
Integrated Clinical Genotype-Phenotype Characteristics of Blastic Plasmacytoid Dendritic Cell Neoplasm


 ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Group
2.2. Immunophenotypic Analysis
2.3. Conventional Cytogenetic and FISH Analysis
2.4. Next-Generation Sequencing
2.5. Statistical Analysis
3. Results
3.1. Clinical Findings
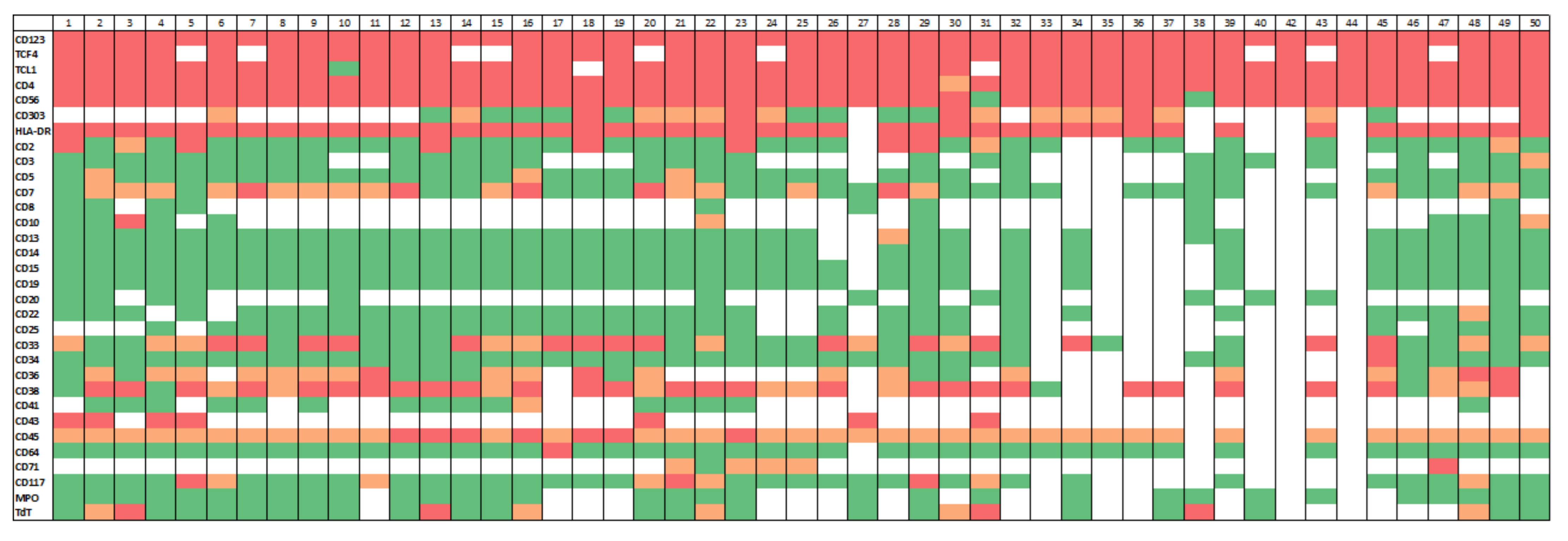
3.2. Immunophenotypic Findings
3.3. Cytogenetic Findings
3.4. Gene Mutation Results
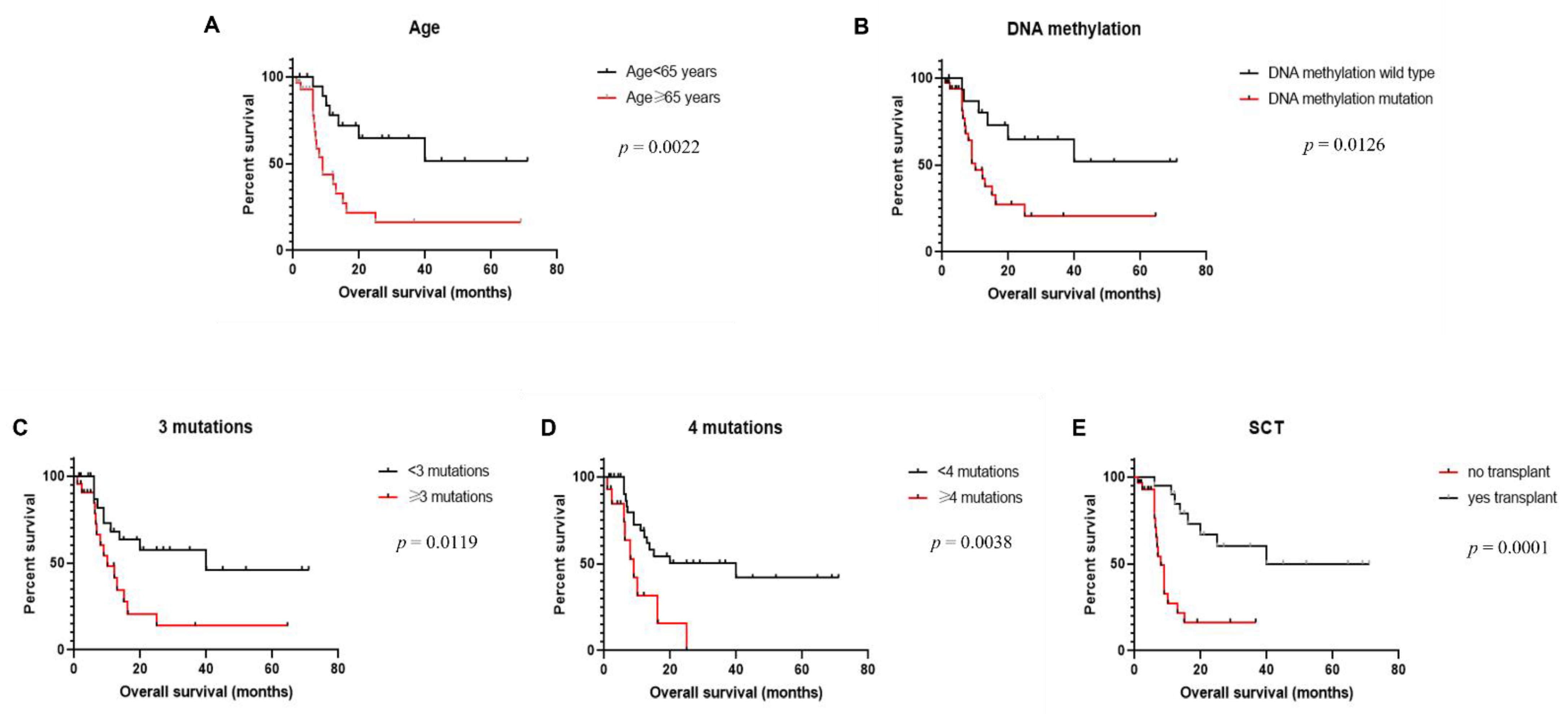
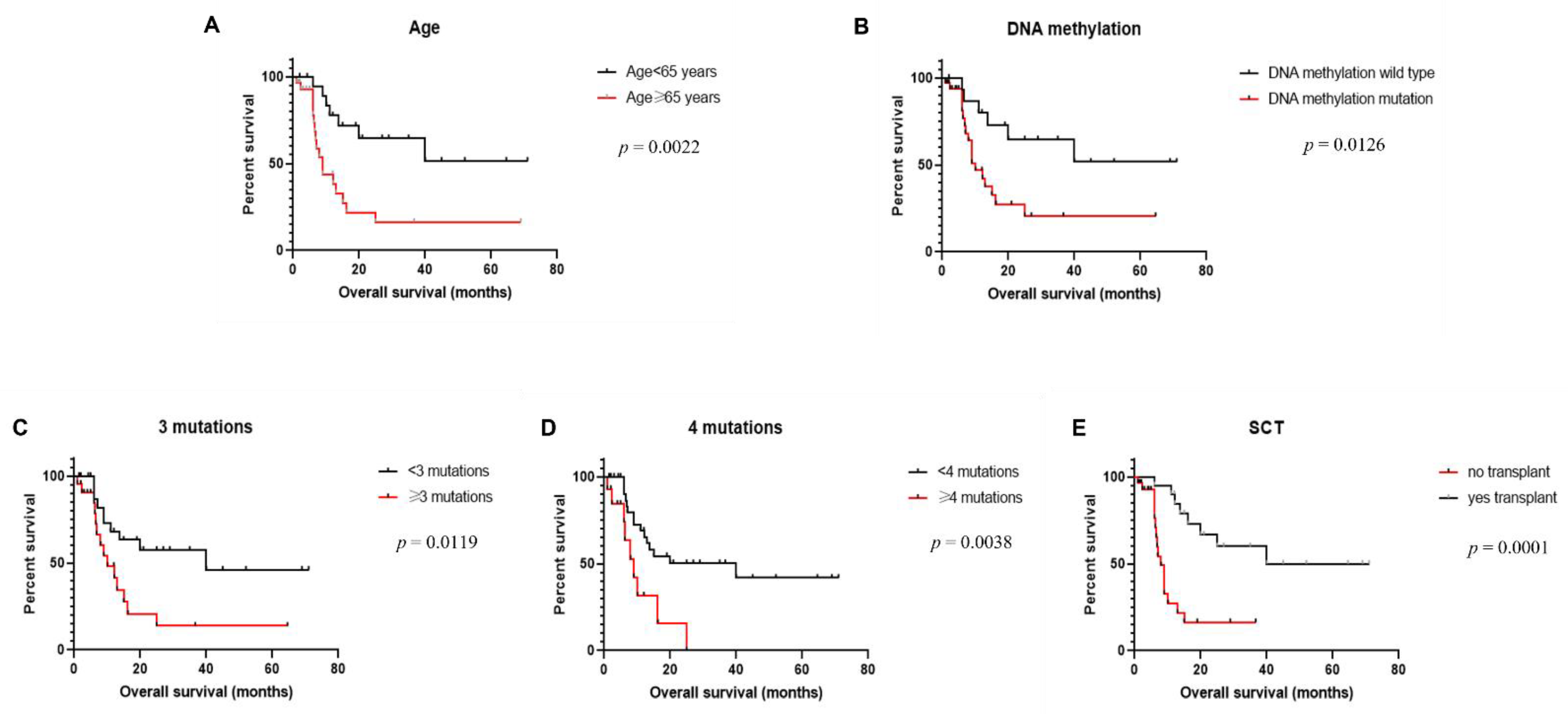
3.5. Clinical Outcome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khoury, J.D. Blastic Plasmacytoid Dendritic Cell Neoplasm. Curr. Hematol. Malig. Rep. 2018, 13, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.R.; Konopleva, M.; Khoury, J.D.; Pemmaraju, N. Novel Therapeutic Approaches in Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN): Era of Targeted Therapy. Clin. Lymphoma Myeloma Leuk. 2021. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Kantarjian, H.M.; Cortes, J.E.; Duvic, M.; Khoury, J.D.; Patel, K.; Daver, N.; O’Brien, S.; Pierce, S.; Garcia-Manero, G.; et al. Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN): A Large Single-Center Experience: Analysis of Clinical and Molecular Characteristics and Patient Outcomes. Blood 2015, 126, 3746. [Google Scholar] [CrossRef]
- Facchetti, F.; Petrella, T.; Pileri, S.A. Blastic plasmacytoid dendritic cell neoplasm. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 174–177. [Google Scholar]
- Wang, W.; Khoury, J.D.; Miranda, R.N.; Jorgensen, J.L.; Xu, J.; Loghavi, S.; Li, S.; Pemmaraju, N.; Nguyen, T.; Medeiros, L.J.; et al. Immunophenotypic characterization of reactive and neoplastic plasmacytoid dendritic cells permits establishment of a 10-color flow cytometric panel for initial workup and residual disease evaluation of blastic plasmacytoid dendritic cell neoplasm. Haematologica 2021, 106, 1047–1055. [Google Scholar] [CrossRef] [Green Version]
- Sukswai, N.; Aung, P.P.; Yin, C.C.; Li, S.; Wang, W.; Wang, S.A.; Ortega, V.; Lyapichev, K.; Nagarajan, P.; Alfattal, R.; et al. Dual Expression of TCF4 and CD123 Is Highly Sensitive and Specific For Blastic Plasmacytoid Dendritic Cell Neoplasm. Am. J. Surg. Pathol. 2019, 43, 1429–1437. [Google Scholar] [CrossRef]
- Wang, X.J.; Medeiros, L.J.; Bueso-Ramos, C.E.; Tang, G.; Wang, S.; Oki, Y.; Desai, P.; Khoury, J.D.; Miranda, R.N.; Tang, Z.; et al. P53 expression correlates with poorer survival and augments the negative prognostic effect of MYC rearrangement, expression or concurrent MYC/BCL2 expression in diffuse large B-cell lymphoma. Mod. Pathol. 2017, 30, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Loghavi, S.; Al-Ibraheemi, A.; Zuo, Z.; Garcia-Manero, G.; Yabe, M.; Wang, S.A.; Kantarjian, H.M.; Yin, C.C.; Miranda, R.N.; Luthra, R.; et al. TP53 overexpression is an independent adverse prognostic factor in de novo myelodysplastic syndromes with fibrosis. Br. J. Haematol. 2015, 171, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Xia, M.; Sun, X.; Han, X.; Zu, Y.; Jabbour, E.J.; You, M.J.; Lin, P.; Li, S.; Xu, J.; et al. High levels of immunoglobulin expression predict shorter overall survival in patients with acute myeloid leukemia. Eur. J. Haematol. 2020, 105, 449–459. [Google Scholar] [CrossRef]
- Jardin, F.; Ruminy, P.; Parmentier, F.; Troussard, X.; Vaida, I.; Stamatoullas, A.; Lepretre, S.; Penther, D.; Duval, A.B.; Picquenot, J.M.; et al. TET2 and TP53 mutations are frequently observed in blastic plasmacytoid dendritic cell neoplasm. Br. J. Haematol. 2011, 153, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Menezes, J.; Acquadro, F.; Wiseman, M.; Gomez-Lopez, G.; Salgado, R.N.; Talavera-Casanas, J.G.; Buno, I.; Cervera, J.V.; Montes-Moreno, S.; Hernandez-Rivas, J.M.; et al. Exome sequencing reveals novel and recurrent mutations with clinical impact in blastic plasmacytoid dendritic cell neoplasm. Leukemia 2014, 28, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Sapienza, M.R.; Fuligni, F.; Agostinelli, C.; Tripodo, C.; Righi, S.; Laginestra, M.A.; Pileri, A., Jr.; Mancini, M.; Rossi, M.; Ricci, F.; et al. Molecular profiling of blastic plasmacytoid dendritic cell neoplasm reveals a unique pattern and suggests selective sensitivity to NF-kB pathway inhibition. Leukemia 2014, 28, 1606–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenzinger, A.; Endris, V.; Pfarr, N.; Andrulis, M.; Johrens, K.; Klauschen, F.; Siebolts, U.; Wolf, T.; Koch, P.S.; Schulz, M.; et al. Targeted ultra-deep sequencing reveals recurrent and mutually exclusive mutations of cancer genes in blastic plasmacytoid dendritic cell neoplasm. Oncotarget 2014, 5, 6404–6413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapienza, M.R.; Abate, F.; Melle, F.; Orecchioni, S.; Fuligni, F.; Etebari, M.; Tabanelli, V.; Laginestra, M.A.; Pileri, A.; Motta, G.; et al. Blastic plasmacytoid dendritic cell neoplasm: Genomics mark epigenetic dysregulation as a primary therapeutic target. Haematologica 2019, 104, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Yannakou, C.K.; Jones, K.; McBean, M.; Thompson, E.R.; Ryland, G.L.; Doig, K.; Markham, J.; Westerman, D.; Blombery, P. ASXL1 c.1934dup;p.Gly646Trpfs*12-a true somatic alteration requiring a new approach. Blood Cancer J. 2017, 7, 656. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Chamoun, K.; Loghavi, S.; Pemmaraju, N.; Konopleva, M.; Kroll, M.; Nguyen-Cao, M.; Hornbaker, M.; DiNardo, C.D.; Kadia, T.; Jorgensen, J.; et al. Early detection of transformation to BPDCN in a patient with MDS. Exp. Hematol. Oncol. 2018, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, A.; Machnicki, M.; Gniot, M.; Pepek, M.; Rydzanicz, M.; Ploski, R.; Kazmierczak, M.; Stoklosa, T.; Lewandowski, K. Germline missense NF1 mutation in an elderly patient with a blastic plasmacytoid dendritic cell neoplasm. Int. J. Hematol. 2019, 110, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzykson, R.; Kosmider, O.; Renneville, A.; Gelsi-Boyer, V.; Meggendorfer, M.; Morabito, M.; Berthon, C.; Ades, L.; Fenaux, P.; Beyne-Rauzy, O.; et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J. Clin. Oncol. 2013, 31, 2428–2436. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015, 29, 910–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, P.A.; Delhommeau, F.; LeCouedic, J.P.; Dawson, M.A.; Chen, E.; Bareford, D.; Kusec, R.; McMullin, M.F.; Harrison, C.N.; Vannucchi, A.M.; et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 2010, 115, 2891–2900. [Google Scholar] [CrossRef] [Green Version]
- Chou, W.C.; Chou, S.C.; Liu, C.Y.; Chen, C.Y.; Hou, H.A.; Kuo, Y.Y.; Lee, M.C.; Ko, B.S.; Tang, J.L.; Yao, M.; et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 2011, 118, 3803–3810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaub, F.X.; Looser, R.; Li, S.; Hao-Shen, H.; Lehmann, T.; Tichelli, A.; Skoda, R.C. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood 2010, 115, 2003–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nibourel, O.; Kosmider, O.; Cheok, M.; Boissel, N.; Renneville, A.; Philippe, N.; Dombret, H.; Dreyfus, F.; Quesnel, B.; Geffroy, S.; et al. Incidence and prognostic value of TET2 alterations in de novo acute myeloid leukemia achieving complete remission. Blood 2010, 116, 1132–1135. [Google Scholar] [CrossRef]
- Renosi, F.; Roggy, A.; Giguelay, A.; Soret, L.; Viailly, P.J.; Cheok, M.; Biichle, S.; Angelot-Delettre, F.; Asnafi, V.; Macintyre, E.; et al. Transcriptomic and genomic heterogeneity in blastic plasmacytoid dendritic cell neoplasms: From ontogeny to oncogenesis. Blood Adv. 2021, 5, 1540–1551. [Google Scholar] [CrossRef]
- Summerer, I.; Walter, W.; Meggendorfer, M.; Kern, W.; Haferlach, T.; Haferlach, C.; Stengel, A. Comprehensive analysis of the genetic landscape of 21 cases with blastic plasmacytoid dendritic cell neoplasm by whole genome and whole transcriptome sequencing. Leuk. Lymphoma 2021, 62, 2543–2546. [Google Scholar] [CrossRef] [PubMed]
- Togami, K.; Chung, S.S.; Madan, V.; Booth, C.A.G.; Kenyon, C.M.; Cabal-Hierro, L.; Taylor, J.; Kim, S.S.; Griffin, G.K.; Ghandi, M.; et al. Sex-biased ZRSR2 mutations in myeloid malignancies impair plasmacytoid dendritic cell activation and apoptosis. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Cytlak, U.; Resteu, A.; Bogaert, D.; Kuehn, H.S.; Altmann, T.; Gennery, A.; Jackson, G.; Kumanovics, A.; Voelkerding, K.V.; Prader, S.; et al. Ikaros family zinc finger 1 regulates dendritic cell development and function in humans. Nat. Commun. 2018, 9, 1239. [Google Scholar] [CrossRef] [PubMed]
- Bastidas Torres, A.N.; Cats, D.; Mei, H.; Fanoni, D.; Gliozzo, J.; Corti, L.; Paulli, M.; Vermeer, M.H.; Willemze, R.; Berti, E.; et al. Whole-genome analysis uncovers recurrent IKZF1 inactivation and aberrant cell adhesion in blastic plasmacytoid dendritic cell neoplasm. Genes Chromosomes Cancer 2020, 59, 295–308. [Google Scholar] [CrossRef] [Green Version]
- Leroux, D.; Mugneret, F.; Callanan, M.; Radford-Weiss, I.; Dastugue, N.; Feuillard, J.; Le Mee, F.; Plessis, G.; Talmant, P.; Gachard, N.; et al. CD4(+), CD56(+) DC2 acute leukemia is characterized by recurrent clonal chromosomal changes affecting 6 major targets: A study of 21 cases by the Groupe Francais de Cytogenetique Hematologique. Blood 2002, 99, 4154–4159. [Google Scholar] [CrossRef]
- Emadali, A.; Hoghoughi, N.; Duley, S.; Hajmirza, A.; Verhoeyen, E.; Cosset, F.L.; Bertrand, P.; Roumier, C.; Roggy, A.; Suchaud-Martin, C.; et al. Haploinsufficiency for NR3C1, the gene encoding the glucocorticoid receptor, in blastic plasmacytoid dendritic cell neoplasms. Blood 2016, 127, 3040–3053. [Google Scholar] [CrossRef] [Green Version]
- Lucioni, M.; Novara, F.; Fiandrino, G.; Riboni, R.; Fanoni, D.; Arra, M.; Venegoni, L.; Nicola, M.; Dallera, E.; Arcaini, L.; et al. Twenty-one cases of blastic plasmacytoid dendritic cell neoplasm: Focus on biallelic locus 9p21.3 deletion. Blood 2011, 118, 4591–4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.Y.; Li, Y.; Wang, W.; Yin, C.C.; Tang, G.L.; Aung, P.P.; Hu, S.M.; Lu, X.Y.; Toruner, G.A.; Medeiros, L.J.; et al. Genomic aberrations involving 12p/ETV6 are highly prevalent in blastic plasmacytoid dendritic cell neoplasms and might represent early clonal events. Leuk. Res. 2018, 73, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Katayama, R.; Asaka, R.; Sakata, S.; Baba, S.; Nakasone, H.; Koike, S.; Tsuyama, N.; Dobashi, A.; Sasaki, M.; et al. Recurrent 8q24 rearrangement in blastic plasmacytoid dendritic cell neoplasm: Association with immunoblastoid cytomorphology, MYC expression, and drug response. Leukemia 2018, 32, 2590–2603. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Suzuki, Y.; Hama, A.; Muramatsu, H.; Nakatochi, M.; Gunji, M.; Ichikawa, D.; Hamada, M.; Taniguchi, R.; Kataoka, S.; et al. Recurrent MYB rearrangement in blastic plasmacytoid dendritic cell neoplasm. Leukemia 2017, 31, 1629–1633. [Google Scholar] [CrossRef] [PubMed]
- Kharfan-Dabaja, M.A.; Al Malki, M.M.; Deotare, U.; Raj, R.V.; El-Jurdi, N.; Majhail, N.; Cherry, M.A.; Bashir, Q.; Darrah, J.; Nishihori, T.; et al. Haematopoietic cell transplantation for blastic plasmacytoid dendritic cell neoplasm: A North American multicentre collaborative study. Br. J. Haematol. 2017, 179, 781–789. [Google Scholar] [CrossRef]
- Bashir, Q.; Milton, D.R.; Popat, U.R.; Kebriaei, P.; Hosing, C.; Khouri, I.F.; Rezvani, K.; Nieto, Y.; Oran, B.; Srour, S.A.; et al. Allogeneic hematopoietic cell transplantation for patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN). Bone Marrow Transplant. 2021, 1–6. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Case | Karyotype |
|---|---|
| 1. | 46,XY[19]/47,XY,+1[1] |
| 2. | 46,XY,t(1;6)(p21;p36.3),del(5)(q13q33),der(7)t(1;7)(q12;p22),del(11)(q13q23),del(12)(p11.2p13),add(15)(q15)[10] |
| 3. | 42,XY,-5,-12,-15,add(17)(p11.2),add(17)(p13),-18,add(20)(q13.3)[3]/43,idem,+mar[8]/42,idem,-13,+mar[2]/46,XY[4] |
| 4. | 46,XY,+1,add(1)(p13),der(1)dup(1)(q21q32)add(1)(q42),-2,-4,+11,add(12)(p11.2), -13,-21,+2mar[8]/48,XY,+1,der(1)dup(1)(q21q32)add(1)(q42),-2,-4,+11,add(12)(p11.2),-13,+3mar[1]/46,XY[11] |
| 5. | 46,XY[20] |
| 6. | 46,XY[20] |
| 7. | 46,XY,t(2;7)(p23;p15),add(9)(p13),del(9)(p21)[20] |
| 8. | 46,XX[20] |
| 9. | 46, XY[20] |
| 10. | 45,XY,der(6)del(6)(q13q22)t(6;9)(q25;q34),-7,der(9)del(9)(p21)t(6;9),der(9;15)(q10;q10),-13,-14,+2~3mar[cp6]/46,XY[14] |
| 11. | 46,XY,t(6;17)(q23;q22),der(7)add(7)(q11.2)t(?;21)(?;q11.2),add(21)(p12)[cp4]/47,XY,+mar[1]/46,XY[15] |
| 12. | 46,X,-Y,+12[3]/46,XY[17] |
| 13. | 44~45,XX,del(5)(q33q35),del(6)(q13q23),der(7;9)(q10;q10),-13[cp12]/46,XX[8] |
| 14. | 44,XX,add(1)(p36.1),del(9)(p21),add(12)(p12),-13,add(17)(p12),-21,-22,add(22)(q12),+mar[5]/46,XX[15] |
| 16. | 48,XY,t(6;8)(p12;q24.2),add(7)(p11.2),del(13)(q12q22),+16,add(20)(q11.2),+21[10]/46,XY[10] |
| 17. | 46, XY[20] |
| 18. | 46,XY[20] |
| 19. | 86~87,XXYY,-5,add(6)(q21)x2,-11,add(11)(q23),-12,i(12)(q10),add(19)(p13.3),+3mar[cp3]/46,XY[17] |
| 20. | 46,XY[20] |
| 21. | 44~47,XY,add(3)(p13),add(5)(p15.1),del(5)(q13q33),-8,add(8)(q13),del(9)(p21),del(9)(q13q22), del(10)(q24),del(11)(q13q23),del(12)(p12),- 13,del(13)(q12q22),+del(17)(p11.2),add(20)(q13.3),add(21)(p13),+mar[cp11]/46,XY[9] |
| 22. | 42,XY,-6,-7,-8,add(9)(q13),der(11)add(11)(p15)add(11)(q24),der(13;15)(q10;q10),add(17)(p11.2),-22,+2mar[4]/46,XY[16] |
| 23. | 46~48,XX,del(6)(q13q23),+der(7)add(7)(p13)del(7)(q11.2q22)add(7)(q32)x2,der(7)add(7)(p13)del(7)(q11.2q22), add(7)(q32)x2,-8,add(9)(p24),add(12)(p13),-14,del(16)(q23),+21,+22,+mar[cp19]/46,XX[1] |
| 24. | 46,XY,del(12)(p11.2p13)[1]/46,XY[19] |
| 25. | 46,XY,del(3)(p11.2)[1]/46,XY[19] |
| 26. | 47,XY,+Y[1]/46,XY[19] |
| 27. | 46,XY,inv(9)(p12q13)[20] |
| 28. | 45,XY,?del(6)(q24),-7,?i(7)(q10),+?12,-14,15,+mar[3]/46,XY[12] |
| 29. | 46,XY,del(12)(q14)[1]/46,XY[19] |
| 30. | 46,XX[20] |
| 31. | 46,XX[20] |
| 32. | 46,XY[20] |
| 33. | 46,XY[19] |
| 46. | 46,XY,t(3;9)(q25;q34)[1]/46,XY[19] |
| 47. | 45,XY,i(7)(q10),i(14)(q10),-21[2]/46,XY[18] |
| 48. | 46,XY[4] |
| 49. | 81~82,XXYY,+1,+1,add(1)(p22),del(1)(p31),del(1)(q21),add(2)(p25),add(2)(q37),-3,-4,-5,-5,-6,-9,-10,-12,-13,-15,+16,+16,-17,-18,-20,-21,+2~4mar[cp2]/46,XY[18] |
| Features | p Value | HR Ratio | 95% CI of HR |
|---|---|---|---|
| Age (<65 years vs. ≥65 years) | 0.0022 * | 0.287 | 0.1273–0.6473 |
| Sex (men vs. women) | 0.8444 | 1.112 | 0.3941–3.136 |
| WBC (low vs. normal vs. high) | 0.0561 | NA | NA |
| Hb (low vs. normal) | 0.6007 | 1.293 | 0.5159–3.243 |
| Platelet (low vs. normal) | 0.4 | 1.403 | 0.6285–3.133 |
| LDH (high vs. normal) | 0.6043 | 1.24 | 0.5522–2.784 |
| β2M (high vs. normal) | 0.7633 | 0.8077 | 0.2143–3.044 |
| Karyotype (abnormal vs. diploid) | 0.0969 | 0.516 | 0.2094–1.272 |
| Mutation (yes vs. no) | 0.2902 | 0.5724 | 0.2296–1.427 |
| ASXL1 (mut vs. wt) | 0.2618 | 0.6382 | 0.2845–1.432 |
| DNMT3A (mut vs. wt) | 0.1098 | 0.4403 | 0.1016–1.909 |
| NRAS (mut vs. wt) | 0.8036 | 1.198 | 0.3122–4.595 |
| TET2 (mut vs. wt) | 0.0743 | 0.4878 | 0.2182–1.090 |
| ZRSR2 (mut vs. wt) | 0.3374 | 0.4977 | 0.0692–3.581 |
| DNA methylation (mut vs. wt) | 0.0126* | 0.3399 | 0.1527–0.7567 |
| Histone modification (mut vs. wt) | 0.2697 | 0.6429 | 0.2867–1.442 |
| Signal transduction (mut vs. wt) | 0.4435 | 1.505 | 0.5825–3.886 |
| Transcription factors (mut vs. wt) | 0.2226 | 0.5627 | 0.1843–1.718 |
| RNA splicing factors (mut vs. wt) | 0.5414 | 0.7159 | 0.2080–2.464 |
| #mutation (<3 vs. ≥3) | 0.0119 * | 0.3766 | 0.1624–0.8737 |
| #mutation (<4 vs. ≥4) | 0.0038 * | 0.3246 | 0.1099–0.9589 |
| Transplantation (yes vs. no) | 0.0001 * | 0.2378 | 0.1010–0.5597 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, C.C.; Pemmaraju, N.; You, M.J.; Li, S.; Xu, J.; Wang, W.; Tang, Z.; Alswailmi, O.; Bhalla, K.N.; Qazilbash, M.H.; et al. Integrated Clinical Genotype-Phenotype Characteristics of Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancers 2021, 13, 5888. https://doi.org/10.3390/cancers13235888
Yin CC, Pemmaraju N, You MJ, Li S, Xu J, Wang W, Tang Z, Alswailmi O, Bhalla KN, Qazilbash MH, et al. Integrated Clinical Genotype-Phenotype Characteristics of Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancers. 2021; 13(23):5888. https://doi.org/10.3390/cancers13235888
Chicago/Turabian StyleYin, C. Cameron, Naveen Pemmaraju, M. James You, Shaoying Li, Jie Xu, Wei Wang, Zhenya Tang, Omar Alswailmi, Kapil N. Bhalla, Muzaffar H. Qazilbash, and et al. 2021. "Integrated Clinical Genotype-Phenotype Characteristics of Blastic Plasmacytoid Dendritic Cell Neoplasm" Cancers 13, no. 23: 5888. https://doi.org/10.3390/cancers13235888
APA StyleYin, C. C., Pemmaraju, N., You, M. J., Li, S., Xu, J., Wang, W., Tang, Z., Alswailmi, O., Bhalla, K. N., Qazilbash, M. H., Konopleva, M., & Khoury, J. D. (2021). Integrated Clinical Genotype-Phenotype Characteristics of Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancers, 13(23), 5888. https://doi.org/10.3390/cancers13235888