
Epigenetic Deregulation of Telomere-Related Genes in Newly Diagnosed Multiple Myeloma Patients



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Background
2. Methods
2.1. Sample Information and Genomic DNA/RNA Preparation
2.2. Library Preparation and Sequence Annotations for Reduced Representation Bisulfite Sequencing
2.3. Gene Expression Analysis
2.4. Analysis of Chromatin Modifications
2.5. Determine G-Quadruplex–DMR Overlaps
2.6. Statistical Analysis
3. Results
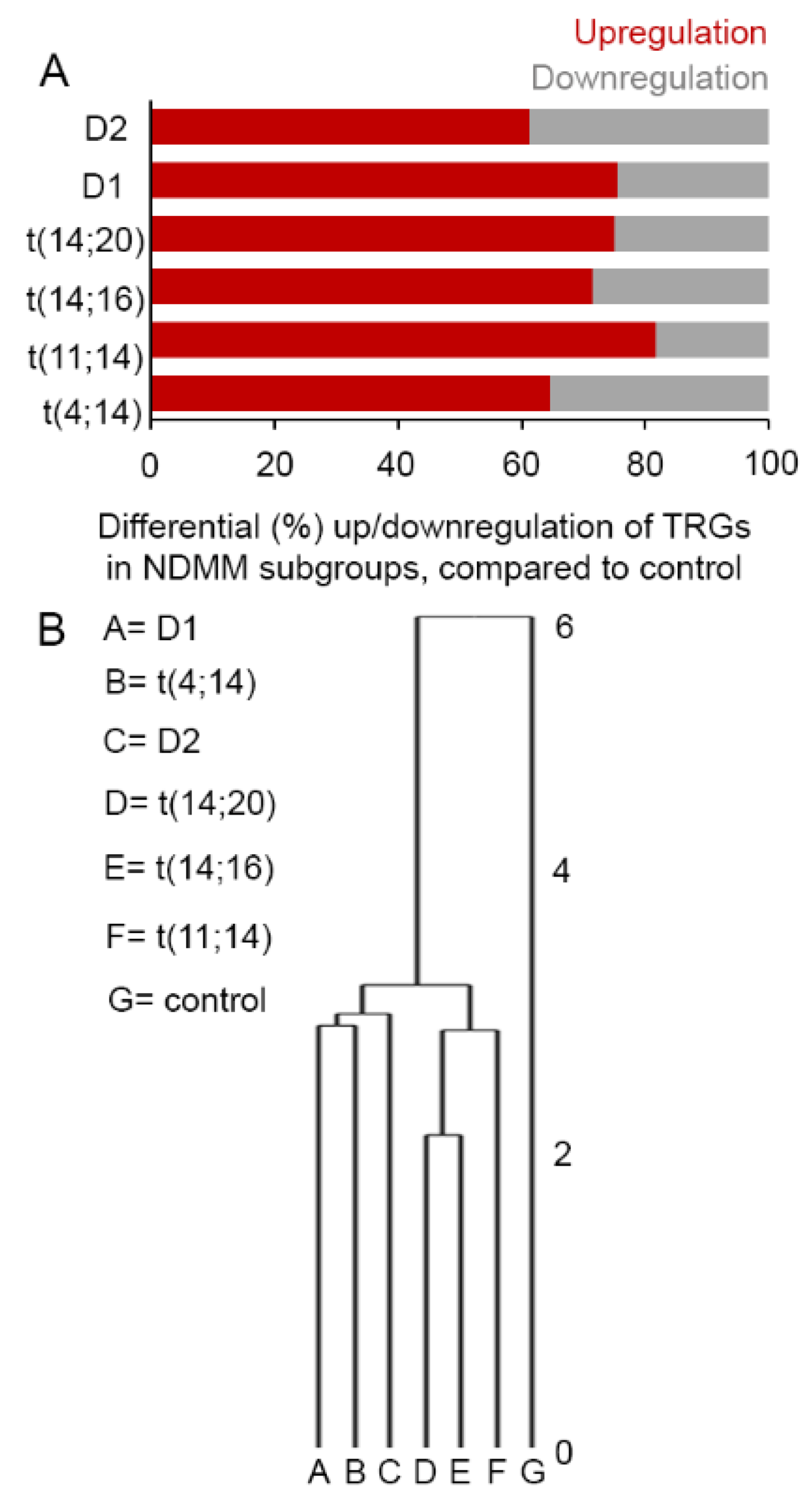
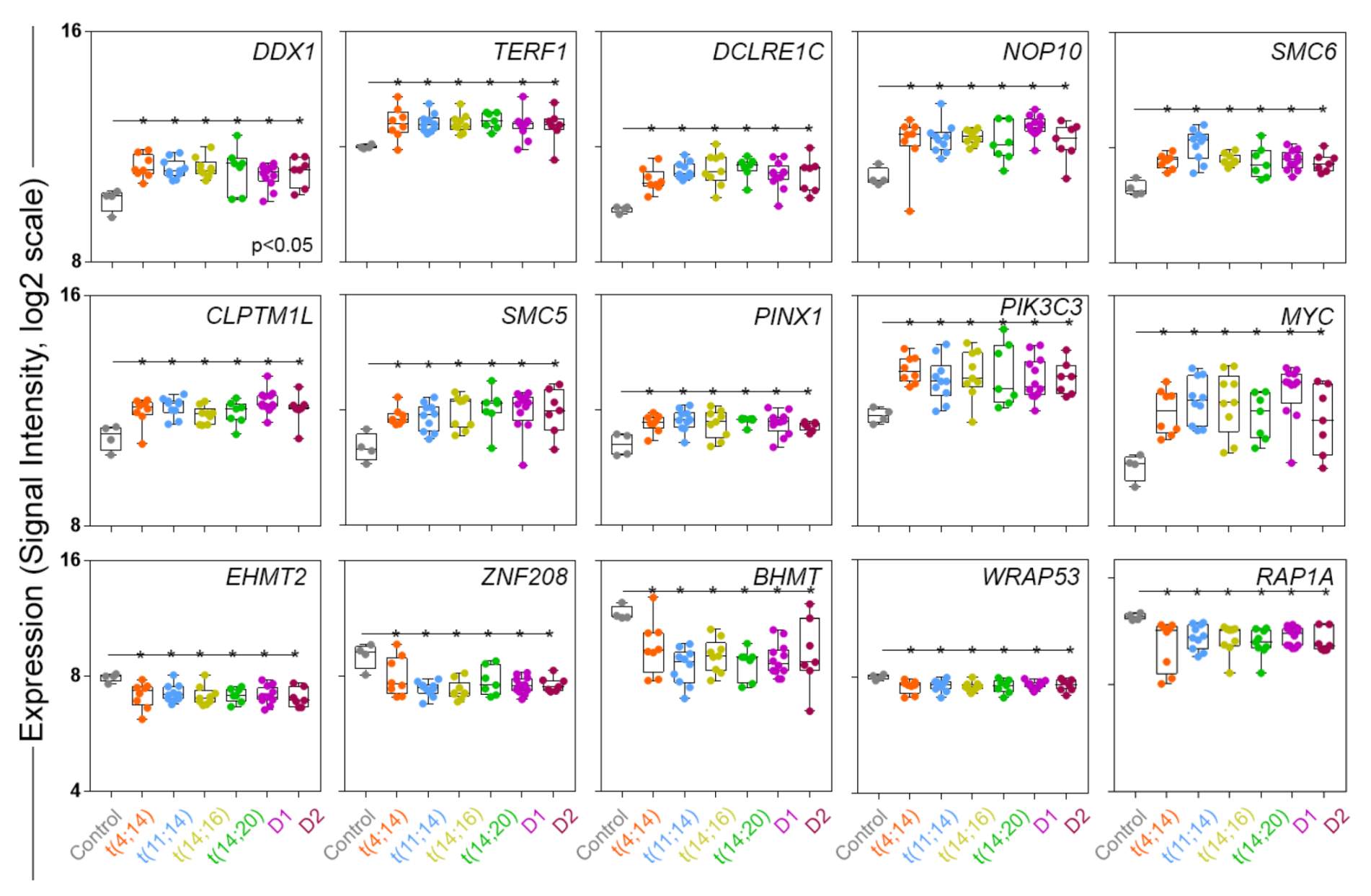
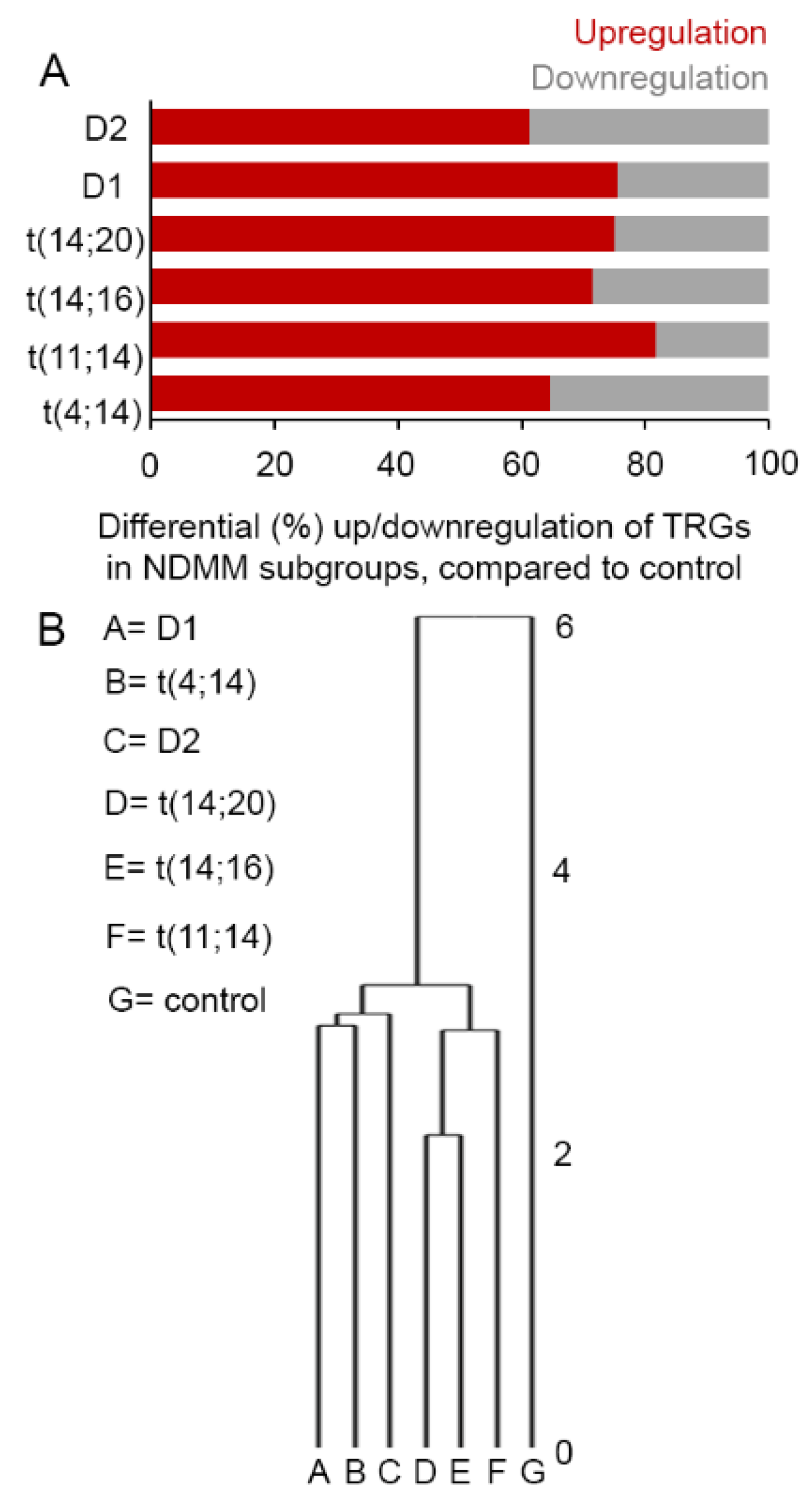
3.1. Selective TRGs Are Differentially Expressed in NDMM
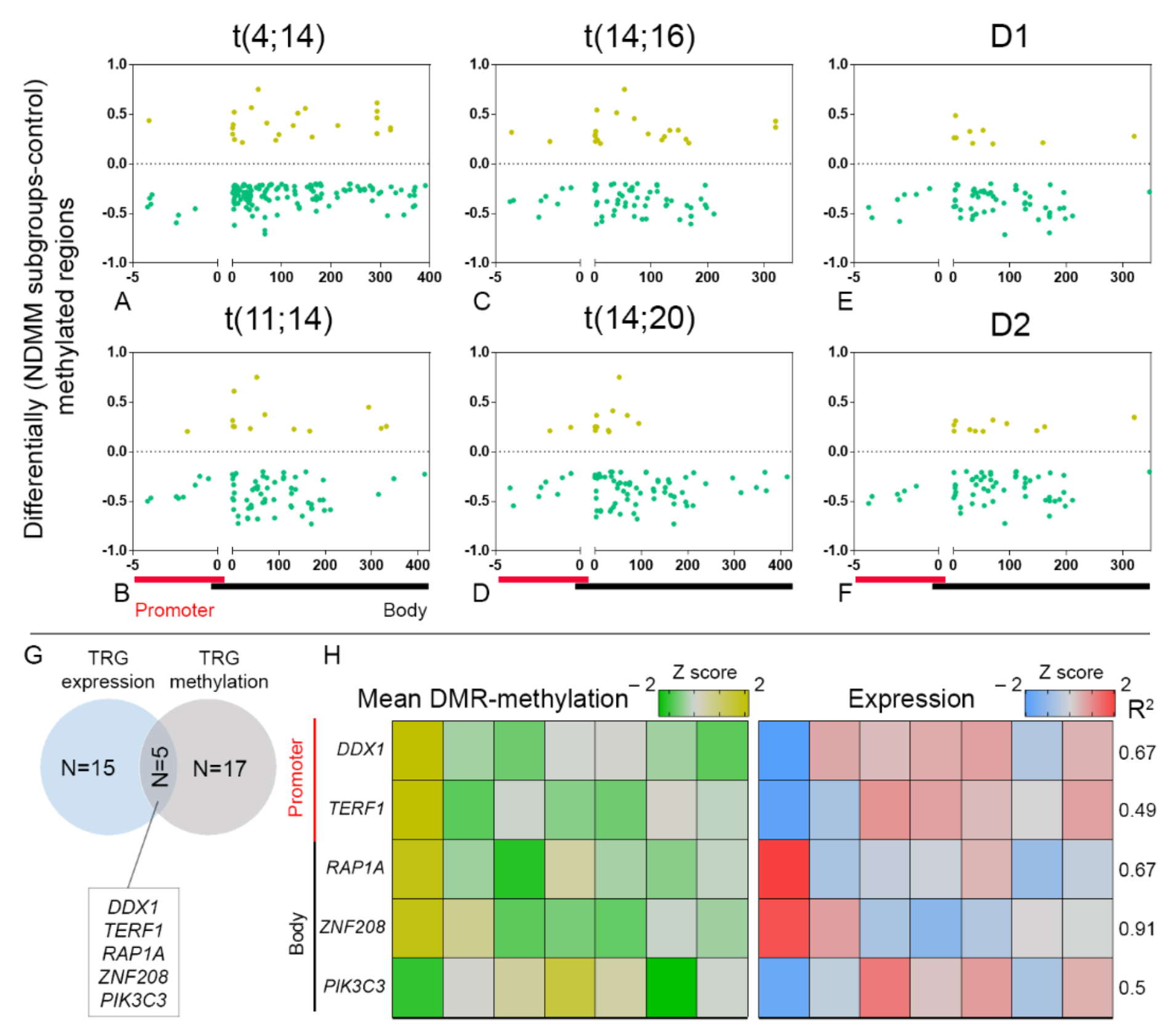
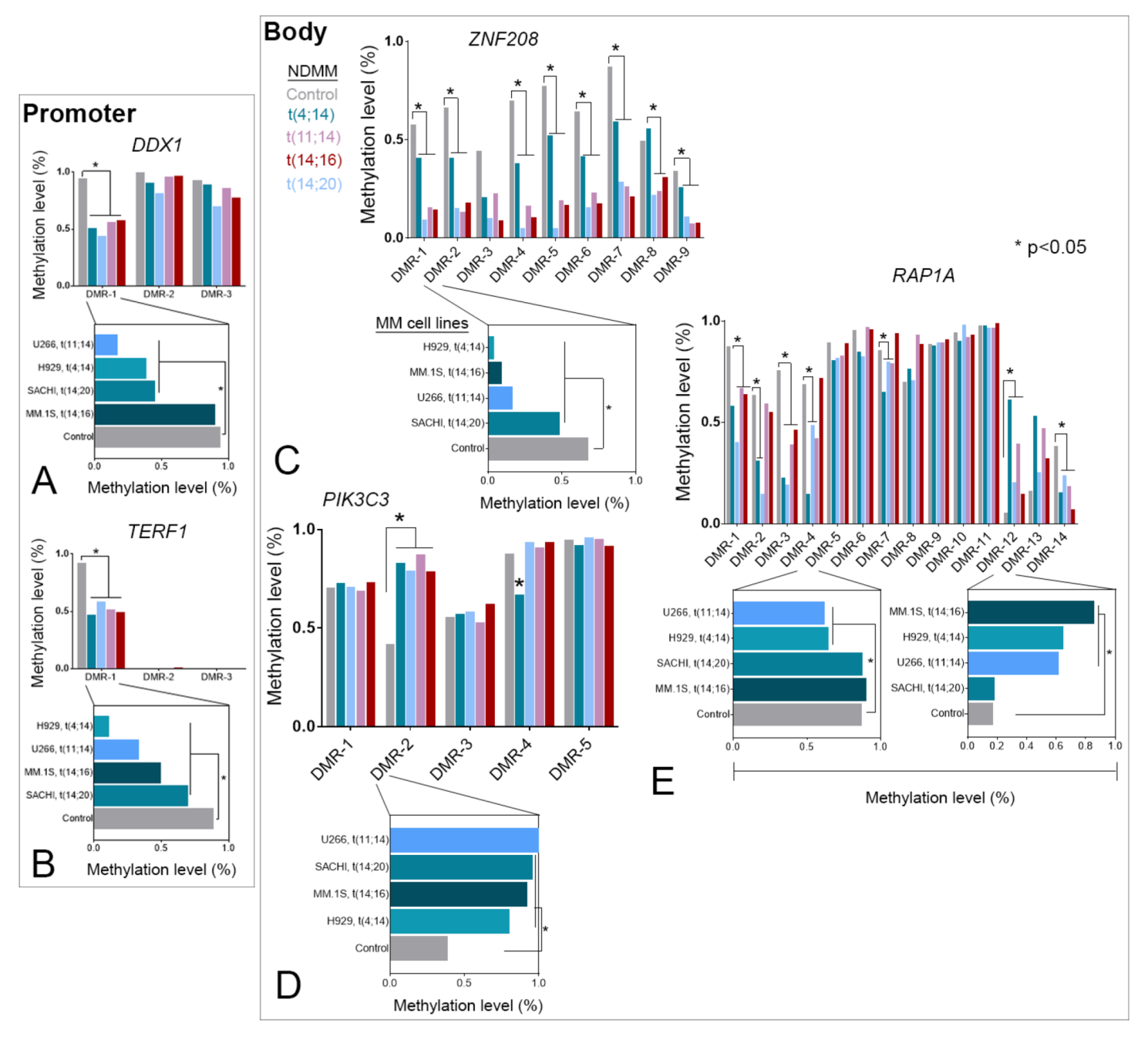
3.2. Selective TRGs Are Differentially Methylated and Tightly Correlated to Expression in NDMM
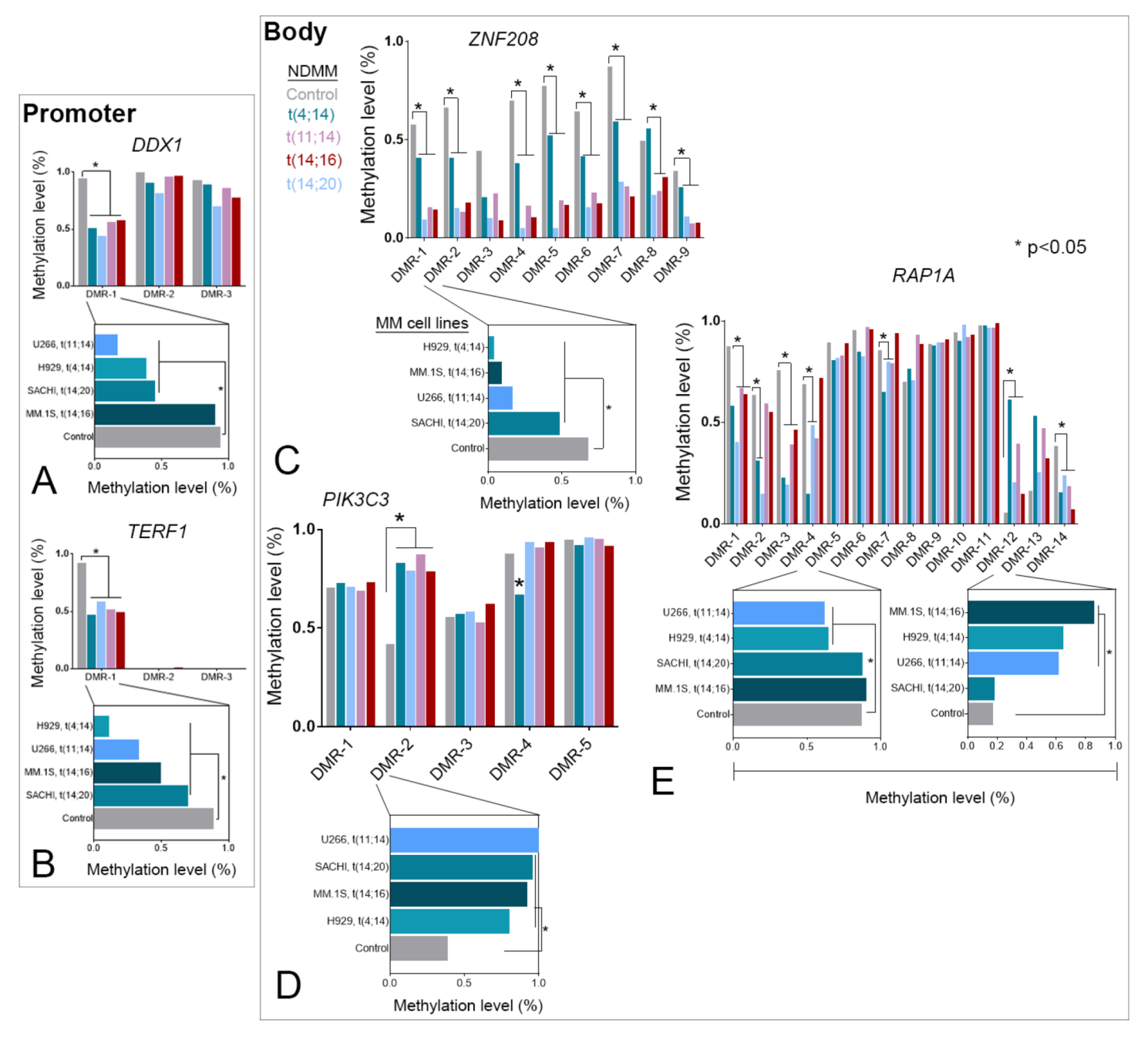
3.3. TRG-Methylation Is Fairly Conserved between Patients with NDMM and MM Cell Lines
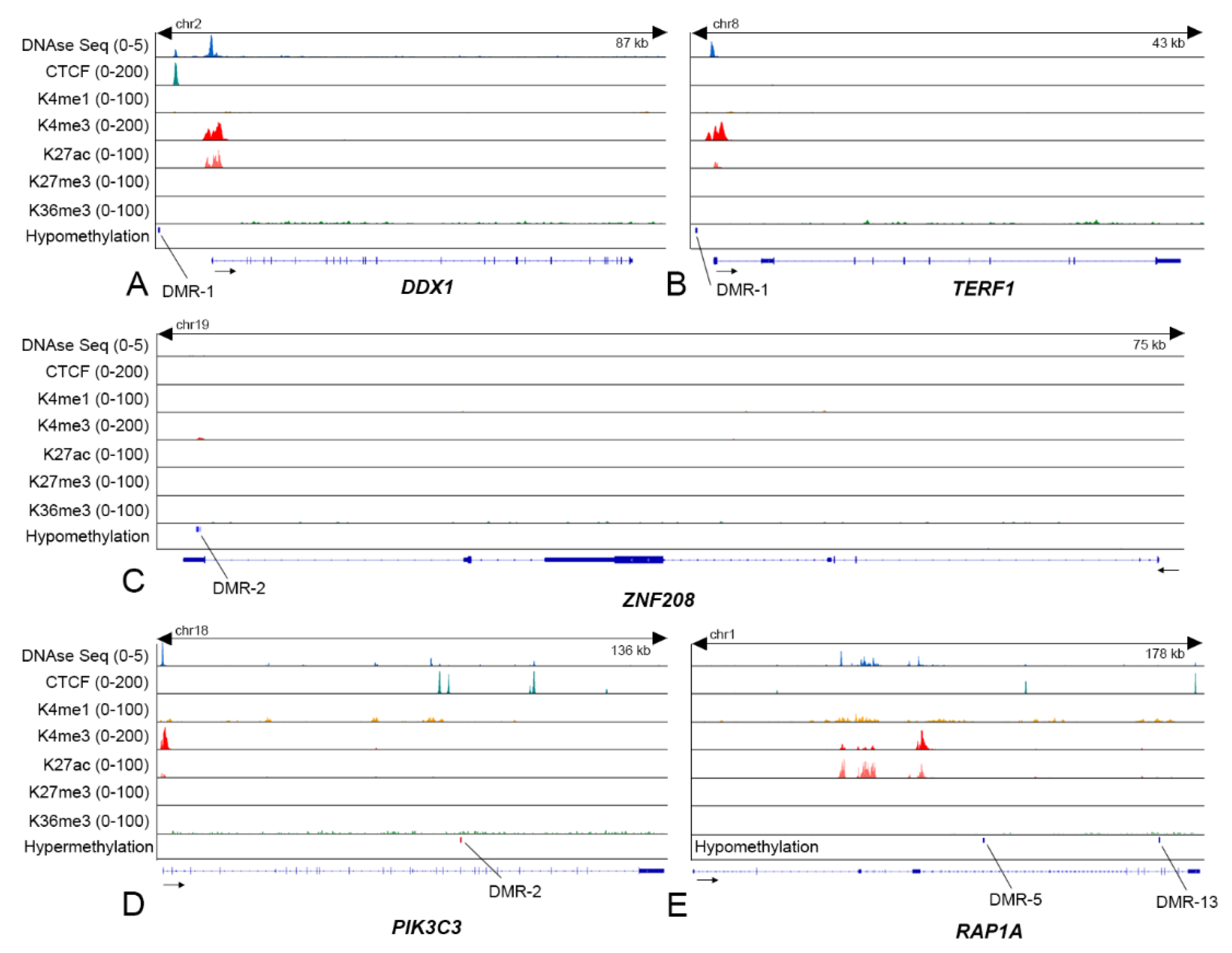
3.4. DNA Methylation Alone or Cooperatively with Chromatin Marks Regulates TRG Expression
3.5. TRG Hypomethylated DMRs Are Stabilized with G-Quadruplexes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ChIP-seq | Chromatin immunoprecipitation sequencing |
| CTCF | CCCTC binding factor |
| DMR | Differentially methylated region |
| FC | Fold change |
| FDR | False discovery rate |
| G4FS | G-quadruplex forming sequences |
| hTERT | Telomerase reverse transcriptase encoding gene |
| HY | Hyperdiploidy |
| IgH | Immunoglobulin heavy chain locus |
| MDM | mean DMR methylation |
| MGUS | Monoclonal gammopathy of undetermined significance |
| MM | Multiple Myeloma |
| NDMM | Newly diagnosed multiple myeloma |
| QGRS | Quadraples forming G-rich sequences |
| RRBS | Reduced representation bisulfite sequencing |
| SMM | Smoldering multiple myeloma |
| TC | Translocation |
| TF | Transcription factor |
| TL | Telomere length |
| TRG | Telomere-related gene |
| TSS | Transcription start site |
References
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Giaccherini, M.; Macauda, A.; Orciuolo, E.; Rymko, M.; Gruenpeter, K.; Dumontet, C.; Raźny, M.; Moreno, V.; Buda, G.; Beider, K.; et al. Genetically determined telomere length and multiple myeloma risk and outcome. Blood Cancer J. 2021, 11, 74. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Ashby, C.; Tytarenko, R.; Bauer, M.; Wang, Y.; Deshpande, S.; Den, J.; Schinke, C.; Zangari, M.; Thanendrarajan, S. The functional epigenetic landscape of aberrant gene expression in molecular subgroups of newly diagnosed multiple myeloma. J. Hematol. Oncol. 2020, 13, 1–15. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Gimsing, P.; Grønbæk, K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 2014, 4, e207–e207. [Google Scholar] [CrossRef]
- Dumontet, C.; Landi, S.; Reiman, T.; Perry, T.; Plesa, A.; Bellini, I.; Barale, R.; Pilarski, L.; Troncy, J.; Tavtigian, S. Genetic polymorphisms associated with outcome in multiple myeloma patients receiving high-dose melphalan. Bone Marrow Transplant. 2010, 45, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, E.H. Switching and signaling at the telomere. Cell 2001, 106, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Bekaert, S.; De Meyer, T.I.M.; Van Oostveldt, P. Telomere Attrition as Ageing Biomarker. Anticancer Res. 2005, 25, 3011. [Google Scholar]
- Lee, M.; Hills, M.; Conomos, D.; Stutz, M.D.; Dagg, R.A.; Lau, L.M.; Reddel, R.R.; Pickett, H.A. Telomere extension by telomerase and ALT generates variant repeats by mechanistically distinct processes. Nucleic Acids Res. 2014, 42, 1733–1746. [Google Scholar] [CrossRef] [Green Version]
- Reddel, R.R. Alternative lengthening of telomeres, telomerase, and cancer. Cancer Lett. 2003, 194, 155–162. [Google Scholar] [CrossRef]
- Hou, L.; Zhang, X.; Gawron, A.J.; Liu, J. Surrogate tissue telomere length and cancer risk Shorter or Longer? Cancer Lett. 2012, 319, 130–135. [Google Scholar] [CrossRef]
- Wentzensen, I.M.; Mirabello, L.; Pfeiffer, R.M.; Savage, S.A. The association of telomere length and cancer a meta-analysis. Cancer Epidemiol. Prev. Biomark. 2011, 20, 1238–1250. [Google Scholar] [CrossRef] [Green Version]
- Campa, D.; Mergarten, B.; De Vivo, I.; Boutron-Ruault, M.-C.; Racine, A.; Severi, G.; Nieters, A.; Katzke, V.A.; Trichopoulou, A.; Yiannakouris, N. Leukocyte telomere length in relation to pancreatic cancer risk a prospective study. Cancer Epidemiol. Prev. Biomark. 2014, 23, 2447–2454. [Google Scholar] [CrossRef] [Green Version]
- Campa, D.; Martino, A.; Varkonyi, J.; Lesueur, F.; Jamroziak, K.; Landi, S.; Jurczyszyn, A.; Marques, H.; Andersen, V.; Jurado, M. Risk of multiple myeloma is associated with polymorphisms within telomerase genes and telomere length. Int. J. Cancer 2015, 136, E351–E358. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, S.; Jones, R.E.; Heppel, N.H.; Grimstead, J.W.; Fegan, C.; Jackson, G.H.; Hills, R.; Allan, J.M.; Pratt, G.; Pepper, C.; et al. Telomere length is a critical determinant for survival in multiple myeloma. Br. J. Haematol. 2017, 178, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.D.; Orme, L.M.; Shaughnessy, J.; Jacobson, J., Jr.; Barlogie, B.; Moore, M.A. Telomerase and telomere length in multiple myeloma correlations with disease heterogeneity, cytogenetic status, and overall survival. Blood 2003, 101, 4982–4989. [Google Scholar] [CrossRef]
- Cottliar, A.; Pedrazzini, E.; Corrado, C.; Engelberger, M.I.; Narbaitz, M.; Slavutsky, I. Telomere shortening in patients with plasma cell disorders. Eur. J. Haematol. 2003, 71, 334–340. [Google Scholar] [CrossRef]
- Rangel-Pozzo, A.; Yu, P.L.I.; LaL, S.; Asbaghi, Y.; Sisdelli, L.; Tammur, P.; Tamm, A.; Punab, M.; Klewes, L.; Louis, S.; et al. Telomere Architecture Correlates with Aggressiveness in Multiple Myeloma. Cancers 2021, 13, 1969. [Google Scholar] [CrossRef]
- Brennan, S.K.; Wang, Q.; Tressler, R.; Harley, C.; Go, N.; Bassett, E.; Huff, C.A.; Jones, R.J.; Matsui, W. Telomerase Inhibition Targets Clonogenic Multiple Myeloma Cells through Telomere Length-Dependent and Independent Mechanisms. PLoS ONE 2010, 5, e12487. [Google Scholar] [CrossRef]
- Shammas, M.A.; Koley, H.; Bertheau, R.C.; Neri p Fulciniti, M.; Tassone p Blotta, S.; Protopopov, A.; Mitsiades, C.; Batchu, R.B. Telomerase inhibitor GRN163L inhibits myeloma cell growth in vitro and in vivo. Leukemia 2008, 22, 1410–1418. [Google Scholar] [CrossRef]
- Akiyama, M.; Hideshima, T.; Hayashi, T.; Tai, Y.T.; Mitsiades, C.S.; Mitsiades, N.; Chauhan, D.; Richardson p Munshi, N.C.; Anderson, K.C. Cytokines modulate telomerase activity in a human multiple myeloma cell line. Cancer Res. 2002, 62, 3876–3882. [Google Scholar]
- Weiss, C.; Uziel, O.; Wolach, O.; Nordenberg, J.; Beery, E.; Bulvick, S.; Kanfer, G.; Cohen, O.; Ram, R.; Bakhanashvili, M.; et al. Differential downregulation of telomerase activity by bortezomib in multiple myeloma cells-multiple regulatory pathways in vitro and ex vivo. Br. J. Cancer 2012, 107, 1844–1852. [Google Scholar] [CrossRef]
- Mirabello, L.; Yu, K.; Kraft p De Vivo, I.; Hunter, D.J.; Prescott, J.; Wong, J.Y.; Chatterjee, N.; Hayes, R.B.; Savage, S.A. The association of telomere length and genetic variation in telomere biology genes. Hum. Mutat. 2010, 31, 1050–1058. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, S.R.; Cui, Y.; Milton, J.R.; Li, J.; Irudayaraj, J. Selective increase in subtelomeric DNA methylation an epigenetic biomarker for malignant glioma. Clin. Epigenet. 2015, 7, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buxton, J.L.; Suderman, M.; Pappas, J.J.; Borghol, N.; McArdle, W.; Blakemore, A.I.F.; Hertzman, C.; Power, C.; Szyf, M.; Pembrey, M. Human leukocyte telomere length is associated with DNA methylation levels in multiple subtelomeric and imprinted loci. Sci. Rep. 2014, 4, 4954. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.Y.Y.; De Vivo, I.; Lin, X.; Grashow, R.; Cavallari, J.; Christiani, D.C. The association between global DNA methylation and telomere length in a longitudinal study of boilermakers. Genet. Epidemiol. 2014, 38, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Zhang, F.; Guo, X.; Tang, L.; Peng, L.; Luo, X.; Xu, X.; Wang, S.; Dai, L.; Wang, J. Integrated analysis of promoter methylation and expression of telomere related genes in breast cancer. Oncotarget 2017, 8, 25442–25454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz de la Guardia, R.; Catalina p Panero, J.; Elosua, C.; Pulgarin, A.; López, M.B.; Ayllón, V.; Ligero, G.; Slavutsky, I.; Leone, P.E. Expression profile of telomere-associated genes in multiple myeloma. J. Cell Mol. Med. 2012, 16, 3009–3021. [Google Scholar] [CrossRef]
- Panero, J.; Stella, F.; Schutz, N.; Fantl, D.B.; Slavutsky, I. Differential Expression of Non-Shelterin Genes Associated with High Telomerase Levels and Telomere Shortening in Plasma Cell Disorders. PLoS ONE 2015, 10, e0137972. [Google Scholar] [CrossRef] [PubMed]
- Joyce, B.T.; Zheng, Y.; Nannini, D.; Zhang, Z.; Liu, L.; Gao, T.; Kocherginsky, M.; Murphy, R.; Yang, H.; Achenbach, C.J.; et al. DNA Methylation of Telomere-Related Genes and Cancer Risk. Cancer Prev. Res. 2018, 11, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, S.R.; Walker, B.A. A Rapid and Robust Protocol for Reduced Representation Bisulfite Sequencing in Multiple Myeloma. In Multiple Myeloma; Springer: Berlin/Heidelberg, Germany, 2018; pp. 179–191. [Google Scholar]
- Schreiber, J.; Bilmes, J.; Noble, W.S. Completing the ENCODE3 compendium yields accurate imputations across a variety of assays and human biosamples. Genome Biol. 2020, 21, 82. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef]
- Storti, C.B.; de Oliveira, R.A.; de Carvalho, M.; Hasimoto, E.N.; Cataneo, D.C.; Cataneo, A.J.M.; De Faveri, J.; Vasconcelos, E.J.R.; Dos Reis, P.P.; Cano, M.I.N. Telomere-associated genes and telomeric lncRNAs are biomarker candidates in lung squamous cell carcinoma (LUSC). Exp. Mol. Pathol. 2020, 112, 104354. [Google Scholar] [CrossRef]
- Cui, H.; Onyango, P.; Brandenburg, S.; Wu, Y.; Hsieh, C.-L.; Feinberg, A.P. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002, 62, 6442–6446. [Google Scholar]
- Palumbo, D.; Affinito, O.; Monticelli, A.; Cocozza, S. DNA Methylation variability among individuals is related to CpGs cluster density and evolutionary signatures. BMC Genom. 2018, 19, 229. [Google Scholar] [CrossRef]
- Chambwe, N.; Kormaksson, M.; Geng, H.; De, S.; Michor, F.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Godley, L.A.; Gascoyne, R.D.; et al. Variability in DNA methylation defines novel epigenetic subgroups of DLBCL associated with different clinical outcomes. Blood 2014, 123, 1699–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, K.W.; Karunasena, E.; Ahuja, N. The Roles of DNA Methylation in the Stages of Cancer. Cancer J. 2017, 23, 257–261. [Google Scholar] [CrossRef]
- De, S.; Michor, F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat. Struct. Mol. Biol. 2011, 18, 950–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedström, E.; Pederiva, C.; Farnebo, J.; Nodin, B.; Jirström, K.; Brennan, D.J.; Farnebo, M. Downregulation of the cancer susceptibility protein WRAP53β in epithelial ovarian cancer leads to defective DNA repair and poor clinical outcome. Cell Death Dis. 2015, 6, e1892. [Google Scholar] [CrossRef]
- Kim, D.S.; Lee, W.K.; Park, J.Y. Promoter methylation of Wrap53α, an antisense transcript of p53, is associated with the poor prognosis of patients with non-small cell lung cancer. Oncol. Lett. 2018, 16, 5823–5828. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Oue, N.; Yoshida, K.; Mitani, Y.; Naka, K.; Nakayama, H.; Yasui, W. Expression of POT1 is associated with tumor stage and telomere length in gastric carcinoma. Cancer Res. 2004, 64, 523–529. [Google Scholar] [CrossRef] [Green Version]
- Pal, D.; Sharma, U.; Singh, S.K.; Kakkar, N.; Prasad, R. Over-Expression of Telomere Binding Factors (TRF1 & TRF2) in Renal Cell Carcinoma and Their Inhibition by Using SiRNA Induce Apoptosis, Reduce Cell Proliferation and Migration Invitro. PLoS ONE 2015, 10, e0115651. [Google Scholar]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Kauffmann, A.; Rosselli, F.; Lazar, V.; Winnepenninckx, V.; Mansuet-Lupo, A.; Dessen, P.; Van den Oord, J.; Spatz, A.; Sarasin, A. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 2008, 27, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Trolle, C.; Nielsen, M.M.; Skakkebæk, A.; Lamy, P.; Vang, S.; Hedegaard, J.; Nordentoft, I.; Ørntoft, T.F.; Pedersen, J.S.; Gravholt, C.H. Widespread DNA hypomethylation and differential gene expression in Turner syndrome. Sci. Rep. 2016, 6, 34220. [Google Scholar] [CrossRef] [Green Version]
- Holwerda, S.J.B.; de Laat, W. CTCF the protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120369. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, R.; Russo, N.; Liu, M.; Van Tubergen, E.; D’Silva, N.J. Rap1 and its regulatory proteins the tumor suppressor, oncogene, tumor suppressor gene axis in head and neck cancer. Small GTPases 2012, 3, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Shang, S.; Yang, Y.; Lu p Wang, T.; Cui, X.; Tang, X. Identification of DNA methylation-driven genes by integrative analysis of DNA methylation and transcriptome data in pancreatic adenocarcinoma. Exp. Ther. Med. 2020, 19, 2963–2972. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, J.Y.; Chen, Y.N.; Yuan, F.; Zhang, H.; Yan, F.H.; Wang, M.J.; Wang, G.; Su, M.; Lu, G.; et al. Whole genome and transcriptome sequencing of matched primary and peritoneal metastatic gastric carcinoma. Sci. Rep. 2015, 5, 13750. [Google Scholar] [CrossRef] [Green Version]
- Jara-Espejo, M.; Line, S.R. DNA G-quadruplex stability, position and chromatin accessibility are associated with CpG island methylation. FEBS J. 2020, 287, 483–495. [Google Scholar] [CrossRef]
- Mao, S.-Q.; Ghanbarian, A.T.; Spiegel, J.; Martínez Cuesta, S.; Beraldi, D.; Di Antonio, M.; Marsico, G.; Hänsel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. DNA G-quadruplex structures mold the DNA methylome. Nat. Struct. Mol. Biol. 2018, 25, 951–957. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy Choudhury, S.; Ashby, C.; Zhan, F.; van Rhee, F. Epigenetic Deregulation of Telomere-Related Genes in Newly Diagnosed Multiple Myeloma Patients. Cancers 2021, 13, 6348. https://doi.org/10.3390/cancers13246348
Roy Choudhury S, Ashby C, Zhan F, van Rhee F. Epigenetic Deregulation of Telomere-Related Genes in Newly Diagnosed Multiple Myeloma Patients. Cancers. 2021; 13(24):6348. https://doi.org/10.3390/cancers13246348
Chicago/Turabian StyleRoy Choudhury, Samrat, Cody Ashby, Fenghuang Zhan, and Frits van Rhee. 2021. "Epigenetic Deregulation of Telomere-Related Genes in Newly Diagnosed Multiple Myeloma Patients" Cancers 13, no. 24: 6348. https://doi.org/10.3390/cancers13246348
APA StyleRoy Choudhury, S., Ashby, C., Zhan, F., & van Rhee, F. (2021). Epigenetic Deregulation of Telomere-Related Genes in Newly Diagnosed Multiple Myeloma Patients. Cancers, 13(24), 6348. https://doi.org/10.3390/cancers13246348