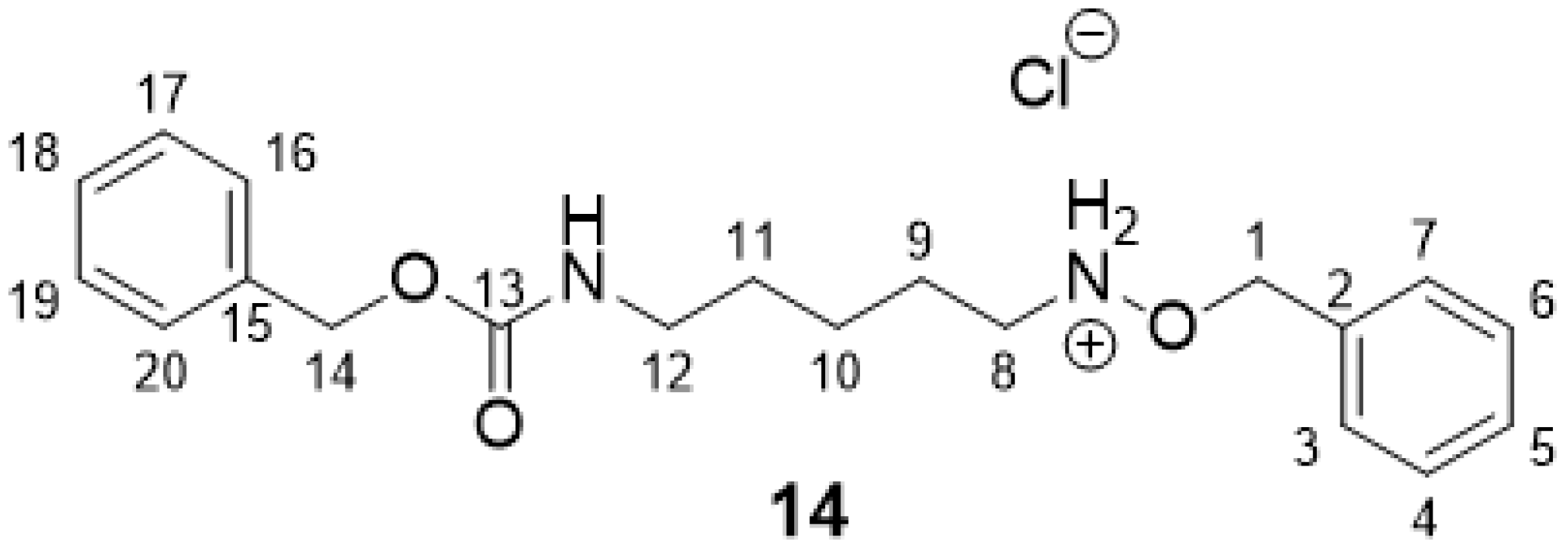
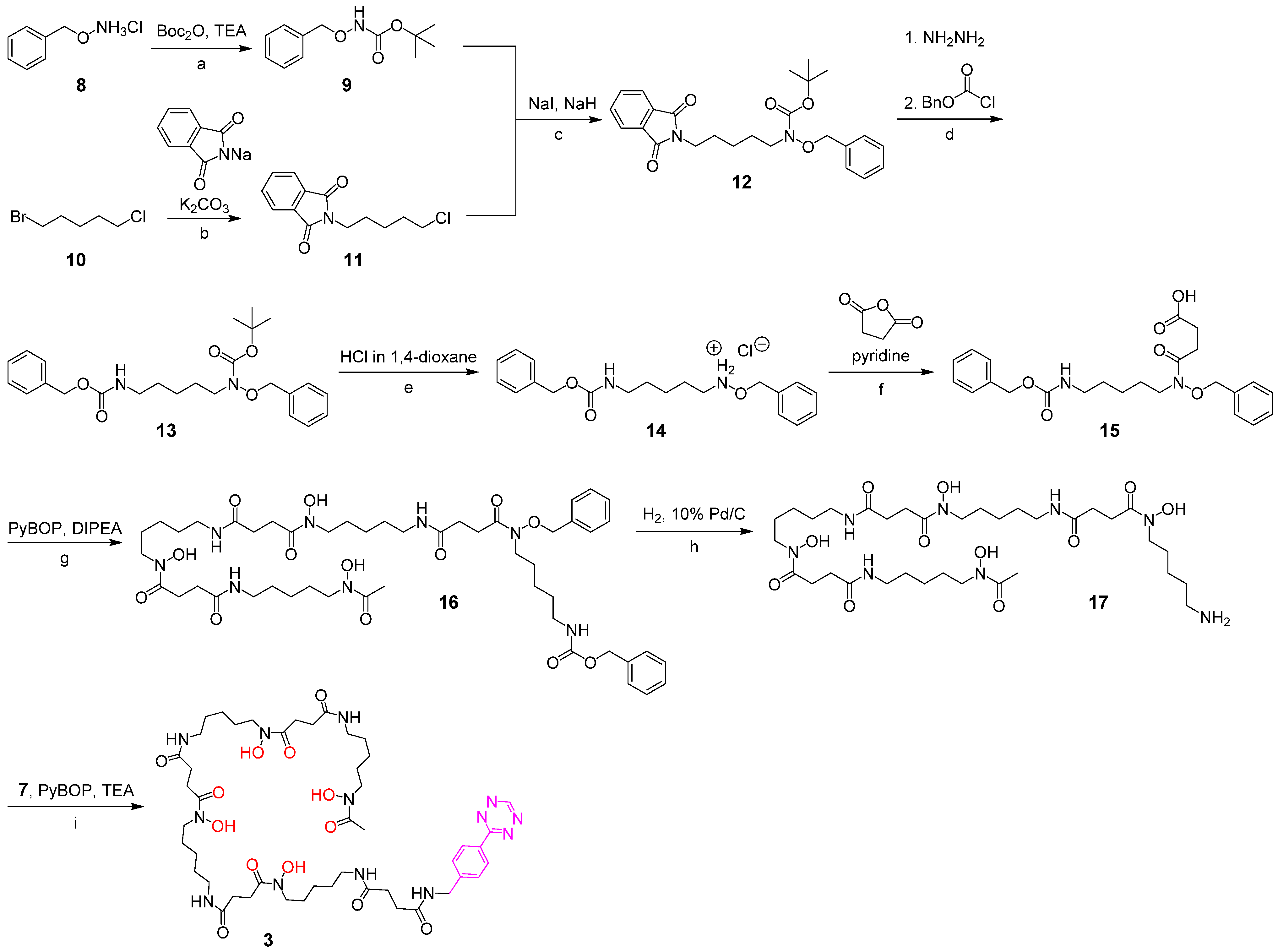
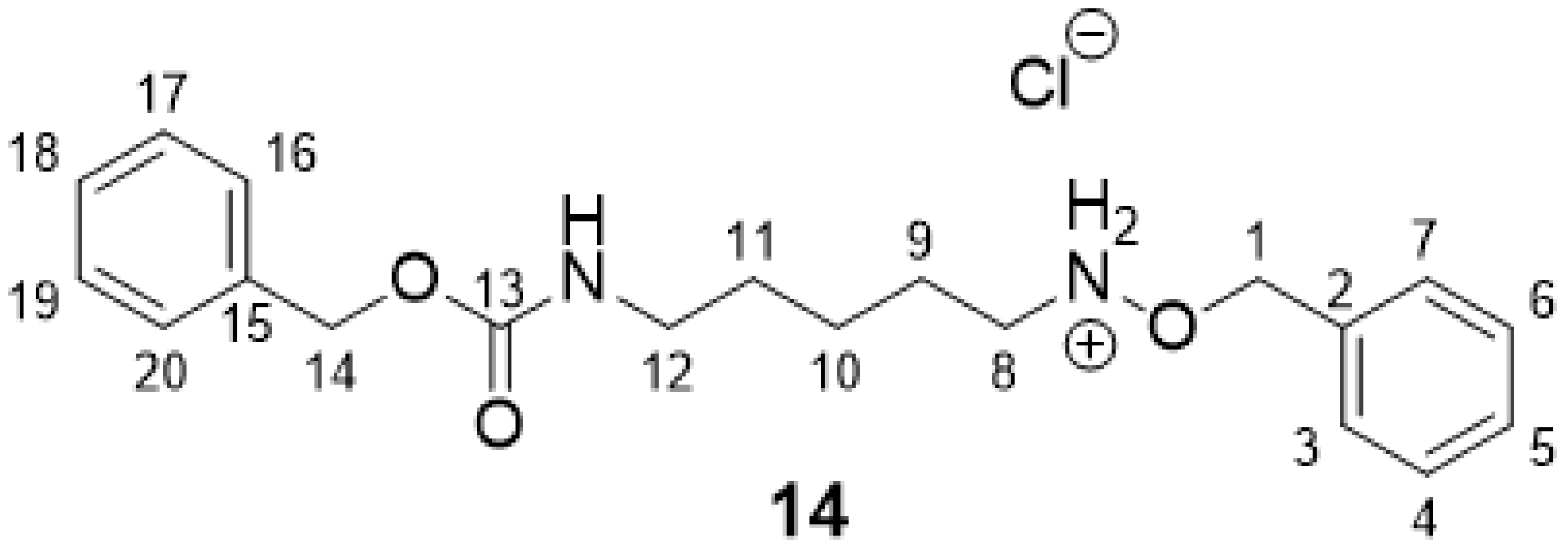
2.2.3. O-Benzyl-N-(5-(((benzyloxy)carbonyl)amino)pentyl)hydroxylammonium Chloride 14
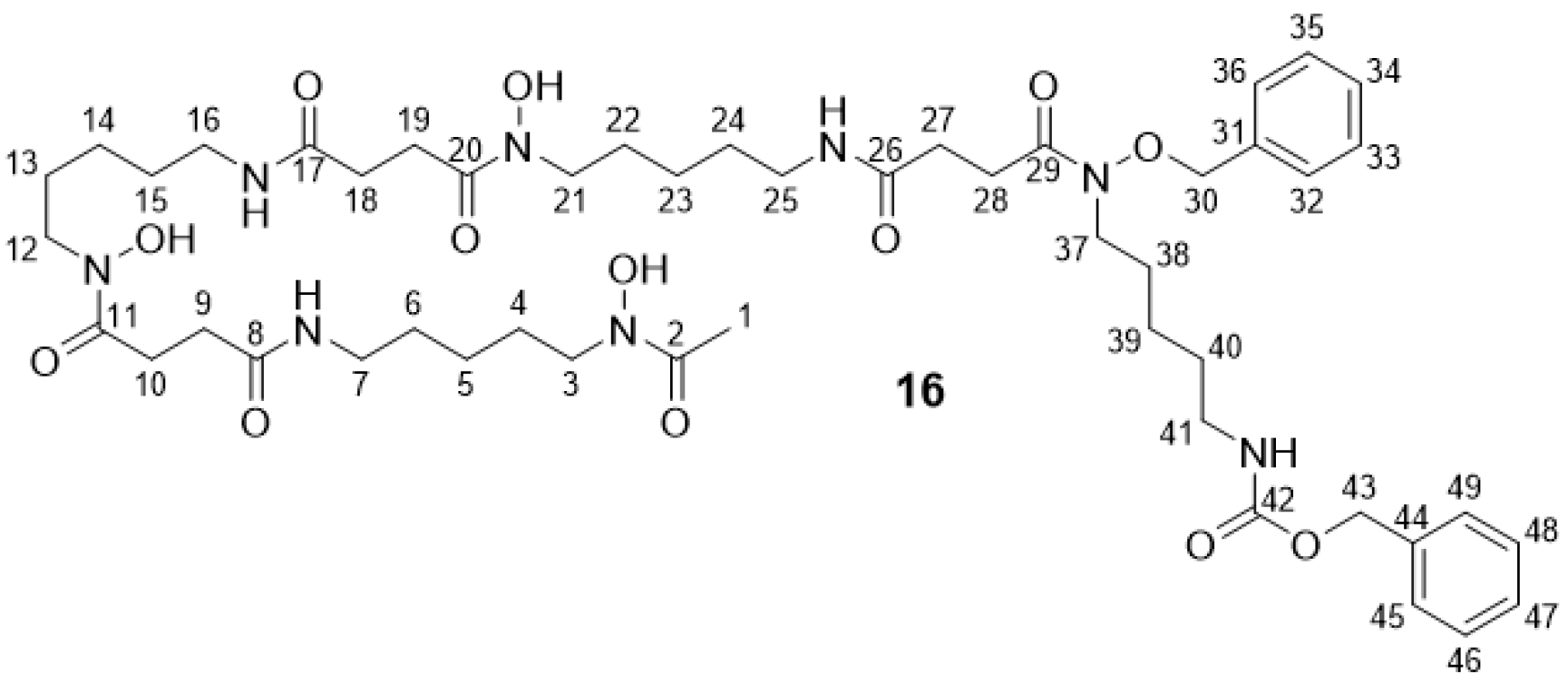
2.2.4. Bn-Cbz-DFO* 16
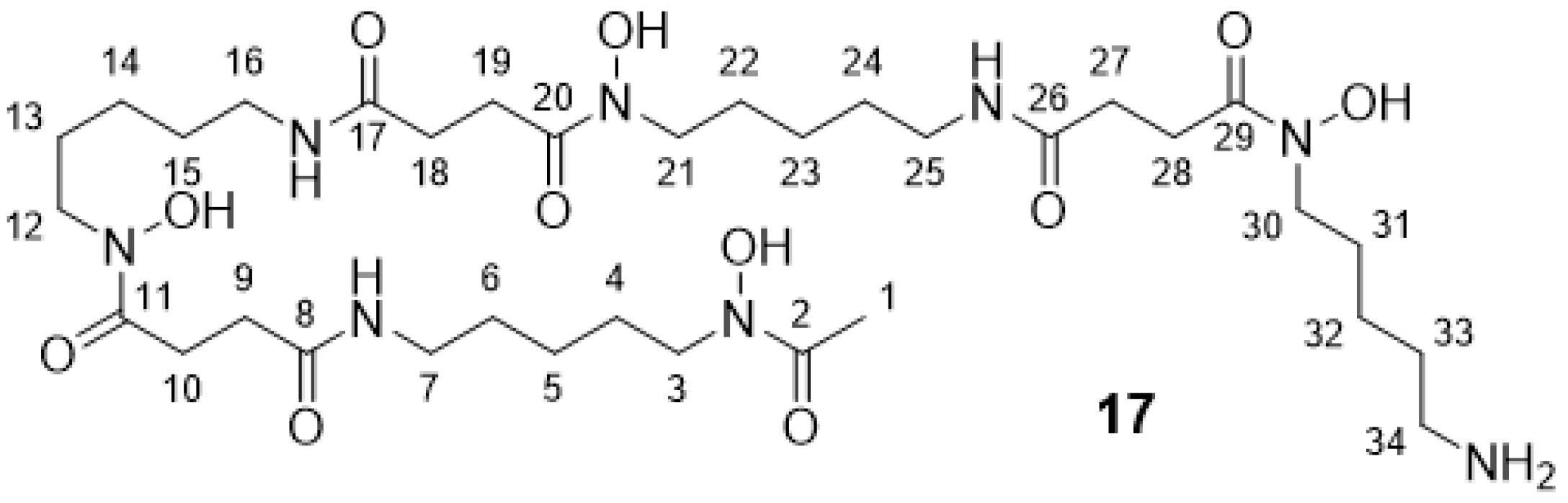
2.2.5. DFO* 17
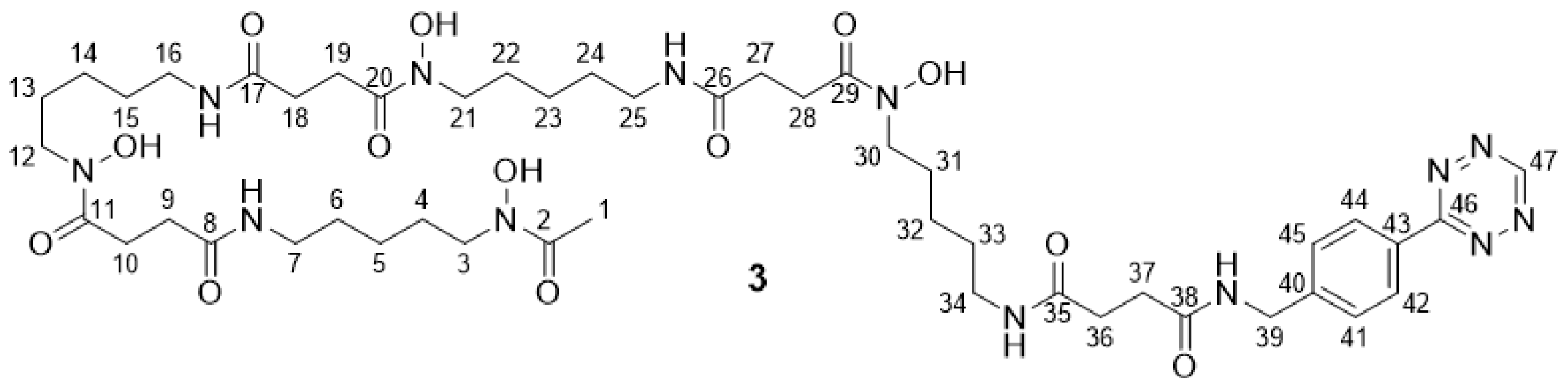
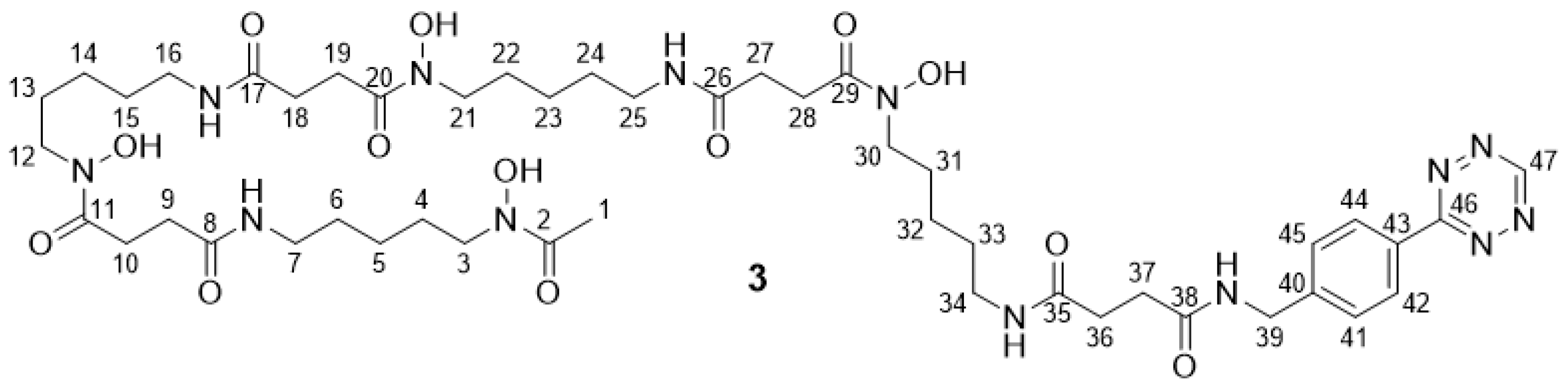
2.2.6. DFO* Tetrazine 3
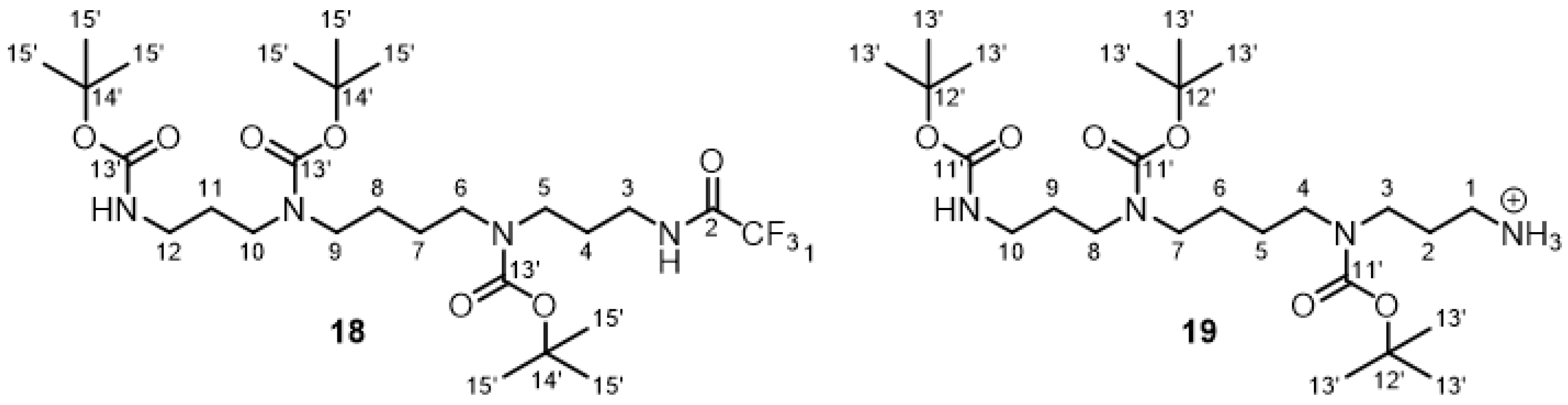
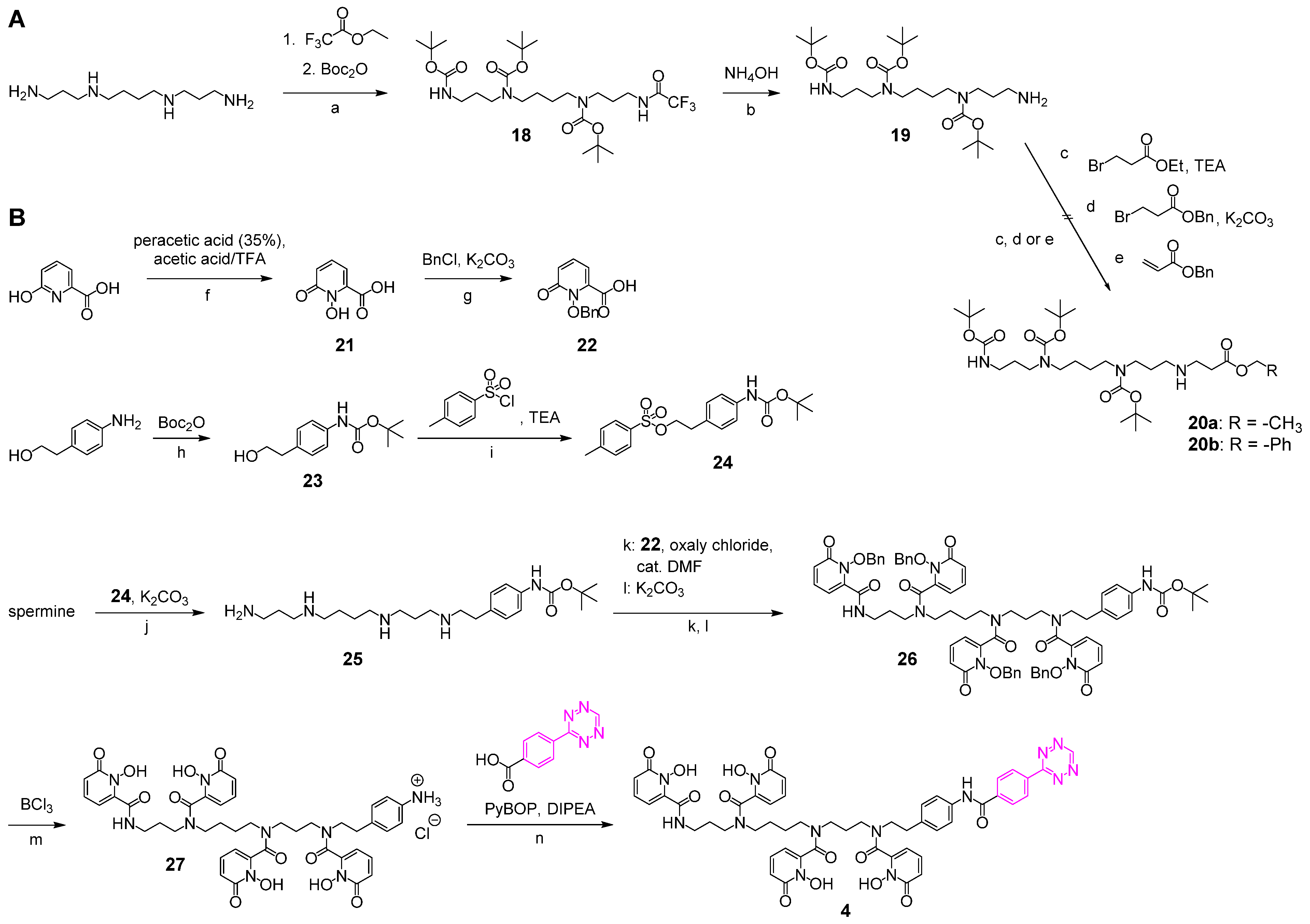
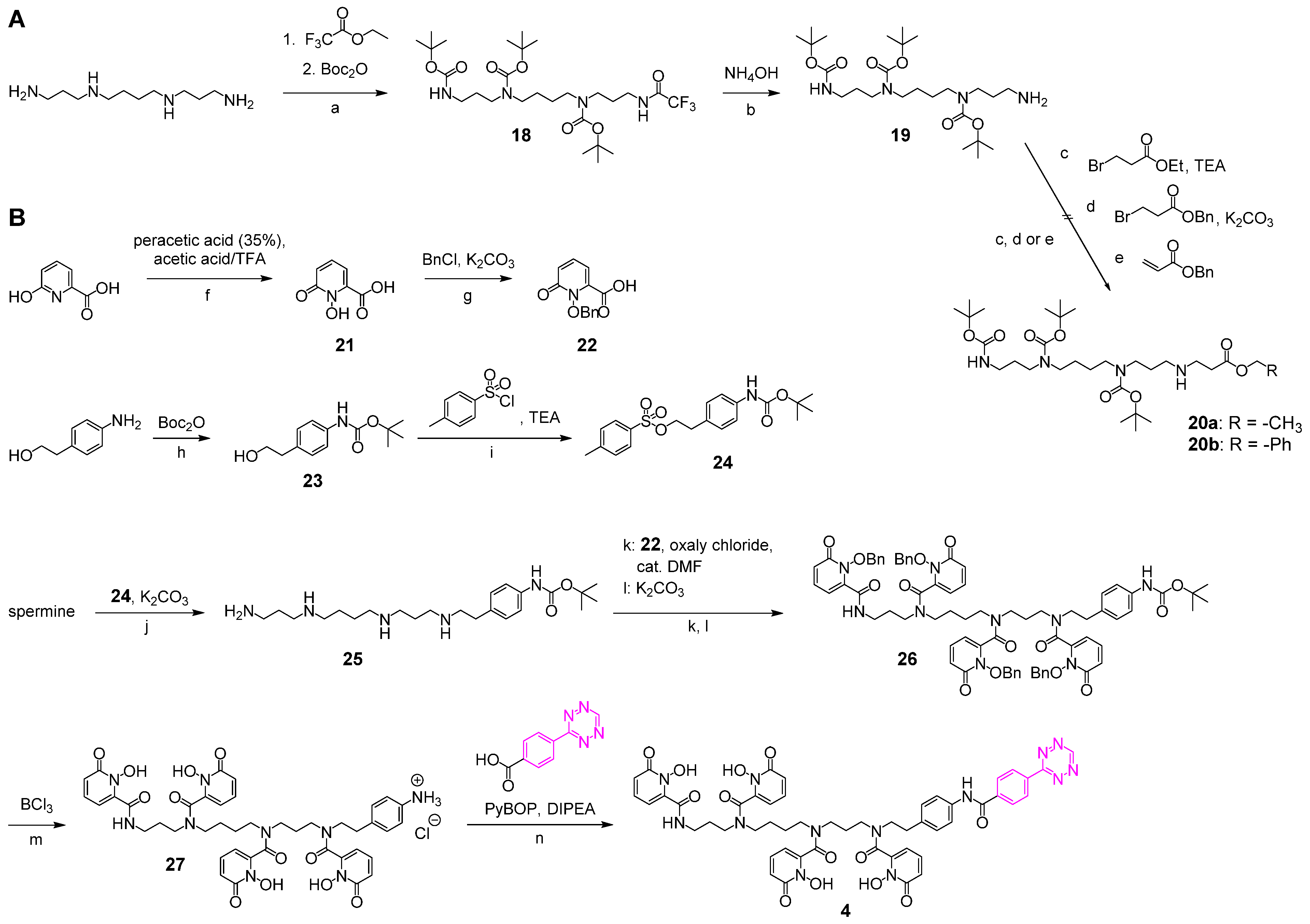
2.2.7. Tris-Boc-spermine-trifluoroacetamide 18 and Tris-Boc-spermine 19
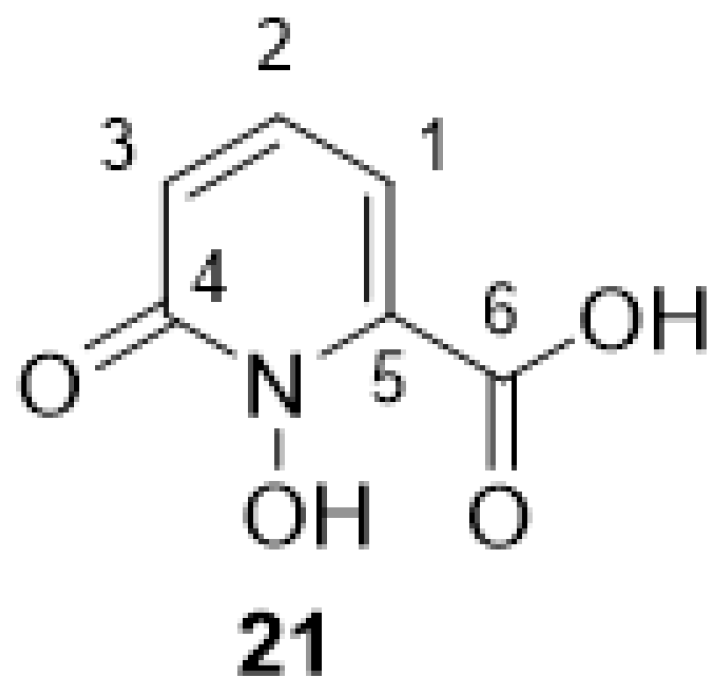
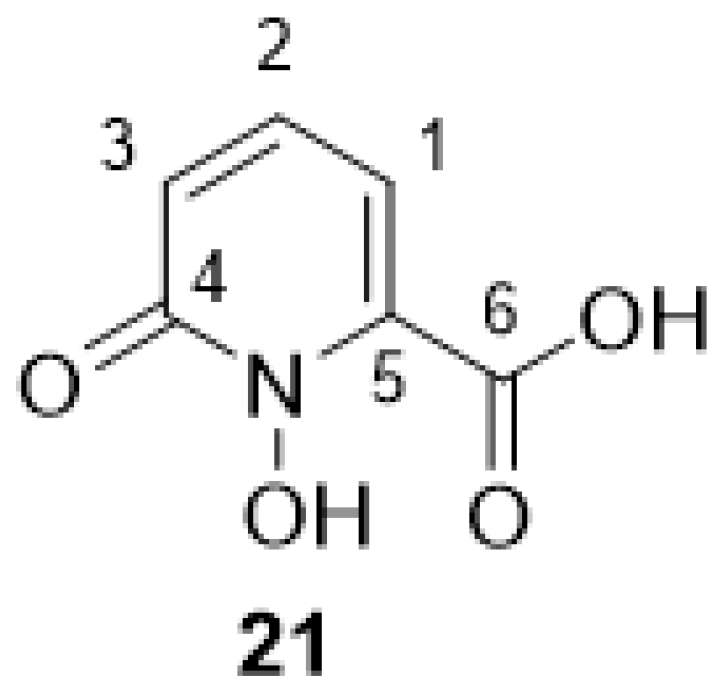
2.2.8. 1-Hydroxy-6-oxo-1,6-dihydropyridine-2-carbonic Acid 21
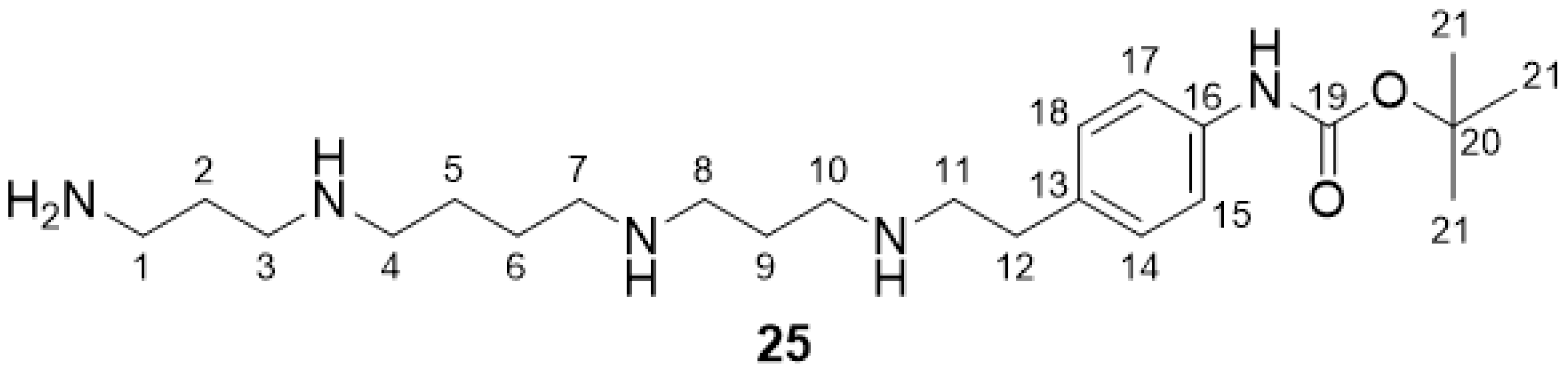
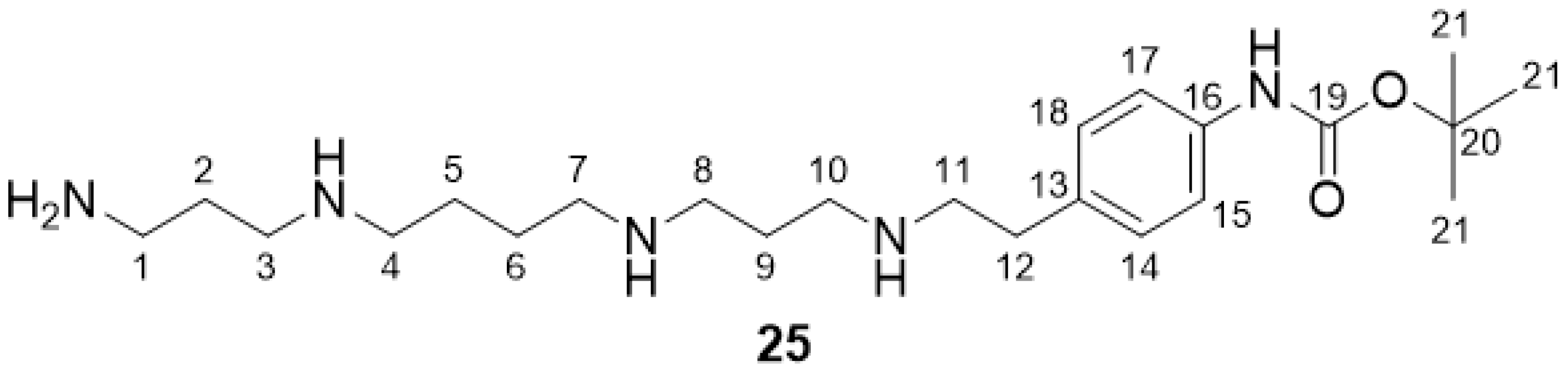
2.2.9. tButyl-(4-(2-((3-((4-((3-aminopropyl)amino)butyl)amino)propyl)amino)ethyl)phenyl)-carbamate 25
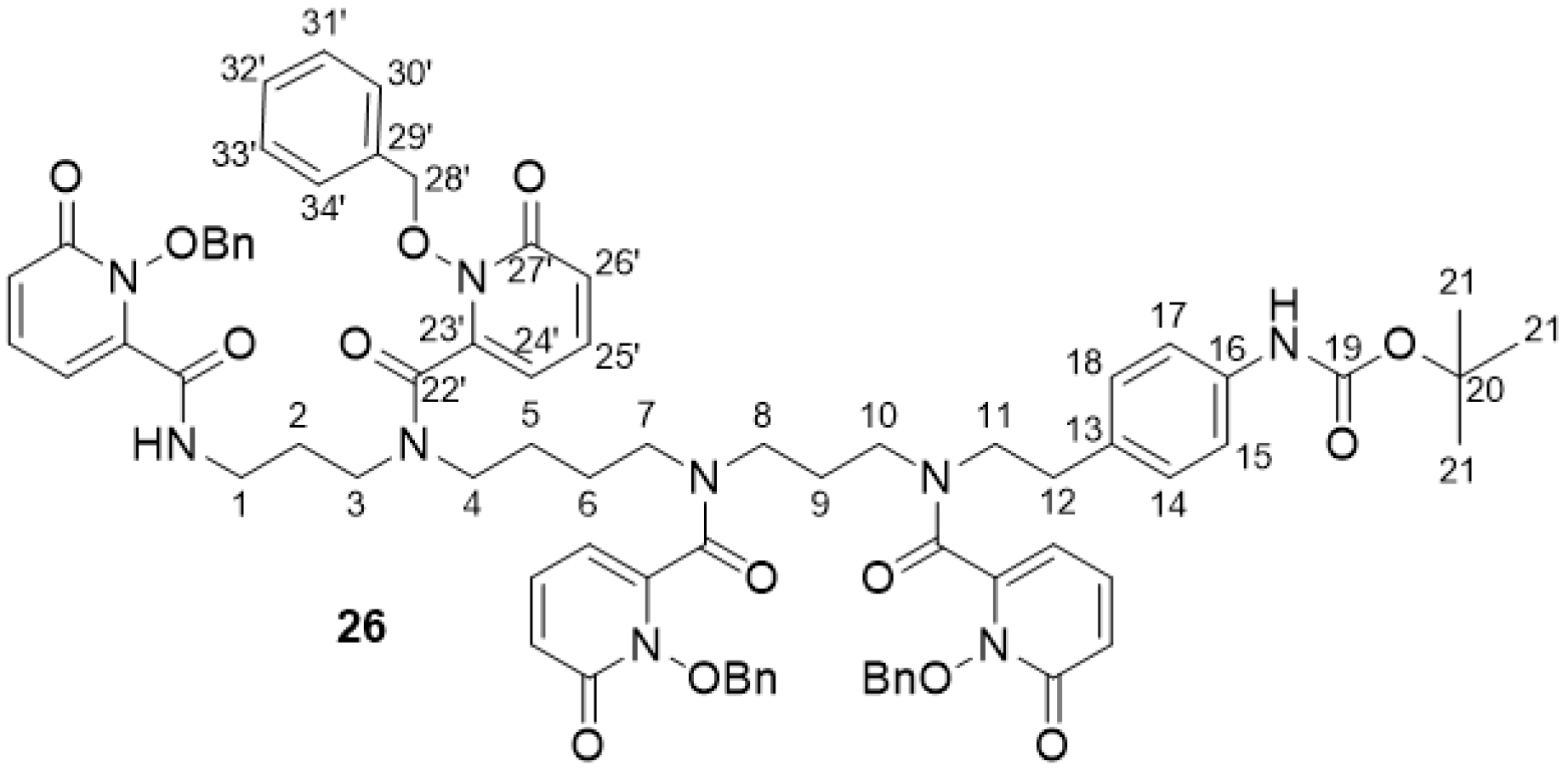
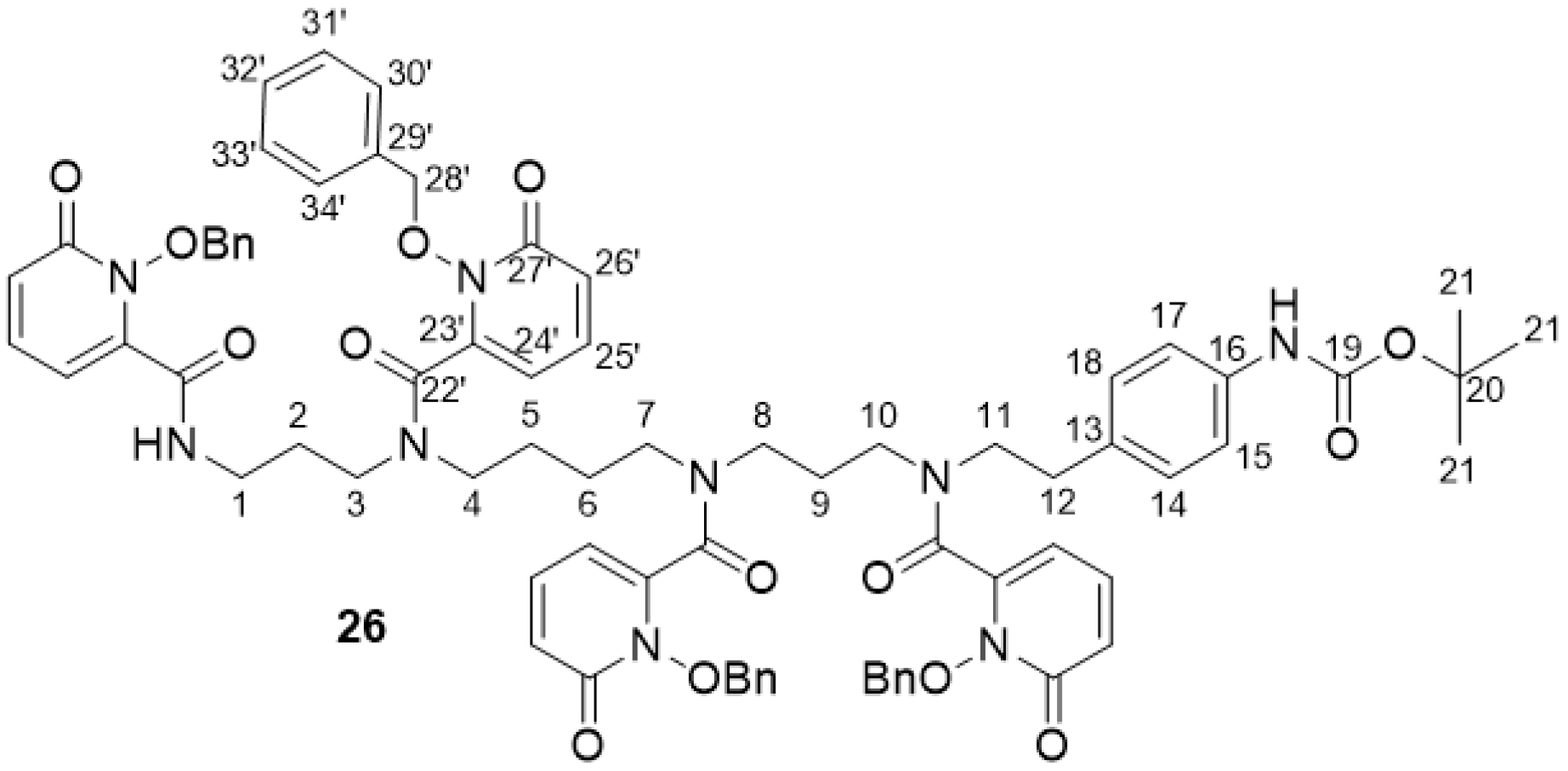
2.2.10. 3,4,3-(LI-1,2-HOPOBn)-NH-Boc 26
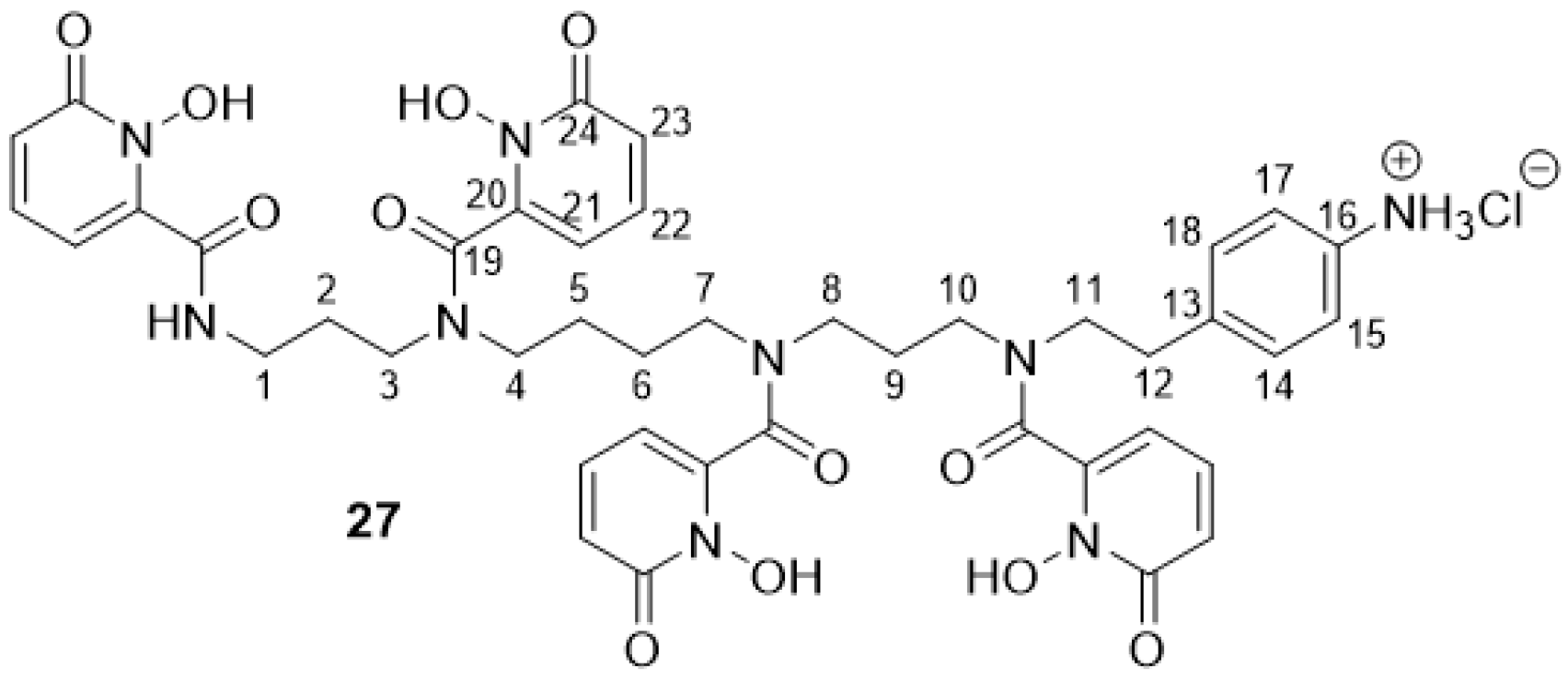
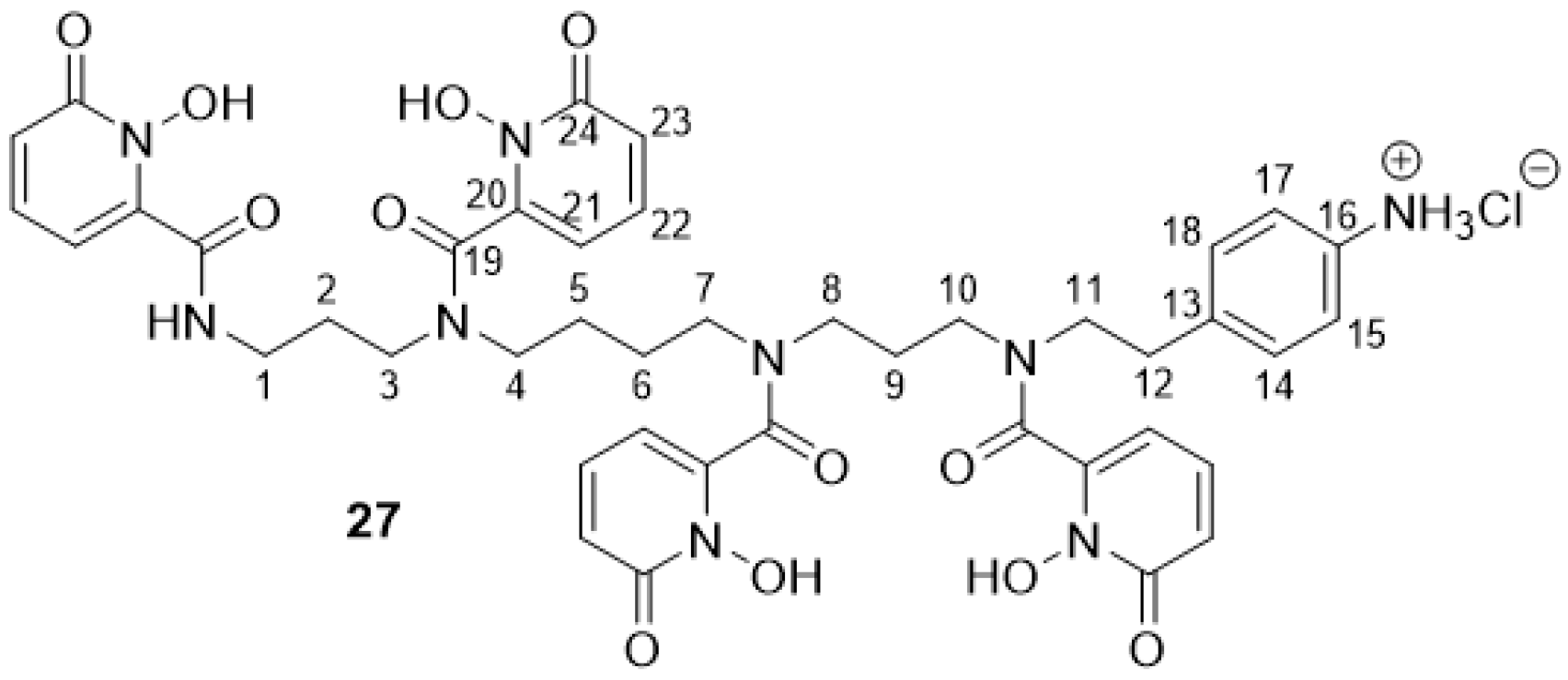
2.2.11. 3,4,3-(LI-1,2-HOPOBn)-NH3Cl 27
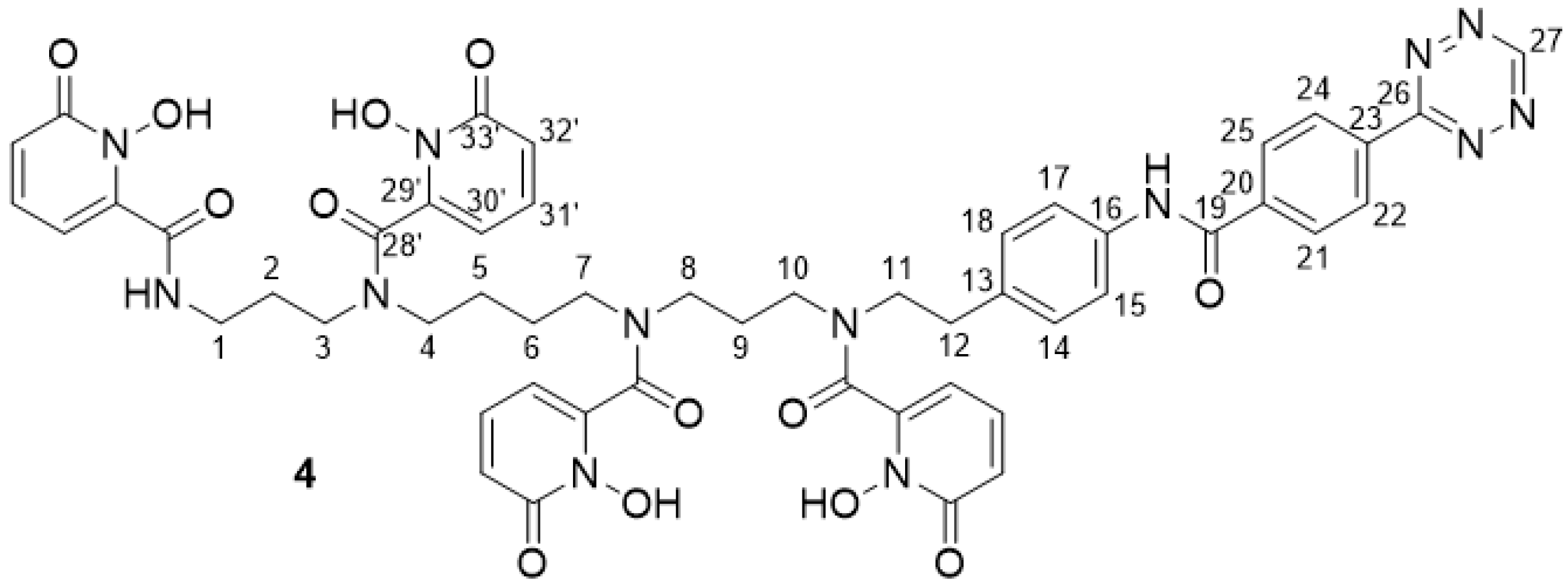
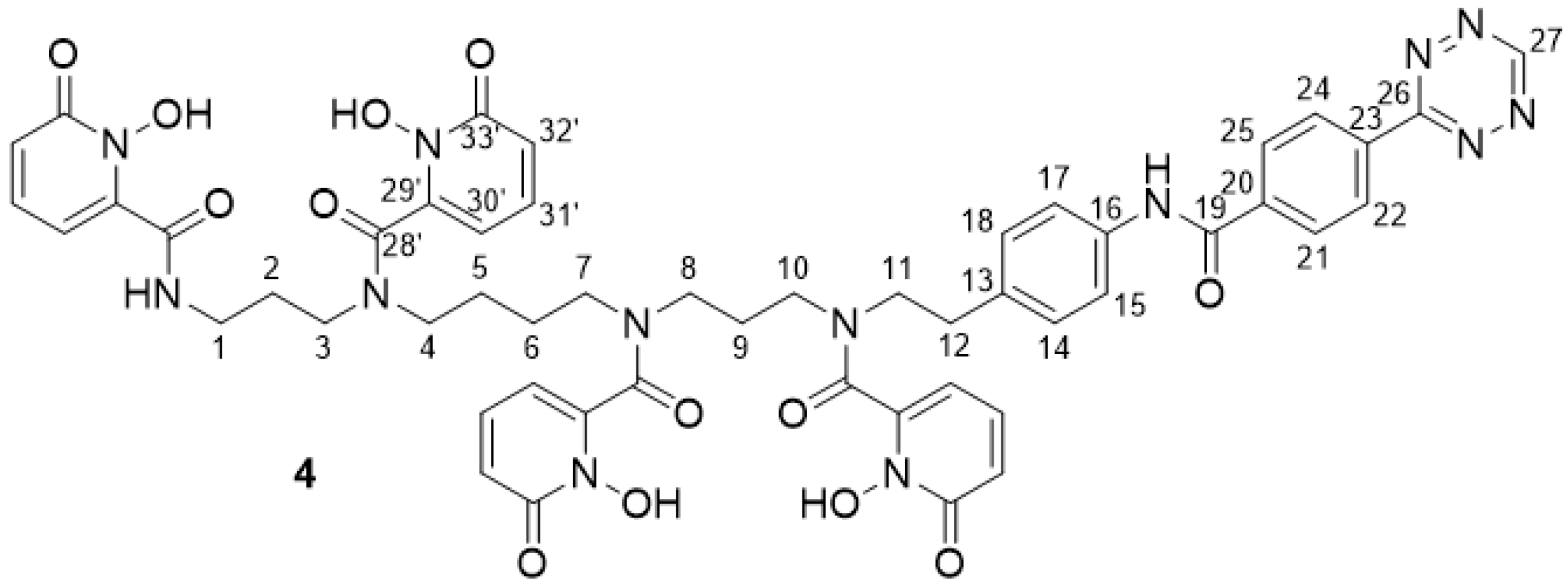
2.2.12. 3,4,3-(LI-1,2-HOPOBn) Tetrazine 4
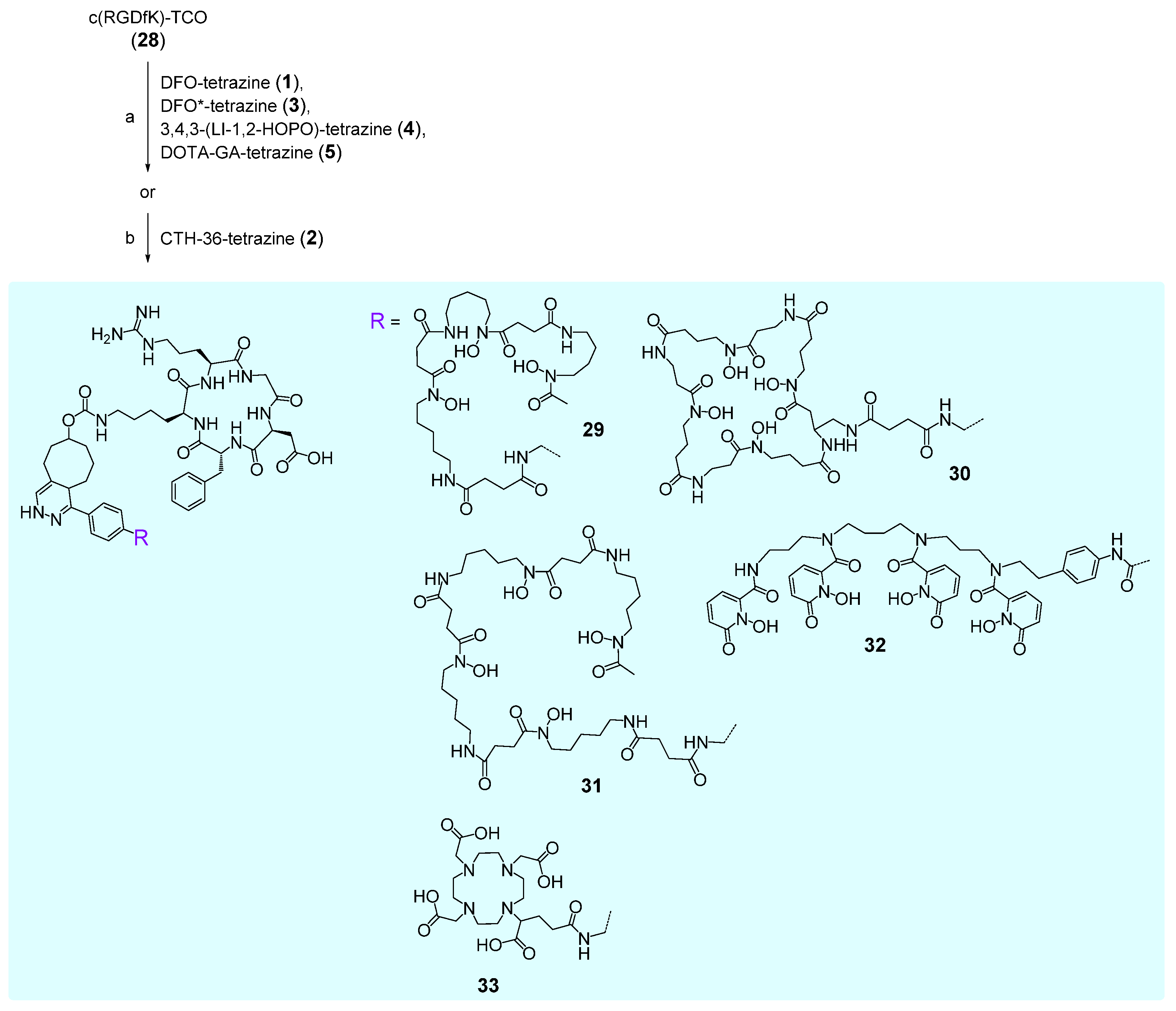
2.3. Syntheses of Chelator Peptide Bioconjugates
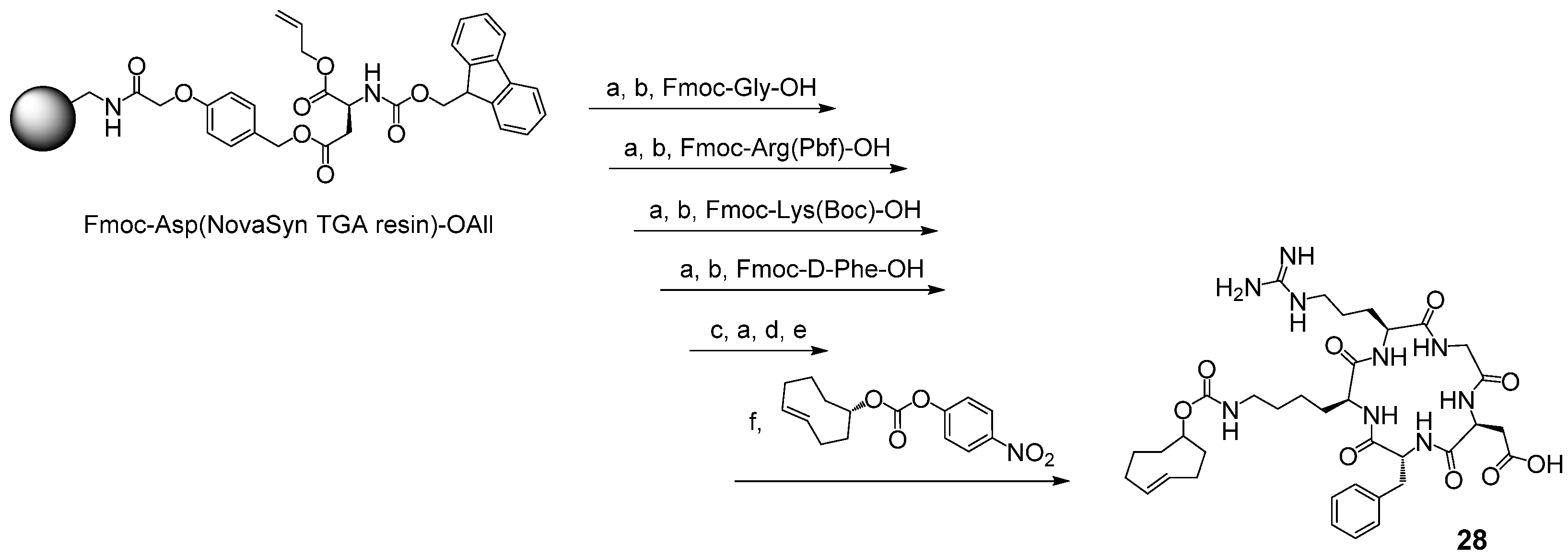
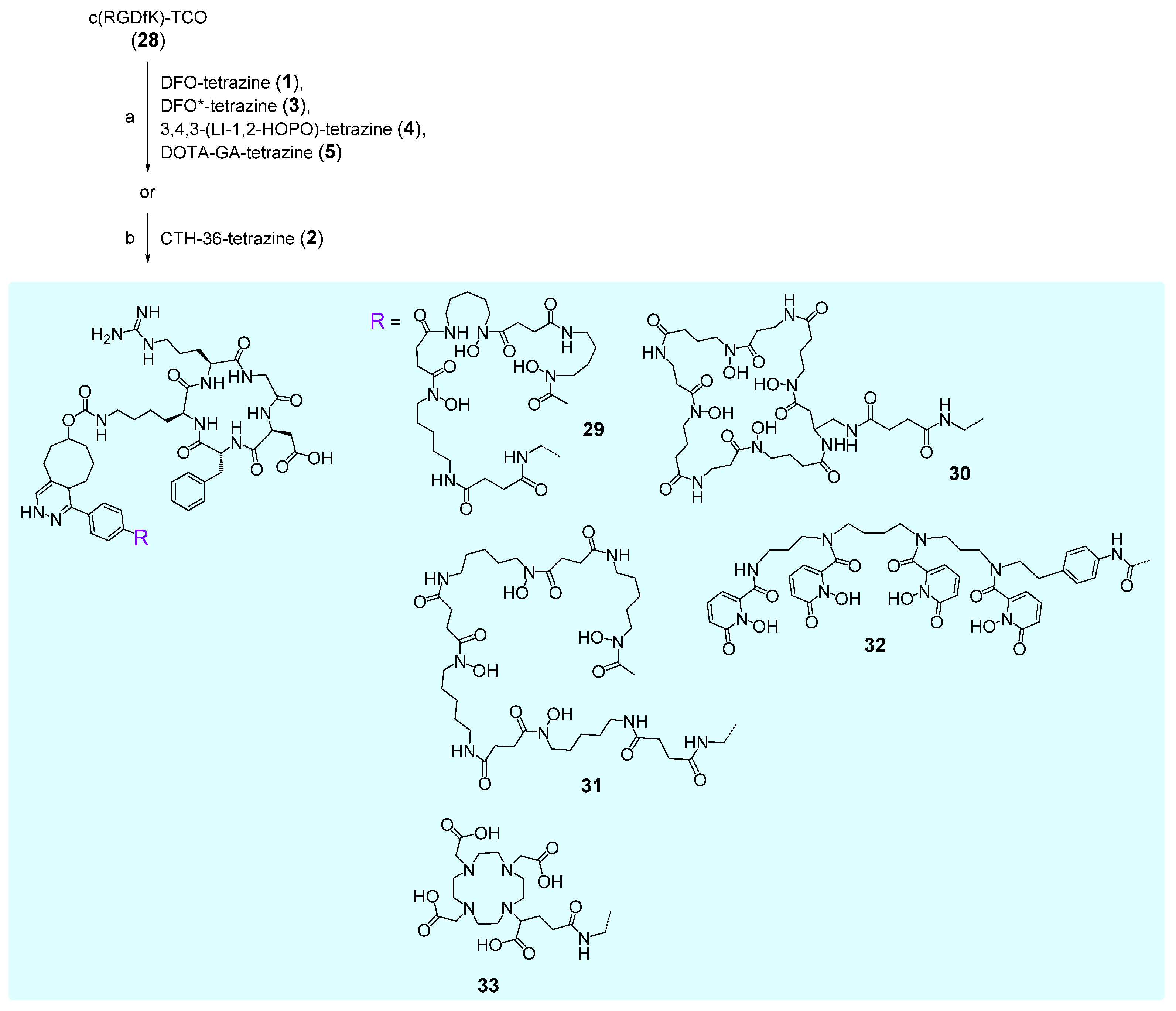
2.3.1. DFO-c(RGDfK) 29
2.3.2. CTH36-c(RGDfK) 30
2.3.3. DFO*-c(RGDfK) 31
2.3.4. 3,4,3-(LI-1,2-HOPOBn)-c(RGDfK) 32
2.3.5. DOTA-GA-c(RGDfK) 33
2.4. Radiochemistry
2.5. Determination of logD(7.4)
2.6. EDTA-Based 89Zr Complex Challenge Experiments
2.7. DFT Calculations
3. Results and Discussion
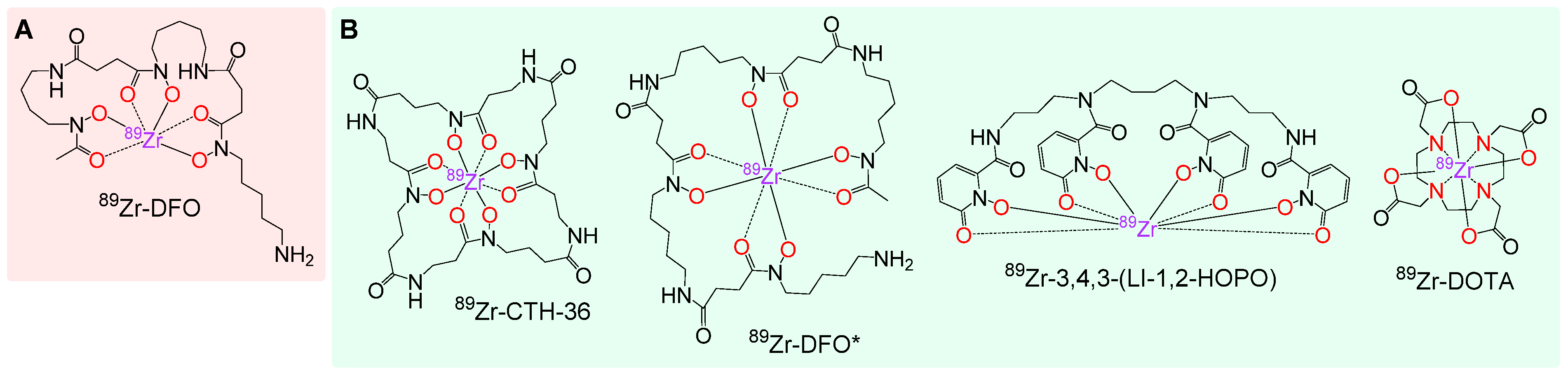
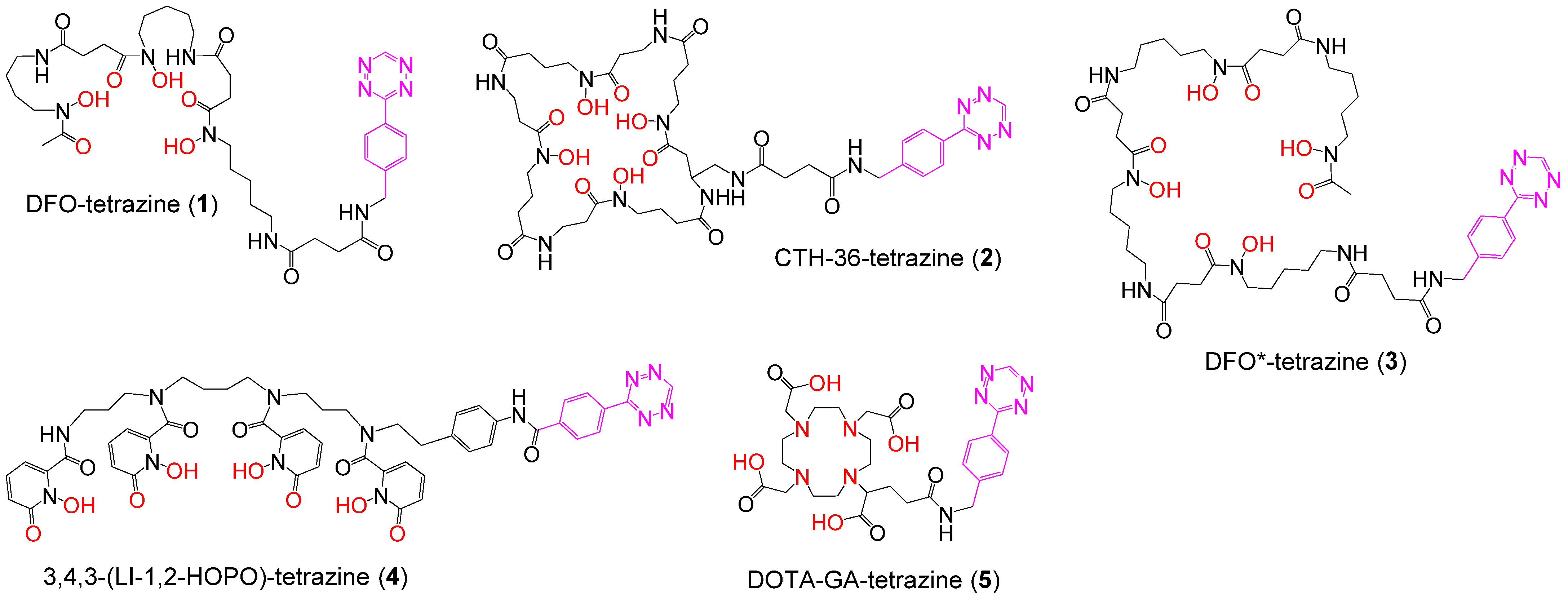
3.1. Synthesis of the Backbone Tetrazine-Modified Chelator Analogs 1–5
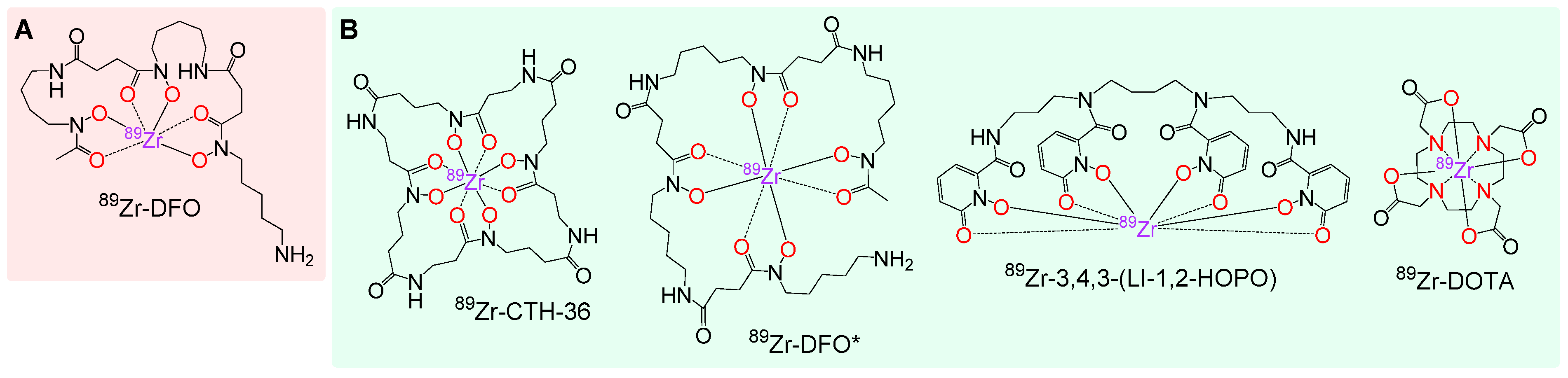
3.2. Investigation of the Properties of the Different 89Zr Complexes by DFT Calculations
3.3. Syntheses of Chelator Bioconjugates 29–33
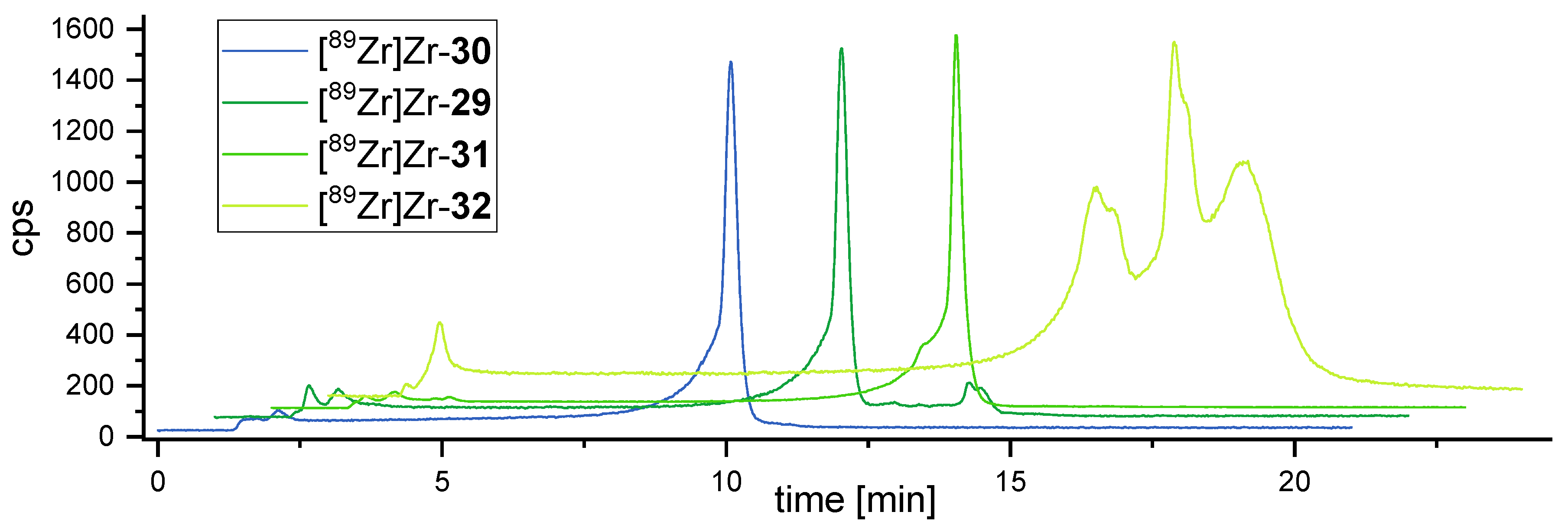
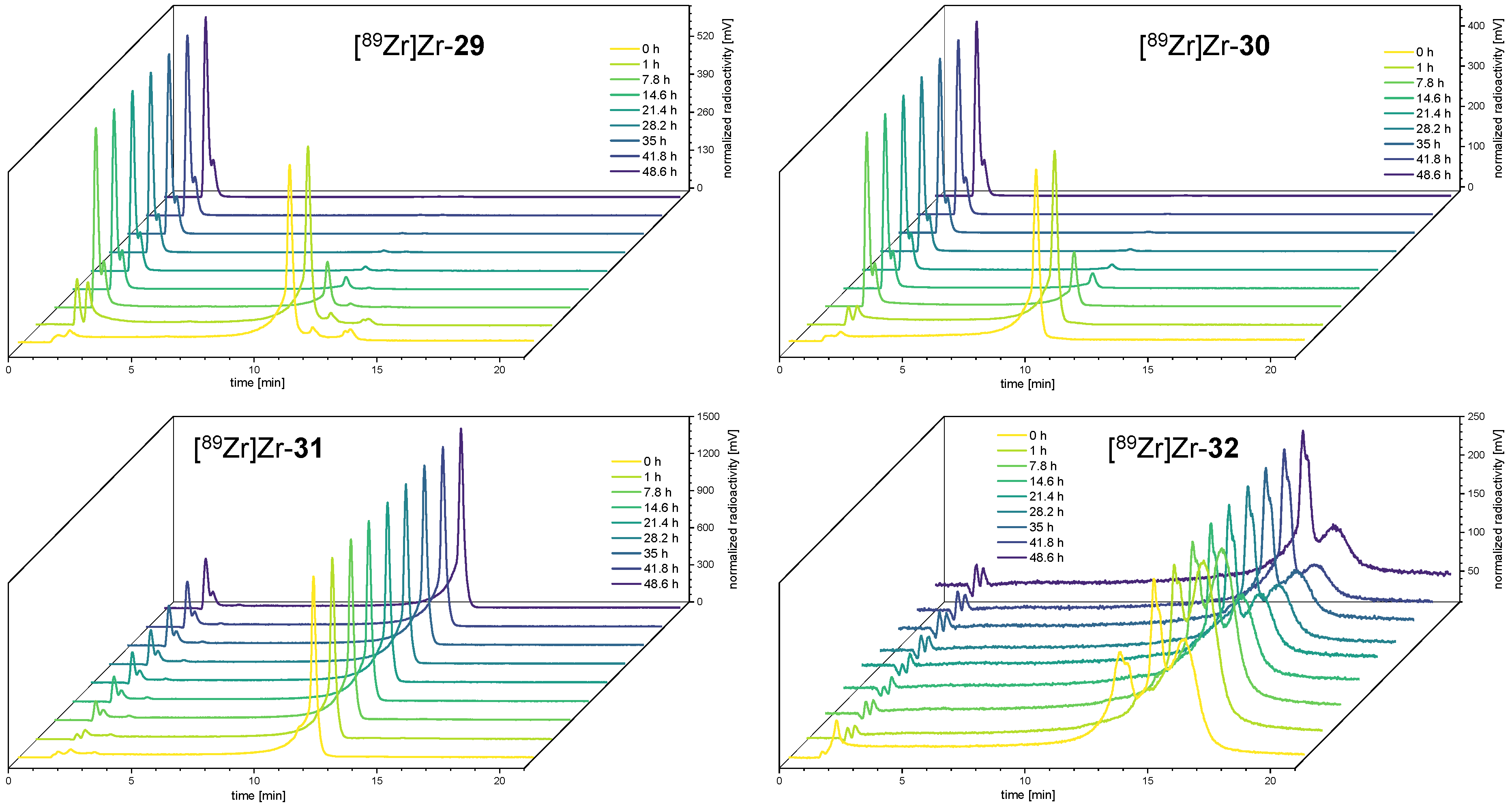
3.4. 89Zr-Radiolabeling of the Bioconjugates 29–33 and Determination of the logD(7.4)s of [89Zr]Zr-29–[89Zr]Zr-32
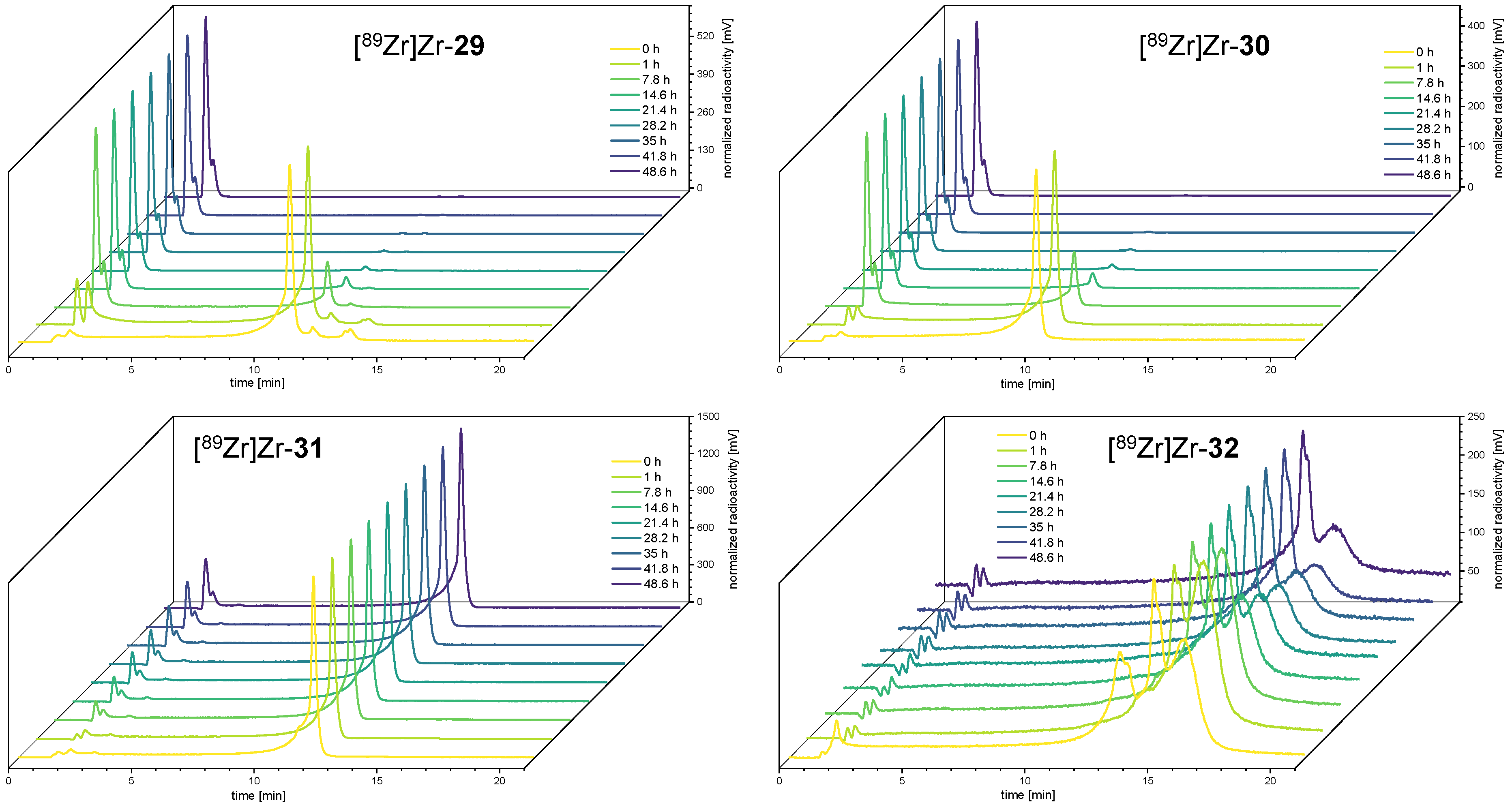
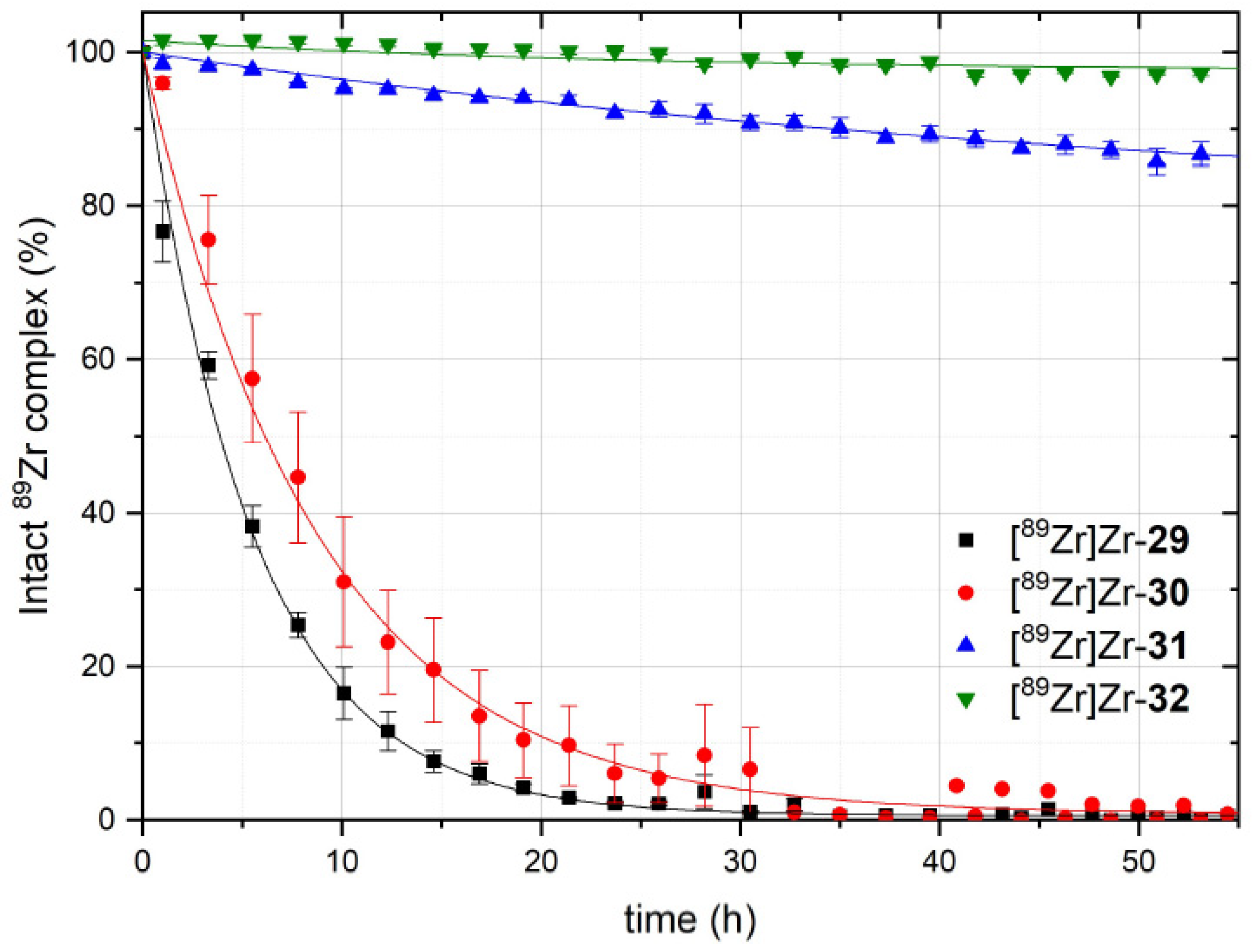
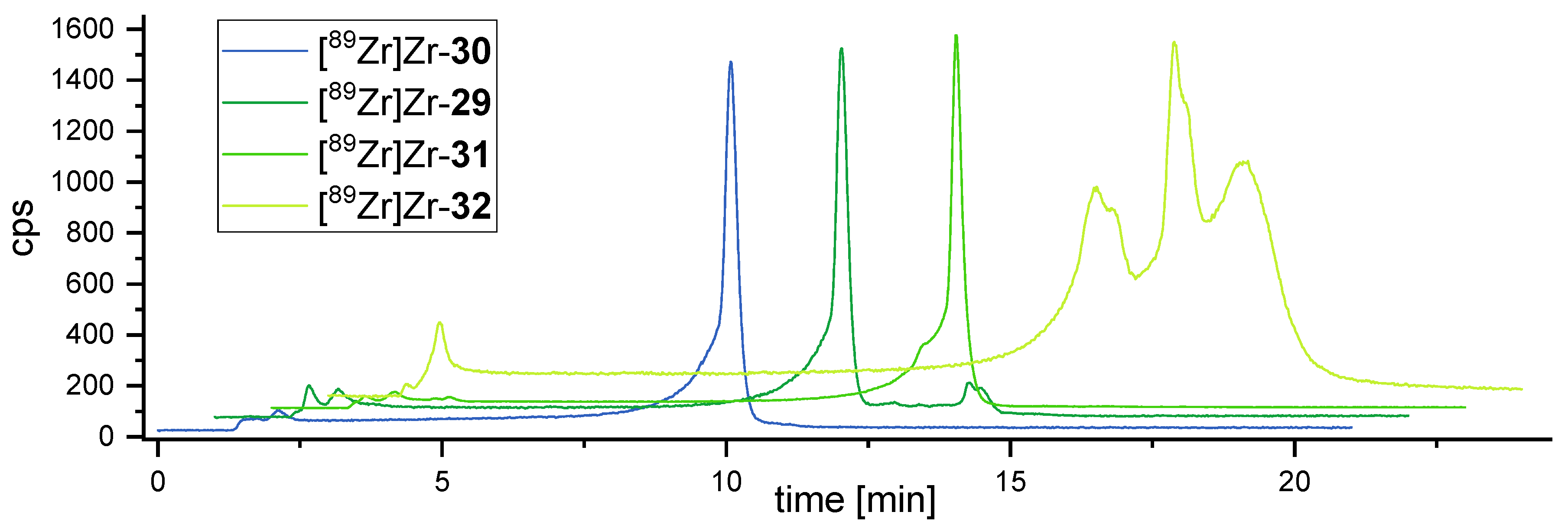
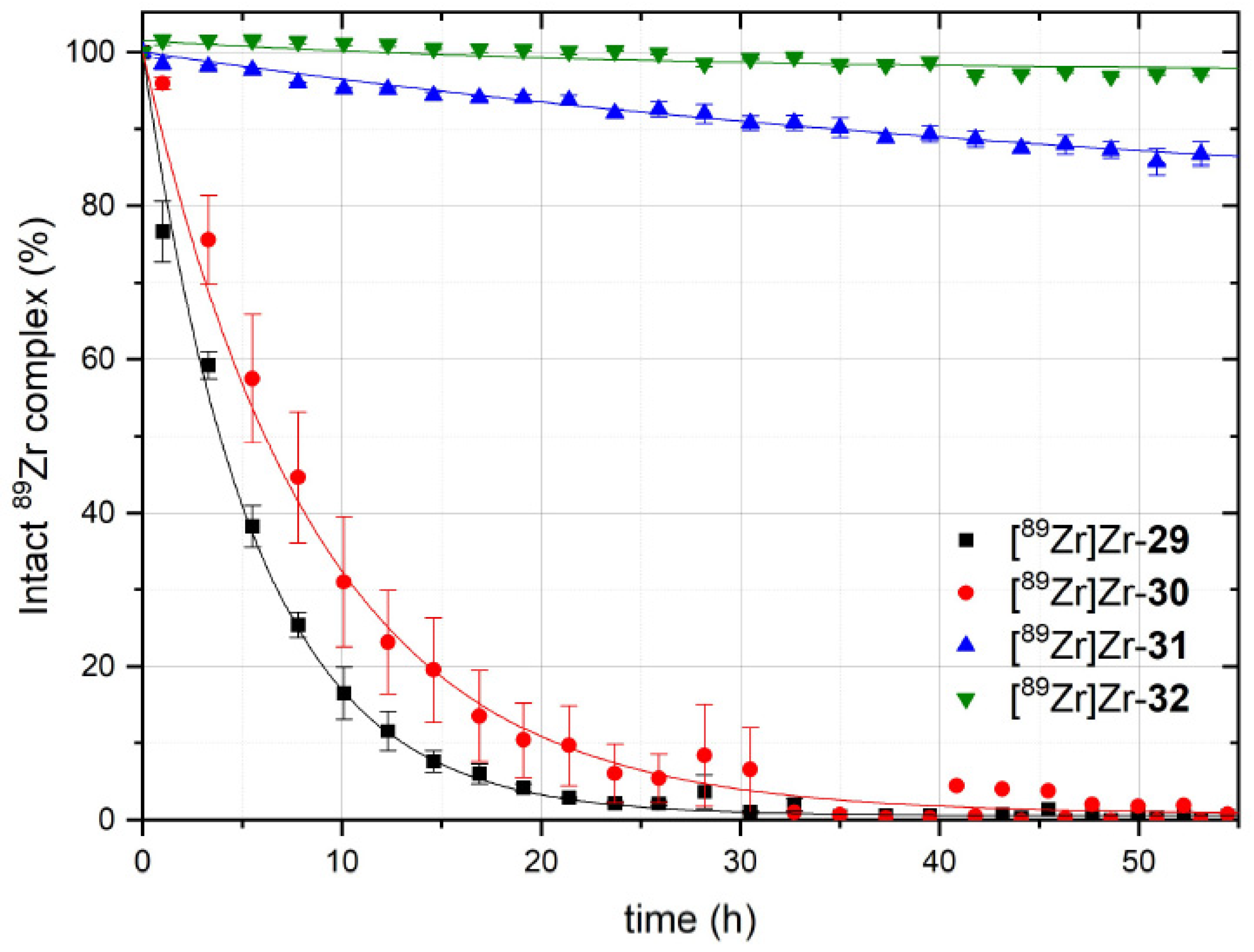
3.5. Comparative Assessment of the Relative Kinetic Inertness of the Complexes [89Zr]Zr-29–[89Zr]Zr-32 by Challenge Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoon, J.K.; Park, B.N.; Ryu, E.K.; An, Y.S.; Lee, S.J. Current Perspectives on 89Zr-PET Imaging. Int. J. Mol. Sci. 2020, 21, 4309. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, G.A.M.S.; Beaino, W.; Windhorst, A.D.; Zwezerijnen, G.J.C.; Oprea-Lager, D.E.; Hendrikse, N.H.; van Kuijk, C.; Boellaard, R.; Huisman, M.C.; Vugts, D.J. The Role of 89Zr-Immuno-PET in Navigating and Derisking the Development of Biopharmaceuticals. J. Nucl. Med. 2021, 62, 438–445. [Google Scholar] [CrossRef]
- Bhatt, N.B.; Pandya, D.N.; Wadas, T.J. Recent Advances in Zirconium-89 Chelator Development. Molecules 2018, 23, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, G.; Seibold, U.; Schirrmacher, R.; Wängler, B.; Wängler, C. Zr-89, a Radiometal Nuclide with High Potential for Molecular Imaging with PET: Chemistry, Applications and Remaining Challenges. Molecules 2013, 18, 6469–6490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feiner, I.V.J.; Brandt, M.; Cowell, J.; Demuth, T.; Vugts, D.; Gasser, G.; Mindt, T.L. The Race for Hydroxamate-Based Zirconium-89 Chelators. Cancers 2021, 13, 4466. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.P. Predicting the Thermodynamic Stability of Zirconium Radiotracers. Inorg. Chem. 2020, 59, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Bauman, A.; Mari, C.; Fischer, C.A.; Blacque, O.; Haussinger, D.; Gasser, G.; Mindt, T.L. An octadentate bifunctional chelating agent for the development of stable zirconium-89 based molecular imaging probes. ChemComm 2014, 50, 11523–11525. [Google Scholar] [CrossRef] [PubMed]
- Chomet, M.; Schreurs, M.; Bolijn, M.J.; Verlaan, M.; Beaino, W.; Brown, K.; Poot, A.J.; Windhorst, A.D.; Gill, H.; Marik, J.; et al. Head-to-head comparison of DFO* and DFO chelators: Selection of the best candidate for clinical (89)Zr-immuno-PET. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 694–707. [Google Scholar] [CrossRef]
- Vugts, D.J.; Klaver, C.; Sewing, C.; Poot, A.J.; Adamzek, K.; Huegli, S.; Mari, C.; Visser, G.W.M.; Valverde, I.E.; Gasser, G.; et al. Comparison of the octadentate bifunctional chelator DFO*-pPhe-NCS and the clinically used hexadentate bifunctional chelator DFO-pPhe-NCS for Zr-89-immuno-PET. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Seibold, U.; Wängler, B.; Wängler, C. Rational Design, Development, and Stability Assessment of a Macrocyclic Four-Hydroxamate-Bearing Bifunctional Chelating Agent for Zr-89. ChemMedChem 2017, 12, 1555–1571. [Google Scholar] [CrossRef] [PubMed]
- Deri, M.A.; Ponnala, S.; Zeglis, B.M.; Pohl, G.; Dannenberg, J.J.; Lewis, J.S.; Francesconi, L.C. Alternative Chelator for Zr-89 Radiopharmaceuticals: Radiolabeling and Evaluation of 3,4,3-(LI-1,2-HOPO). J. Med. Chem. 2014, 57, 4849–4860. [Google Scholar] [CrossRef] [Green Version]
- Deri, M.A.; Ponnala, S.; Kozowski, P.; Burton-Pye, B.P.; Cicek, H.T.; Hu, C.H.; Lewis, J.S.; Francesconi, L.C. p-SCN-Bn-HOPO: A Superior Bifunctional Chelator for Zr-89 ImmunoPET. Bioconjug. Chem. 2015, 26, 2579–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandya, D.N.; Bhatt, N.; Yuan, H.; Day, C.S.; Ehrmann, B.M.; Wright, M.; Bierbach, U.; Wadas, T.J. Zirconium tetraazamacrocycle complexes display extraordinary stability and provide a new strategy for zirconium-89-based radiopharmaceutical development. Chem. Sci. 2017, 8, 2309–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.P.; Adumeau, P.; Lewis, J.S.; Zeglis, B.M. Click Chemistry and Radiochemistry: The First 10 Years. Bioconjug. Chem. 2016, 27, 2791–2807. [Google Scholar] [CrossRef] [Green Version]
- Reiner, T.; Zeglis, B.M. The inverse electron demand Diels-Alder click reaction in radiochemistry. J. Label. Comp. Radiopharm. 2014, 57, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, D.X.; Zeglis, B.M.; Lewis, J.S.; Anderson, C.J. The Growing Impact of Bioorthogonal Click Chemistry on the Development of Radiopharmaceuticals. J. Nucl. Med. 2013, 54, 829–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litau, S.; Seibold, U.; Wängler, B.; Schirrmacher, R.; Wängler, C. iEDDA Conjugation Reaction in Radiometal Labeling of Peptides with Ga-68 and Cu-64: Unexpected Findings. ACS Omega 2018, 3, 14039–14053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadi, N.; Oves-Costales, D.; Barona-Gomez, F.; Challis, G.L. A new family of ATP-dependent oligomerization-macrocyclization biocatalysts. Nat. Chem. Biol. 2007, 3, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Durbin, P.W.; Kullgren, B.; Ebbe, S.N.; Uhlir, L.C.; Raymond, K.N. Synthesis and initial evaluation for in vivo chelation of Pu(IV) of a mixed octadentate spermine-based ligand containing 4-carbamoyl-3-hydroxy-1-methyl-2(1H)-pyridinone and 6-carbamoyl-1-hydroxy-2(1H)-pyridinone. J. Med. Chem. 2002, 45, 3963–3971. [Google Scholar] [CrossRef]
- Bhupathiraju, N.V.S.D.K.; Younes, A.; Cao, M.H.; Ali, J.; Cicek, H.T.; Tully, K.M.; Ponnala, S.; Babich, J.W.; Deri, M.A.; Lewis, J.S.; et al. Improved synthesis of the bifunctional chelator p-SCN-Bn-HOPO. Org. Biomol. Chem. 2019, 17, 6866–6871. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab-Initio Calculation of Vibrational Absorption and Circular-Dichroism Spectra Using Density-Functional Force-Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentealba, P.; Preuss, H.; Stoll, H.; Vonszentpaly, L. A Proper Account of Core-Polarization with Pseudopotentials—Single Valence-Electron Alkali Compounds. Chem. Phys. Lett. 1982, 89, 418–422. [Google Scholar] [CrossRef]
- Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-Adjusted Abinitio Pseudopotentials for the 2nd and 3rd Row Transition-Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Maltsev, O.V.; Cavalcanti-Adam, E.A.; Zarka, R.; Reuning, U.; Notni, J.; Wester, H.J.; Mas-Moruno, C.; et al. A Comprehensive Evaluation of the Activity and Selectivity Profile of Ligands for RGD-binding Integrins. Sci. Rep. 2017, 7, 39805. [Google Scholar] [CrossRef] [Green Version]
- Wängler, C.; Maschauer, S.; Prante, O.; Schafer, M.; Schirrmacher, R.; Bartenstein, P.; Eisenhut, M.; Wängler, B. Multimerization of cRGD Peptides by Click Chemistry: Synthetic Strategies, Chemical Limitations, and Influence on Biological Properties. ChemBioChem 2010, 11, 2168–2181. [Google Scholar] [CrossRef] [PubMed]
- Del Gatto, A.; De Simone, M.; de Paola, I.; Saviano, M.; Zaccaro, L. Investigation of the Best Conditions to Obtain c(RGDfK) Peptide on Solid Phase. Int. J. Pept. Res. Ther. 2011, 17, 39–45. [Google Scholar] [CrossRef]
- Carlson, J.C.T.; Mikula, H.; Weissleder, R. Unraveling Tetrazine-Triggered Bioorthogonal Elimination Enables Chemical Tools for Ultrafast Release and Universal Cleavage. J. Am. Chem. Soc. 2018, 140, 3603–3612. [Google Scholar] [CrossRef] [Green Version]
- Versteegen, R.M.; Rossin, R.; ten Hoeve, W.; Janssen, H.M.; Robillard, M.S. Click to Release: Instantaneous Doxorubicin Elimination upon Tetrazine Ligation. Angew. Chem. Int. Ed. 2013, 52, 14112–14116. [Google Scholar] [CrossRef] [PubMed]
- van Onzen, A.H.A.M.; Versteegen, R.M.; Hoeben, F.J.M.; Filot, I.A.W.; Rossin, R.; Zhu, T.; Wu, J.; Hudson, P.J.; Janssen, H.M.; ten Hoeve, W.; et al. Bioorthogonal Tetrazine Carbamate Cleavage by Highly Reactive trans-Cyclooctene. J. Am. Chem. Soc. 2020, 142, 10955–10963. [Google Scholar] [CrossRef]
- Breda, S.; Reva, I.D.; Lapinski, L.; Nowak, M.J.; Fausto, R. Infrared spectra of pyrazine, pyrimidine and pyridazine in solid argon. J. Mol. Struct. 2006, 786, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, B.L.; Guo, Z.; Bernardes, G.J.L. Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, S.A.; Kutyreff, C.; Barrett, K.E.; Hernandez, R.; Ellison, P.A.; Happel, S.; Aluicio-Sarduy, E.; Barnhart, T.E.; Nickles, R.J.; Engle, J.W. Evaluation of a chloride-based 89Zr isolation strategy using a tributyl phosphate (TBP)-functionalized extraction resin. Nucl. Med. Biol. 2018, 64–65, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Hosseini, M.; Madsen, J.; Jorgensen, T.J.D.; Jensen, K.J.; Kjaer, A.; Ploug, M. Improved PET Imaging of uPAR Expression Using new Cu-64-labeled Cross-Bridged Peptide Ligands: Comparative in vitro and in vivo Studies. Theranostics 2013, 3, 618–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litau, S.; Seibold, U.; Vall-Sagarra, A.; Fricker, G.; Wangler, B.; Wangler, C. Comparative Assessment of Complex Stabilities of Radiocopper Chelating Agents by a Combination of Complex Challenge and in vivo Experiments. ChemMedChem 2015, 10, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Glaser, M.; Morrison, M.; Solbakken, M.; Arukwe, J.; Karlsen, H.; Wiggen, U.; Champion, S.; Kindberg, G.M.; Cuthbertson, A. Radiosynthesis and biodistribution of cyclic RGD peptides conjugated with novel [18F]fluorinated aldehyde-containing prosthetic groups. Bioconjug. Chem. 2008, 19, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Niedermoser, S.; Chin, J.; Wängler, C.; Kostikov, A.; Bernard-Gauthier, V.; Vogler, N.; Soucy, J.P.; McEwan, A.J.; Schirrmacher, R.; Wängler, B. In Vivo Evaluation of F-18-SiFAlin-Modified TATE: A Potential Challenge for Ga-68-DOTATATE, the Clinical Gold Standard for Somatostatin Receptor Imaging with PET. J. Nucl. Med. 2015, 56, 1100–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varasteh, Z.; Rosenstrom, U.; Velikyan, I.; Mitran, B.; Altai, M.; Honarvar, H.; Rosestedt, M.; Lindeberg, G.; Sorensen, J.; Larhed, M.; et al. The Effect of Mini-PEG-Based Spacer Length on Binding and Pharmacokinetic Properties of a Ga-68-Labeled NOTA-Conjugated Antagonistic Analog of Bombesin. Molecules 2014, 19, 10455–10472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, R.; Stammes, M.A.; de Roos, I.H.C.; Stigter-van Walsum, M.; Visser, G.W.M.; van Dongen, G.A.M.S. Inert coupling of IRDye800CW to monoclonal antibodies for clinical optical imaging of tumor targets. EJNMMI Res. 2011, 1, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
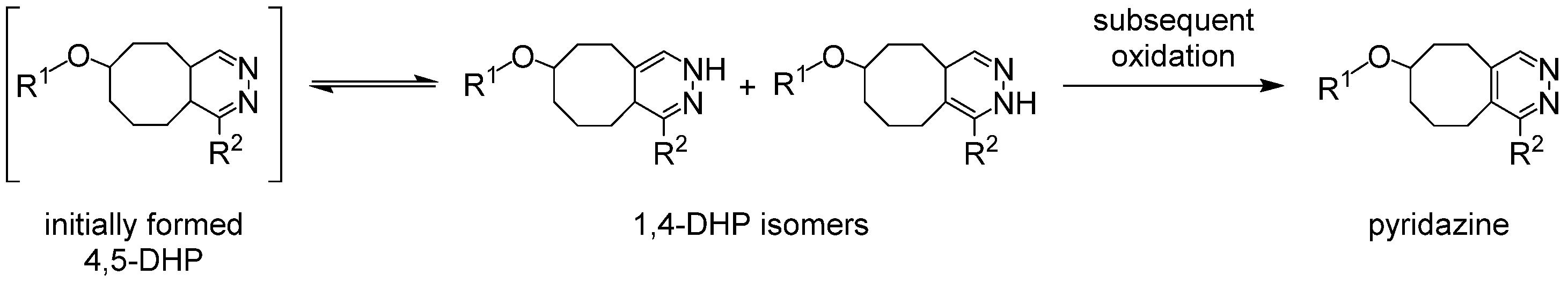
| Optimized Structure | Calculated Bond Lengths [Å] | Literature Values for Bond Lengths [Å] | ||
|---|---|---|---|---|
| product of Zr-1 and TCO-butyl carbamate | 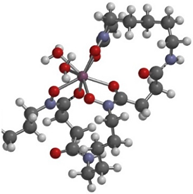 | Zr-DFO(H2O)2 [6] | ||
| 2.2260 (Zr-O (C=O)) | 2.2317 | |||
| 2.1563 (Zr-O (N-O)) | 2.1736 | |||
| 2.2232 (Zr-O (C=O)) | 2.2305 | |||
| 2.1321 (Zr-O (N-O)) | 2.1447 | |||
| 2.1899 (Zr-O (C=O)) | 2.1993 | |||
| 2.1829 (Zr-O (N-O)) | 2.2023 | |||
| 2.4080 (Zr-O (H2O)) | 2.4335 | |||
| 2.3523 (Zr-O (H2O)) | 2.3656 | |||
| product of Zr-2 and TCO-butyl carbamate | 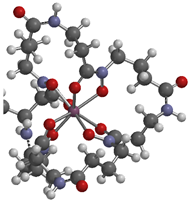 | Zr-CTH-36 [10] | Zr-CTH-36 [6] | |
| 2.2749 (Zr-O (C=O)) | 2.3250 | 2.2862 | ||
| 2.1746 (Zr-O (N-O)) | 2.2411 | 2.1979 | ||
| 2.2787 (Zr-O (C=O)) | 2.3250 | 2.2862 | ||
| 2.1780 (Zr-O (N-O)) | 2.2411 | 2.1978 | ||
| 2.2778 (Zr-O (C=O)) | 2.3250 | 2.2862 | ||
| 2.1816 (Zr-O (N-O)) | 2.2411 | 2.1979 | ||
| 2.2871 (Zr-O (C=O)) | 2.3250 | 2.2865 | ||
| 2.1714 (Zr-O (N-O)) | 2.2411 | 2.1979 | ||
| product of Zr-3 and TCO-methyl carbamate |  | Zr-DFO* [6] | ||
| 2.2454 (Zr-O (C=O)) | 2.2549 | |||
| 2.2079 (Zr-O (N-O)) | 2.2241 | |||
| 2.3099 (Zr-O (C=O)) | 2.3216 | |||
| 2.1475 (Zr-O (N-O)) | 2.1651 | |||
| 2.2533 (Zr-O (C=O)) | 2.2945 | |||
| 2.2345 (Zr-O (N-O)) | 2.2289 | |||
| 2.2853 (Zr-O (C=O)) | 2.3249 | |||
| 2.1845 (Zr-O (N-O)) | 2.1954 | |||
| product of Zr-4 and TCO-butyl carbamate |  | Zr-3,4,3-(LI-1,2-HOPO) [11] | Zr-3,4,3-(LI-1,2-HOPO) [6] | |
| 2.2446 (Zr-O (C=O)) | 2.2394 | 2.2316 | ||
| 2.2043 (Zr-O (N-O)) | 2.2876 | 2.2574 | ||
| 2.2099 (Zr-O (C=O)) | 2.2714 | 2.2410 | ||
| 2.2094 (Zr-O (N-O)) | 2.2324 | 2.2139 | ||
| 2.2528 (Zr-O (C=O)) | 2.2820 | 2.2765 | ||
| 2.2173 (Zr-O (N-O)) | 2.2258 | 2.2079 | ||
| 2.1734 (Zr-O (C=O)) | 2.1979 | 2.1871 | ||
| 2.3182 (Zr-O (N-O)) | 2.3215 | 2.3012 | ||
| product of Zr-5 and TCO-butyl carbamate | 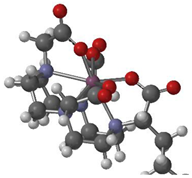 | Zr-DOTA [13] | Zr-DOTA [6] | |
| 2.5652 (Zr-N) | 2.415 | 2.5470 | ||
| 2.1107 (Zr-O) | 2.133 | 2.1570 | ||
| 2.4766 (Zr-N) | 2.415 | 2.4952 | ||
| 2.1655 (Zr-O) | 2.133 | 2.1690 | ||
| 2.5244 (Zr-N) | 2.415 | 2.5471 | ||
| 2.1207 (Zr-O) | 2.133 | 2.1569 | ||
| 2.4762 (Zr-N) | 2.415 | 2.4952 | ||
| 2.1595 (Zr-O) | 2.133 | 2.1690 | ||
| N(1)-N(2) | N(1)-C(1) | C(1)-C(2) | C(2)-C(3) | C(3)-C(4) | C(4)-N(2) | |
|---|---|---|---|---|---|---|
| product of Zr-1 and TCO-butyl carbamate | 1.334 | 1.346 | 1.420 | 1.400 | 1.402 | 1.332 |
| product of Zr-2 and TCO-butyl carbamate | 1.337 | 1.330 | 1.403 | 1.398 | 1.420 | 1.343 |
| product of Zr-3 and TCO-methyl carbamate | 1.335 | 1.331 | 1.403 | 1.400 | 1.421 | 1.345 |
| product of Zr-4 and TCO-butyl carbamate | 1.333 | 1.345 | 1.418 | 1.400 | 1.402 | 1.332 |
| product of Zr-5 and TCO-butyl carbamate | 1.334 | 1.345 | 1.422 | 1.400 | 1.403 | 1.331 |
| pyridazine | 1.337 | 1.338 | 1.400 | 1.385 | 1.400 | 1.338 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damerow, H.; Hübner, R.; Judmann, B.; Schirrmacher, R.; Wängler, B.; Fricker, G.; Wängler, C. Side-by-Side Comparison of Five Chelators for 89Zr-Labeling of Biomolecules: Investigation of Chemical/Radiochemical Properties and Complex Stability. Cancers 2021, 13, 6349. https://doi.org/10.3390/cancers13246349
Damerow H, Hübner R, Judmann B, Schirrmacher R, Wängler B, Fricker G, Wängler C. Side-by-Side Comparison of Five Chelators for 89Zr-Labeling of Biomolecules: Investigation of Chemical/Radiochemical Properties and Complex Stability. Cancers. 2021; 13(24):6349. https://doi.org/10.3390/cancers13246349
Chicago/Turabian StyleDamerow, Helen, Ralph Hübner, Benedikt Judmann, Ralf Schirrmacher, Björn Wängler, Gert Fricker, and Carmen Wängler. 2021. "Side-by-Side Comparison of Five Chelators for 89Zr-Labeling of Biomolecules: Investigation of Chemical/Radiochemical Properties and Complex Stability" Cancers 13, no. 24: 6349. https://doi.org/10.3390/cancers13246349
APA StyleDamerow, H., Hübner, R., Judmann, B., Schirrmacher, R., Wängler, B., Fricker, G., & Wängler, C. (2021). Side-by-Side Comparison of Five Chelators for 89Zr-Labeling of Biomolecules: Investigation of Chemical/Radiochemical Properties and Complex Stability. Cancers, 13(24), 6349. https://doi.org/10.3390/cancers13246349