
The Estrogen Receptor α Signaling Pathway Controls Alternative Splicing in the Absence of Ligands in Breast Cancer Cells

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
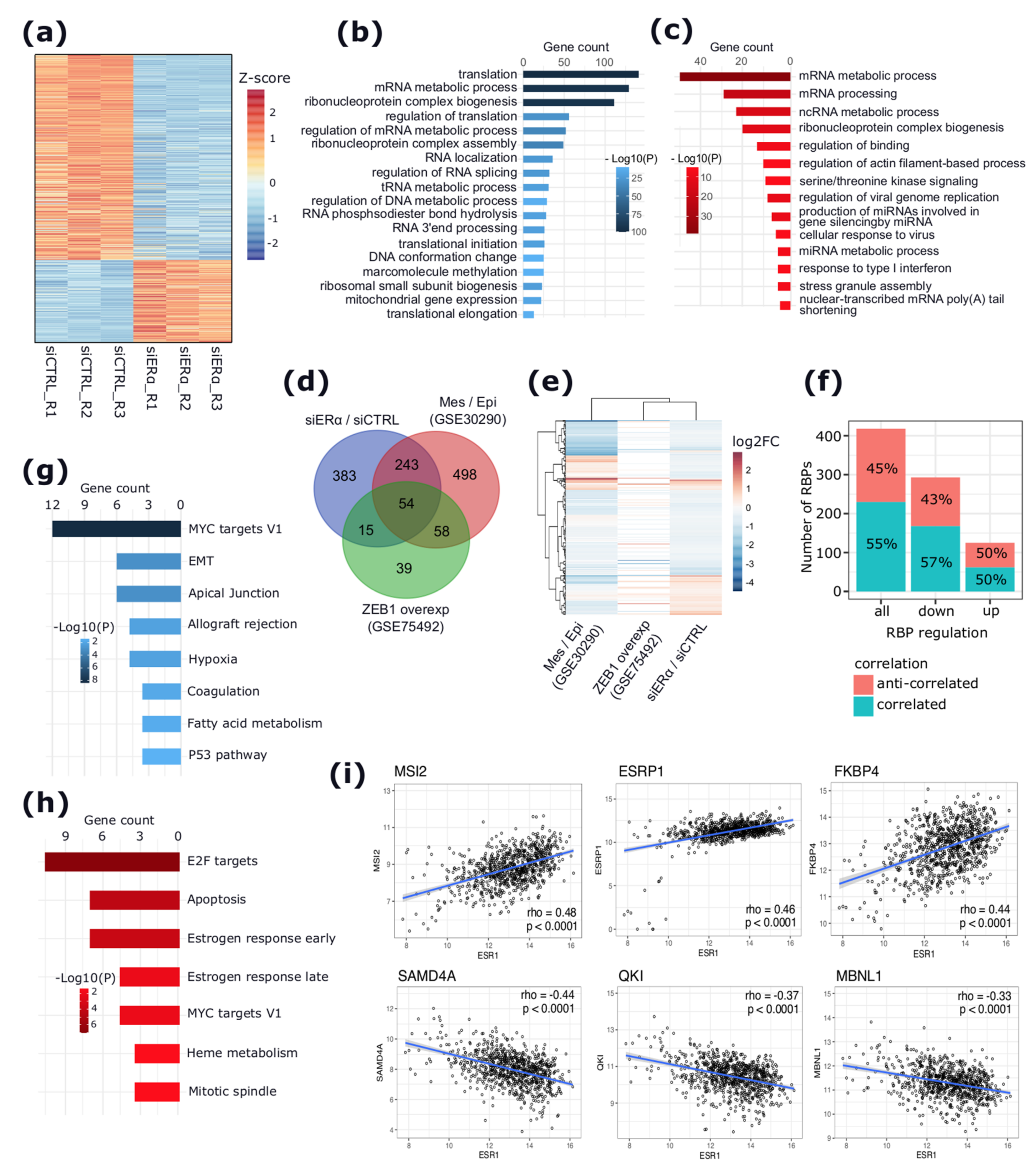
2.1. apoERα Activity Regulates the Expression of RBPs and SFs in the MCF-7 BC Cell Line
2.2. EMT-Related Gene Isoforms Are Differentially Expressed upon apoERα Silencing
2.3. apoERα Depletion Induces Internal ASEs in EMT-Related Genes
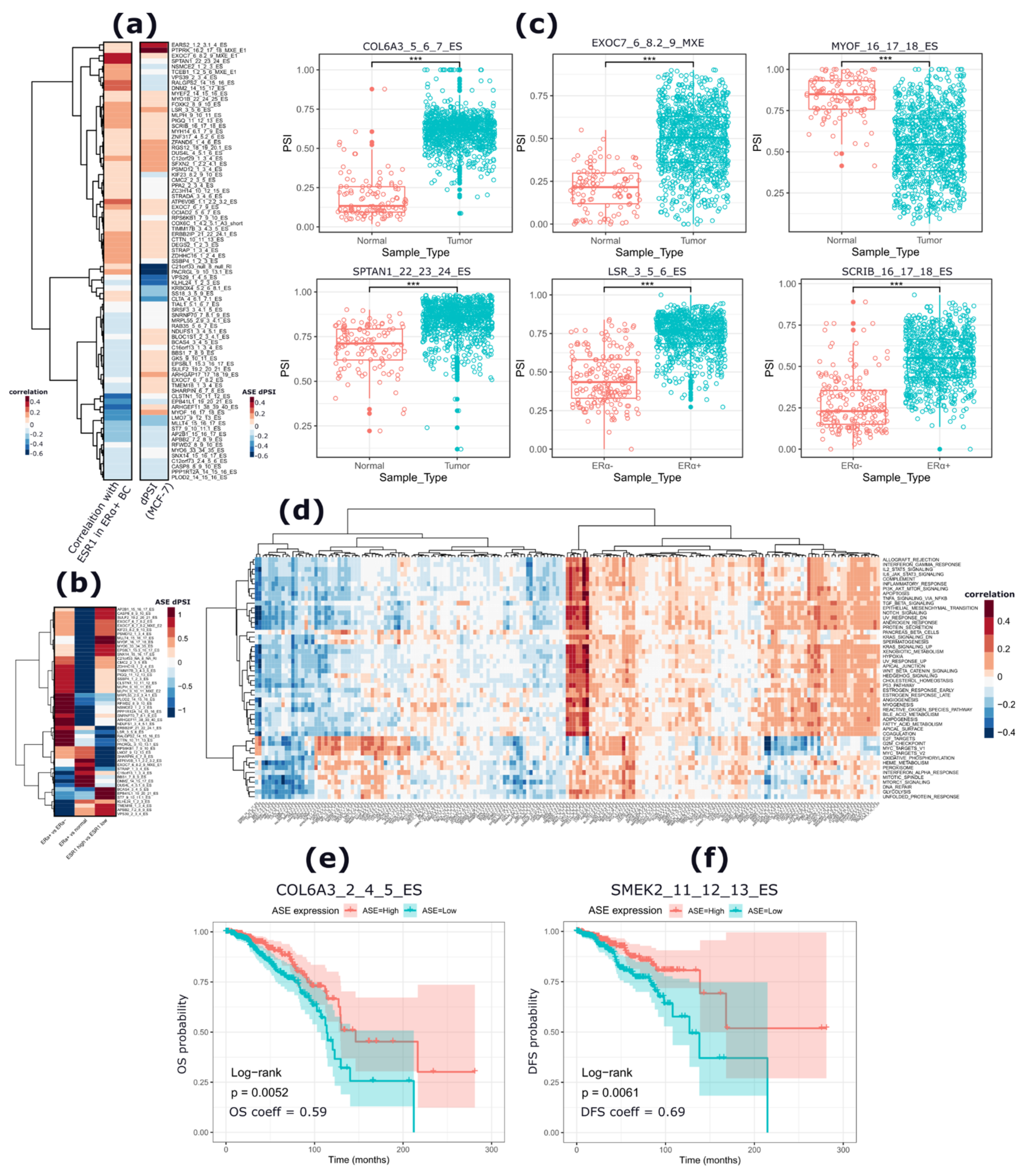
2.4. The apoERα-Regulated RBPs Are Significantly Correlated with ERα mRNA Levels in ERα+ BCs and Are Predicted Regulators of the apoERα-Modulated ASEs
2.5. apoERα-Regulated Exons Are Differentially Included in Primary BCs and Correlate with ERα mRNA Levels
3. Discussion
4. Materials and Methods
4.1. RNA-Seq Read Preprocessing, Alignment, and Expression Quantification
4.2. The Differential Expression Analysis
4.3. The Gene Ontology Enrichment Analysis
4.4. The Isoform Switching Analysis
4.5. The Differential Alternative Splicing Analysis
4.6. The RBP Binding Motif Enrichment Analysis
4.7. An Overlap with Alternative Splicing Events in Primary Tumor Data
4.8. The Correlation Analysis between ERα mRNA Levels and RNA-Binding Proteins Encoding Genes in ERα+ Breast Tumor Samples
4.9. The Overlap with ERα HITS-CLIP Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, B.; Eyras, E. The Role of Alternative Splicing in Cancer. Transcription 2017, 8, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladomery, M. Aberrant Alternative Splicing Is Another Hallmark of Cancer. Int. J. Cell Biol. 2013, 2013, 463786. [Google Scholar] [CrossRef] [PubMed]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and Mechanisms of Alternative Splicing in Cancer—Implications for Care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, J.; Zhang, W.; Chen, D.; Wang, Y. Aberrant Alternative Splicing in Breast Cancer. J. Mol. Cell Biol. 2019, 11, 920–929. [Google Scholar] [CrossRef]
- Park, S.; Brugiolo, M.; Akerman, M.; Das, S.; Urbanski, L.; Geier, A.; Kesarwani, A.K.; Fan, M.; Leclair, N.; Lin, K.-T.; et al. Differential Functions of Splicing Factors in Mammary Transformation and Breast Cancer Metastasis. Cell Rep. 2019, 29, 2672–2688.e7. [Google Scholar] [CrossRef]
- Russnes, H.G.; Lingjærde, O.C.; Børresen-Dale, A.-L.; Caldas, C. Breast Cancer Molecular Stratification: From Intrinsic Subtypes to Integrative Clusters. Am. J. Pathol. 2017, 187, 2152–2162. [Google Scholar] [CrossRef]
- Russo, J. The Molecular Basis of Breast Cancer Subtypes. Pathobiol. Breast Cancer 2016, 1, 111–116. [Google Scholar]
- Yang, S.X.; Polley, E.C. Systemic Treatment and Radiotherapy, Breast Cancer Subtypes, and Survival after Long-Term Clinical Follow-Up. Breast Cancer Res. Treat. 2019, 175, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Anbalagan, M.; Rowan, B.G. Estrogen Receptor Alpha Phosphorylation and Its Functional Impact in Human Breast Cancer. Mol. Cell. Endocrinol. 2015, 418, 264–272. [Google Scholar] [CrossRef]
- Chen, D.; Washbrook, E.; Sarwar, N.; Bates, G.J.; Pace, P.E.; Thirunuvakkarasu, V.; Taylor, J.; Epstein, R.J.; Fuller-Pace, F.V.; Egly, J.-M.; et al. Phosphorylation of Human Estrogen Receptor Alpha at Serine 118 by Two Distinct Signal Transduction Pathways Revealed by Phosphorylation-Specific Antisera. Oncogene 2002, 21, 4921–4931. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhou, J.; Xiao, M.; Zhao, L.; Zhao, Y.; Wang, S.; Gao, S.; Zhuang, Y.; Niu, Y.; Li, S.; et al. Uncovering the Subtype-Specific Molecular Characteristics of Breast Cancer by Multiomics Analysis of Prognosis-Associated Genes, Driver Genes, Signaling Pathways, and Immune Activity. Front. Cell Dev. Biol. 2021, 9, 689028. [Google Scholar] [CrossRef]
- Zhang, X. Estrogen Receptor and Breast Cancer: Celebrating the 60th Anniversary of the Discovery of ER; Springer Nature Switzerland AG: Berlin, Germany, 2018; ISBN 9783319993508. [Google Scholar]
- Caizzi, L.; Ferrero, G.; Cutrupi, S.; Cordero, F.; Ballaré, C.; Miano, V.; Reineri, S.; Ricci, L.; Friard, O.; Testori, A.; et al. Genome-Wide Activity of Unliganded Estrogen Receptor-α in Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 4892–4897. [Google Scholar] [CrossRef] [Green Version]
- Cicatiello, L.; Mutarelli, M.; Grober, O.M.V.; Paris, O.; Ferraro, L.; Ravo, M.; Tarallo, R.; Luo, S.; Schroth, G.P.; Seifert, M.; et al. Estrogen Receptor Alpha Controls a Gene Network in Luminal-like Breast Cancer Cells Comprising Multiple Transcription Factors and microRNAs. Am. J. Pathol. 2010, 176, 2113–2130. [Google Scholar] [CrossRef]
- Grober, O.M.V.; Mutarelli, M.; Giurato, G.; Ravo, M.; Cicatiello, L.; De Filippo, M.R.; Ferraro, L.; Nassa, G.; Papa, M.F.; Paris, O.; et al. Global Analysis of Estrogen Receptor Beta Binding to Breast Cancer Cell Genome Reveals an Extensive Interplay with Estrogen Receptor Alpha for Target Gene Regulation. BMC Genom. 2011, 12, 36. [Google Scholar] [CrossRef]
- Li, W.; Notani, D.; Ma, Q.; Tanasa, B.; Nunez, E.; Chen, A.Y.; Merkurjev, D.; Zhang, J.; Ohgi, K.; Song, X.; et al. Functional Roles of Enhancer RNAs for Oestrogen-Dependent Transcriptional Activation. Nature 2013, 498, 516–520. [Google Scholar] [CrossRef]
- Yang, M.; Lee, J.-H.; Zhang, Z.; Bi, M.; Tan, Y.; Liao, Y.; Hong, J.; Du, B.; Wu, Y.; De La Rosa, R.; et al. Enhancer RNAs Mediate Estrogen-Induced Decommissioning of Selective Enhancers by Recruiting Erα and Its Cofactor. Cell Rep. 2020, 31, 107803. [Google Scholar] [CrossRef]
- Lorent, J.; Kusnadi, E.P.; van Hoef, V.; Rebello, R.J.; Leibovitch, M.; Ristau, J.; Chen, S.; Lawrence, M.G.; Szkop, K.J.; Samreen, B.; et al. Translational Offsetting as a Mode of Estrogen Receptor α-Dependent Regulation of Gene Expression. EMBO J. 2019, 38, e101323. [Google Scholar] [CrossRef]
- Xu, Y.; Huangyang, P.; Wang, Y.; Xue, L.; Devericks, E.; Nguyen, H.G.; Yu, X.; Oses-Prieto, J.A.; Burlingame, A.L.; Miglani, S.; et al. ERα Is an RNA-Binding Protein Sustaining Tumor Cell Survival and Drug Resistance. Cell 2021, 184, 5215–5229. [Google Scholar] [CrossRef]
- Maggi, A. Liganded and Unliganded Activation of Estrogen Receptor and Hormone Replacement Therapies. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 1054–1060. [Google Scholar] [CrossRef]
- Cardamone, M.D.; Bardella, C.; Gutierrez, A.; Di Croce, L.; Rosenfeld, M.G.; Di Renzo, M.F.; De Bortoli, M. ER as Ligand-Independent Activator of CDH-1 Regulates Determination and Maintenance of Epithelial Morphology in Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7420–7425. [Google Scholar] [CrossRef] [Green Version]
- Miano, V.; Ferrero, G.; Rosti, V.; Manitta, E.; Elhasnaoui, J.; Basile, G.; De Bortoli, M. Luminal lncRNAs Regulation by ERα-Controlled Enhancers in a Ligand-Independent Manner in Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 593. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT–Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype. PLoS Genet. 2011, 7, e1002218. [Google Scholar] [CrossRef] [Green Version]
- Warzecha, C.C.; Jiang, P.; Amirikian, K.; Dittmar, K.A.; Lu, H.; Shen, S.; Guo, W.; Xing, Y.; Carstens, R.P. An ESRP-Regulated Splicing Programme Is Abrogated during the Epithelial-Mesenchymal Transition. EMBO J. 2010, 29, 3286–3300. [Google Scholar] [CrossRef]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-Beta-Induced Epithelial to Mesenchymal Transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Yang, Y.; Park, J.W.; Bebee, T.W.; Warzecha, C.C.; Guo, Y.; Shang, X.; Xing, Y.; Carstens, R.P. Determination of a Comprehensive Alternative Splicing Regulatory Network and Combinatorial Regulation by Key Factors during the Epithelial-to-Mesenchymal Transition. Mol. Cell. Biol. 2016, 36, 1704–1719. [Google Scholar] [CrossRef] [Green Version]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Sebestyén, E.; Singh, B.; Miñana, B.; Pagès, A.; Mateo, F.; Pujana, M.A.; Valcárcel, J.; Eyras, E. Large-Scale Analysis of Genome and Transcriptome Alterations in Multiple Tumors Unveils Novel Cancer-Relevant Splicing Networks. Genome Res. 2016, 26, 732–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitting-Seerup, K.; Sandelin, A. The Landscape of Isoform Switches in Human Cancers. Mol. Cancer Res. 2017, 15, 1206–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niknafs, Y.S.; Han, S.; Ma, T.; Speers, C.; Zhang, C.; Wilder-Romans, K.; Iyer, M.K.; Pitchiaya, S.; Malik, R.; Hosono, Y.; et al. The lncRNA Landscape of Breast Cancer Reveals a Role for DSCAM-AS1 in Breast Cancer Progression. Nat. Commun. 2016, 7, 12791. [Google Scholar] [CrossRef] [PubMed]
- Elhasnaoui, J.; Miano, V.; Ferrero, G.; Doria, E.; Leon, A.E.; Fabricio, A.S.C.; Annaratone, L.; Castellano, I.; Sapino, A.; De Bortoli, M. DSCAM-AS1-Driven Proliferation of Breast Cancer Cells Involves Regulation of Alternative Exon Splicing and 3’-End Usage. Cancers 2020, 12, 1453. [Google Scholar] [CrossRef]
- Miano, V.; Ferrero, G.; Reineri, S.; Caizzi, L.; Annaratone, L.; Ricci, L.; Cutrupi, S.; Castellano, I.; Cordero, F.; De Bortoli, M. Luminal Long Non-Coding RNAs Regulated by Estrogen Receptor Alpha in a Ligand-Independent Manner Show Functional Roles in Breast Cancer. Oncotarget 2016, 7, 3201–3216. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.; Wong, W.C.; Brown, R.; Akbani, R.; Su, X.; Broom, B.; Melott, J.; Weinstein, J. TCGASpliceSeq a Compendium of Alternative mRNA Splicing in Cancer. Nucleic Acids Res. 2016, 44, D1018–D1022. [Google Scholar] [CrossRef]
- Phillips, J.W.; Pan, Y.; Tsai, B.L.; Xie, Z.; Demirdjian, L.; Xiao, W.; Yang, H.T.; Zhang, Y.; Lin, C.H.; Cheng, D.; et al. Pathway-Guided Analysis Identifies Myc-Dependent Alternative Pre-mRNA Splicing in Aggressive Prostate Cancers. Proc. Natl. Acad. Sci. USA 2020, 117, 5269–5279. [Google Scholar] [CrossRef] [Green Version]
- Bouris, P.; Skandalis, S.S.; Piperigkou, Z.; Afratis, N.; Karamanou, K.; Aletras, A.J.; Moustakas, A.; Theocharis, A.D.; Karamanos, N.K. Estrogen Receptor Alpha Mediates Epithelial to Mesenchymal Transition, Expression of Specific Matrix Effectors and Functional Properties of Breast Cancer Cells. Matrix Biol. 2015, 43, 42–60. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Chen, D.; Parker, T.M.; Bhat-Nakshatri, P.; Chu, X.; Liu, Y.; Wang, Y.; Nakshatri, H. Nonlinear Relationship between Chromatin Accessibility and Estradiol-Regulated Gene Expression. Oncogene 2021, 40, 1332–1346. [Google Scholar] [CrossRef]
- Dago, D.N.; Scafoglio, C.; Rinaldi, A.; Memoli, D.; Giurato, G.; Nassa, G.; Ravo, M.; Rizzo, F.; Tarallo, R.; Weisz, A. Estrogen Receptor Beta Impacts Hormone-Induced Alternative mRNA Splicing in Breast Cancer Cells. BMC Genom. 2015, 16, 367. [Google Scholar] [CrossRef] [Green Version]
- Vasaikar, S.V.; Deshmukh, A.P.; den Hollander, P.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A Resource for Pan-Cancer Analysis of Epithelial-Mesenchymal Transition Genes and Signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef]
- Wen, J.; Toomer, K.H.; Chen, Z.; Cai, X. Genome-Wide Analysis of Alternative Transcripts in Human Breast Cancer. Breast Cancer Res. Treat. 2015, 151, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Stricker, T.P.; Brown, C.D.; Bandlamudi, C.; McNerney, M.; Kittler, R.; Montoya, V.; Peterson, A.; Grossman, R.; White, K.P. Robust Stratification of Breast Cancer Subtypes Using Differential Patterns of Transcript Isoform Expression. PLoS Genet. 2017, 13, e1006589. [Google Scholar] [CrossRef]
- Koedoot, E.; Smid, M.; Foekens, J.A.; Martens, J.W.M.; Le Dévédec, S.E.; van de Water, B. Co-Regulated Gene Expression of Splicing Factors as Drivers of Cancer Progression. Sci. Rep. 2019, 9, 5484. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.-Z.; Xue, M.-Z.; Shen, H.-J.; Li, X.-G.; Ma, D.; Gong, Y.; Liu, Y.-R.; Qiao, F.; Xie, H.-Y.; Lian, B.; et al. PHF5A Epigenetically Inhibits Apoptosis to Promote Breast Cancer Progression. Cancer Res. 2018, 78, 3190–3206. [Google Scholar] [CrossRef] [Green Version]
- Elsharawy, K.A.; Mohammed, O.J.; Aleskandarany, M.A.; Hyder, A.; El-Gammal, H.L.; Abou-Dobara, M.I.; Green, A.R.; Dalton, L.W.; Rakha, E.A. The Nucleolar-Related Protein Dyskerin Pseudouridine Synthase 1 (DKC1) Predicts Poor Prognosis in Breast Cancer. Br. J. Cancer 2020, 123, 1543–1552. [Google Scholar] [CrossRef]
- Guerrieri, A.N.; Zacchini, F.; Onofrillo, C.; Di Viggiano, S.; Penzo, M.; Ansuini, A.; Gandin, I.; Nobe, Y.; Taoka, M.; Isobe, T.; et al. DKC1 Overexpression Induces a More Aggressive Cellular Behavior and Increases Intrinsic Ribosomal Activity in Immortalized Mammary Gland Cells. Cancers 2020, 12, 3512. [Google Scholar] [CrossRef]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.J.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H.; et al. Alternative Splicing of CD44 mRNA by ESRP1 Enhances Lung Colonization of Metastatic Cancer Cell. Nat. Commun. 2012, 3, 883. [Google Scholar] [CrossRef] [Green Version]
- Fish, L.; Pencheva, N.; Goodarzi, H.; Tran, H.; Yoshida, M.; Tavazoie, S.F. Muscleblind-like 1 Suppresses Breast Cancer Metastatic Colonization and Stabilizes Metastasis Suppressor Transcripts. Genes Dev. 2016, 30, 386–398. [Google Scholar] [CrossRef] [Green Version]
- Masuda, A.; Andersen, H.S.; Doktor, T.K.; Okamoto, T.; Ito, M.; Andresen, B.S.; Ohno, K. CUGBP1 and MBNL1 Preferentially Bind to 3′ UTRs and Facilitate mRNA Decay. Sci. Rep. 2012, 2, 209. [Google Scholar] [CrossRef] [Green Version]
- Batra, R.; Charizanis, K.; Manchanda, M.; Mohan, A.; Li, M.; Finn, D.J.; Goodwin, M.; Zhang, C.; Sobczak, K.; Thornton, C.A.; et al. Loss of MBNL Leads to Disruption of Developmentally Regulated Alternative Polyadenylation in RNA-Mediated Disease. Mol. Cell 2014, 56, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.-X.; Huang, Q.; Park, J.W.; Shen, S.; Lin, L.; Tokheim, C.J.; Henry, M.D.; Xing, Y. Transcriptome-Wide Landscape of Pre-mRNA Alternative Splicing Associated with Metastatic Colonization. Mol. Cancer Res. 2015, 13, 305–318. [Google Scholar] [CrossRef] [Green Version]
- Watermann, D.O.; Tang, Y.; zur Hausen, A.; Jäger, M.; Stamm, S.; Stickeler, E. Splicing Factor Tra2-β1 Is Specifically Induced in Breast Cancer and Regulates Alternative Splicing of the CD44 Gene. Cancer Res. 2006, 66, 4774–4780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaoka, K.; Fujii, K.; Zhang, H.; Usuda, K.; Watanabe, G.; Ivshina, M.; Richter, J.D. CPEB1 Mediates Epithelial-to-Mesenchyme Transition and Breast Cancer Metastasis. Oncogene 2016, 35, 2893–2901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, S.L.; Leonidou, A.; Wai, P.; Marchiò, C.; Ng, C.K.; Sapino, A.; Salomon, A.-V.; Reis-Filho, J.S.; Weigelt, B.; Natrajan, R.C. SF3B1 Mutations Constitute a Novel Therapeutic Target in Breast Cancer. J. Pathol. 2015, 235, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Tian, M.; Gu, J.; Cheng, T.; Ma, D.; Feng, L.; Xin, X. SF3B1 Mutation Is a Poor Prognostic Indicator in Luminal B and Progesterone Receptor-Negative Breast Cancer Patients. Oncotarget 2017, 8, 115018–115027. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.; Chu, C.; Chen, W.; Ren, H.; Cao, Y.; Li, X.; He, J.; Wang, Y.; Jin, Y.; Liu, X.; et al. Prognostic Value of Epithelial-Mesenchymal Transition Related Genes: SLUG and QKI in Breast Cancer Patients. Int. J. Clin. Exp. Pathol. 2019, 12, 2009–2021. [Google Scholar]
- Zhou, M.; Wang, B.; Li, H.; Han, J.; Li, A.; Lu, W. RNA-Binding Protein SAMD4A Inhibits Breast Tumor Angiogenesis by Modulating the Balance of Angiogenesis Program. Cancer Sci. 2021, 112, 3835–3845. [Google Scholar] [CrossRef]
- Cao, Y.; Chu, C.; Li, X.; Gu, S.; Zou, Q.; Jin, Y. RNA-Binding Protein QKI Suppresses Breast Cancer via RASA1/MAPK Signaling Pathway. Ann. Transl Med. 2021, 9, 104. [Google Scholar] [CrossRef]
- Ferrero, G.; Miano, V.; Beccuti, M.; Balbo, G.; De Bortoli, M.; Cordero, F. Dissecting the Genomic Activity of a Transcriptional Regulator by the Integrative Analysis of Omics Data. Sci. Rep. 2017, 7, 8564. [Google Scholar] [CrossRef] [Green Version]
- Stumpf, C.R.; Moreno, M.V.; Olshen, A.B.; Taylor, B.S.; Ruggero, D. The Translational Landscape of the Mammalian Cell Cycle. Mol. Cell 2013, 52, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Chaudhury, A.; Cheema, S.; Fachini, J.M.; Kongchan, N.; Lu, G.; Simon, L.M.; Wang, T.; Mao, S.; Rosen, D.G.; Ittmann, M.M.; et al. CELF1 Is a Central Node in Post-Transcriptional Regulatory Programmes Underlying EMT. Nat. Commun. 2016, 7, 13362. [Google Scholar] [CrossRef]
- Xue, Z.; Warren, R.L.; Gibb, E.A.; MacMillan, D.; Wong, J.; Chiu, R.; Hammond, S.A.; Yang, C.; Nip, K.M.; Ennis, C.A.; et al. Recurrent Tumor-Specific Regulation of Alternative Polyadenylation of Cancer-Related Genes. BMC Genom. 2018, 19, 536. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Kim, S.; Bai, Y.; Diergaarde, B.; Park, H.J. 3’-UTR Shortening Contributes to Subtype-Specific Cancer Growth by Breaking Stable ceRNA Crosstalk of Housekeeping Genes. Front. Bioeng. Biotechnol. 2020, 8, 334. [Google Scholar] [CrossRef]
- Ota, T.; Suzuki, Y.; Nishikawa, T.; Otsuki, T.; Sugiyama, T.; Irie, R.; Wakamatsu, A.; Hayashi, K.; Sato, H.; Nagai, K.; et al. Complete Sequencing and Characterization of 21,243 Full-Length Human cDNAs. Nat. Genet. 2004, 36, 40–45. [Google Scholar] [CrossRef]
- Le Tonquèze, O.; Gschloessl, B.; Legagneux, V.; Paillard, L.; Audic, Y. Identification of CELF1 RNA Targets by CLIP-Seq in Human HeLa Cells. Genom. Data 2016, 8, 97–103. [Google Scholar] [CrossRef]
- Li, C.; Kato, M.; Shiue, L.; Shively, J.E.; Ares, M.; Lin, R.-J. Cell Type and Culture Condition–Dependent Alternative Splicing in Human Breast Cancer Cells Revealed by Splicing-Sensitive Microarrays. Cancer Res. 2006, 66, 1990–1999. [Google Scholar] [CrossRef] [Green Version]
- Lian, H.; Wang, A.; Shen, Y.; Wang, Q.; Zhou, Z.; Zhang, R.; Li, K.; Liu, C.; Jia, H. Identification of Novel Alternative Splicing Isoform Biomarkers and Their Association with Overall Survival in Colorectal Cancer. BMC Gastroenterol. 2020, 20, 171. [Google Scholar] [CrossRef]
- Liu, W.; Li, L.; Ye, H.; Tao, H.; He, H. Role of COL6A3 in Colorectal Cancer. Oncol. Rep. 2018, 39, 2527–2536. [Google Scholar] [CrossRef]
- Arafat, H.; Lazar, M.; Salem, K.; Chipitsyna, G.; Gong, Q.; Pan, T.-C.; Zhang, R.-Z.; Yeo, C.J.; Chu, M.-L. Tumor-Specific Expression and Alternative Splicing of the COL6A3 Gene in Pancreatic Cancer. Surgery 2011, 150, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, L. ggplot2: Elegant Graphics for Data Analysis by WICKHAM, H. Biometrics 2011, 67, 678–679. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Vitting-Seerup, K.; Sandelin, A. IsoformSwitchAnalyzeR: Analysis of Changes in Genome-Wide Patterns of Alternative Splicing and Its Functional Consequences. Bioinformatics 2019, 35, 4469–4471. [Google Scholar] [CrossRef]
- Shen, S.; Park, J.W.; Lu, Z.-X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and Flexible Detection of Differential Alternative Splicing from Replicate RNA-Seq Data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593-601. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Tokheim, C.; Shen, S.; Xing, Y. Identifying Differential Alternative Splicing Events from RNA Sequencing Data Using RNASeq-MATS. In Methods in Molecular Biology; Springer: Berlin, Germany, 2013; pp. 171–179. [Google Scholar]
- Ray, D.; Ha, K.C.H.; Nie, K.; Zheng, H.; Hughes, T.R.; Morris, Q.D. RNAcompete Methodology and Application to Determine Sequence Preferences of Unconventional RNA-Binding Proteins. Methods 2017, 118, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for Occurrences of a given Motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.C.; Cleland, J.; Kim, R.; Wong, W.C.; Weinstein, J.N. SpliceSeq: A Resource for Analysis and Visualization of RNA-Seq Data on Alternative Splicing and Its Functional Impacts. Bioinformatics 2012, 28, 2385–2387. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elhasnaoui, J.; Ferrero, G.; Miano, V.; Cutrupi, S.; De Bortoli, M. The Estrogen Receptor α Signaling Pathway Controls Alternative Splicing in the Absence of Ligands in Breast Cancer Cells. Cancers 2021, 13, 6261. https://doi.org/10.3390/cancers13246261
Elhasnaoui J, Ferrero G, Miano V, Cutrupi S, De Bortoli M. The Estrogen Receptor α Signaling Pathway Controls Alternative Splicing in the Absence of Ligands in Breast Cancer Cells. Cancers. 2021; 13(24):6261. https://doi.org/10.3390/cancers13246261
Chicago/Turabian StyleElhasnaoui, Jamal, Giulio Ferrero, Valentina Miano, Santina Cutrupi, and Michele De Bortoli. 2021. "The Estrogen Receptor α Signaling Pathway Controls Alternative Splicing in the Absence of Ligands in Breast Cancer Cells" Cancers 13, no. 24: 6261. https://doi.org/10.3390/cancers13246261
APA StyleElhasnaoui, J., Ferrero, G., Miano, V., Cutrupi, S., & De Bortoli, M. (2021). The Estrogen Receptor α Signaling Pathway Controls Alternative Splicing in the Absence of Ligands in Breast Cancer Cells. Cancers, 13(24), 6261. https://doi.org/10.3390/cancers13246261