
The Endocannabinoid System as a Pharmacological Target for New Cancer Therapies

Simple Summary
Abstract
1. Introduction
2. Regulation of the Endocannabinoid System in Different Tumour Entities in Context with the Clinical Outcome of Cancer Patients
2.1. Regulation of Endocannabinoids in the Tumour Process
2.2. Regulation of Cannabinoid Receptors in the Tumour Process
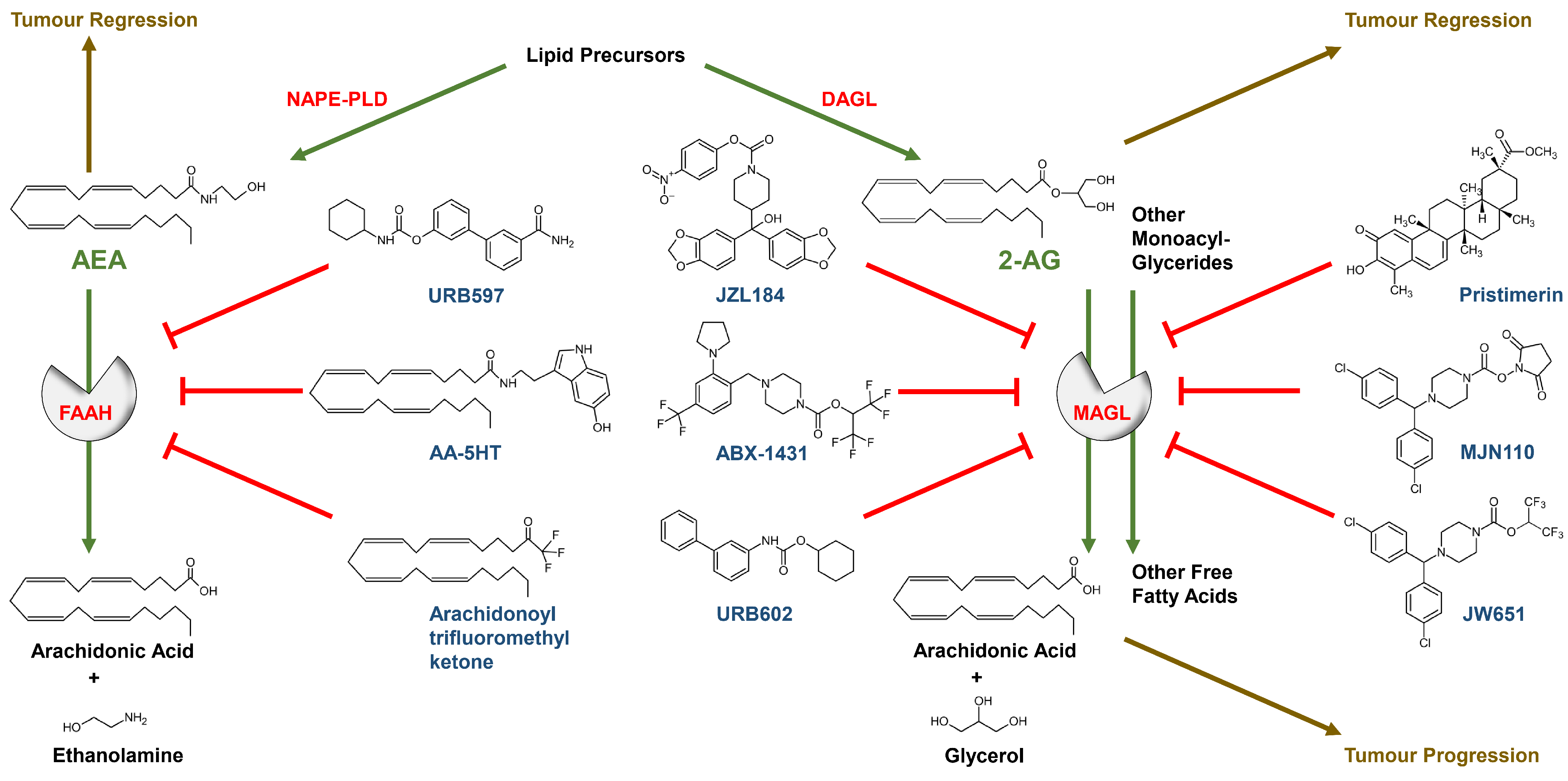
2.3. Regulation of Endocannabinoid-Synthesising and -Degrading Enzymes in the Tumour Process
3. Systemic Effects of Cannabinoid Compounds on Different Levels of Carcinogenesis
3.1. Cancer Cell Proliferation and Viability
3.1.1. Effect of Cannabinoids on Cancer Cell Proliferation and Viability
3.1.2. Effect of FAAH and MAGL Inhibition on Cancer Cell Proliferation and Viability
3.2. Cancer Cell Invasion and Metastasis
3.2.1. Effect of Cannabinoids on Cancer Cell Invasion and Metastasis
3.2.2. Effect of FAAH and MAGL Inhibition on Cancer Cell Invasion and Metastasis
3.3. Tumour Angiogenesis
3.3.1. Effects of Cannabinoid Compounds on Tumour Angiogenesis
3.3.2. Effects of FAAH and MAGL Inhibition on Tumour Angiogenesis
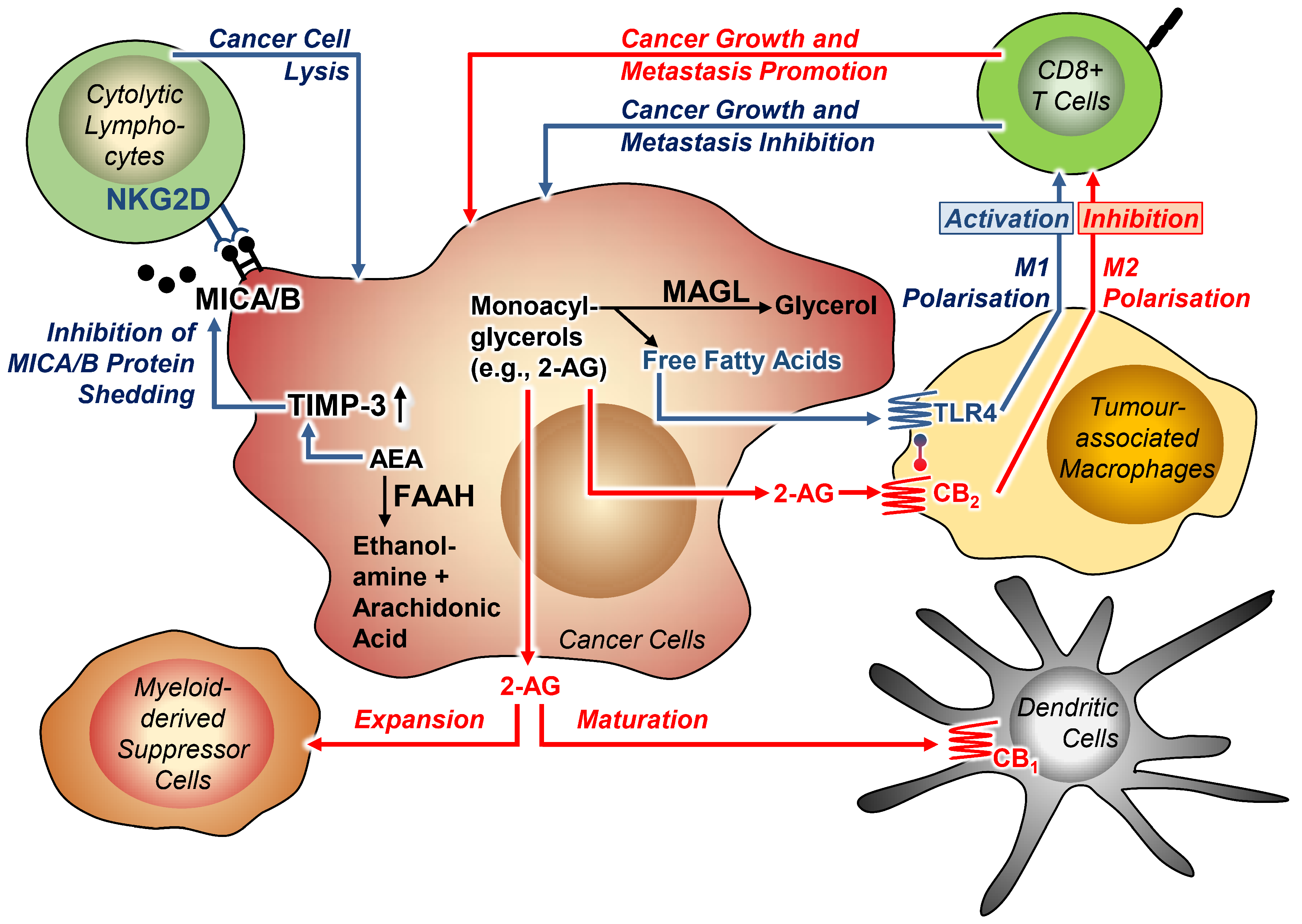
3.4. Tumour-Immune Interactions
3.4.1. Effect of Cannabinoid Compounds on Tumour-Immune Interactions
3.4.2. Effect of FAAH and MAGL Inhibition on Tumour-Immune Interactions
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Devane:, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Hanus, L.; Abu-Lafi, S.; Fride, E.; Breuer, A.; Vogel, Z.; Shalev, D.E.; Kustanovich, I.; Mechoulam, R. 2-arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 3662–3665. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Melck, D.; Bobrov, M.Y.; Gretskaya, N.M.; Bezuglov, V.V.; De Petrocellis, L.; Di Marzo, V. N-acyl-dopamines: Novel synthetic CB1 cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem. J. 2000, 351, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.C.; Sauer, J.M.; Knierman, M.D.; Becker, G.W.; Berna, M.J.; Bao, J.; Nomikos, G.G.; Carter, P.; Bymaster, F.P.; Leese, A.B.; et al. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J. Pharmacol. Exp. Ther. 2002, 301, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.; Sørgård, M.; Di Marzo, V.; Julius, D.; Högestätt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Hanus, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, ΨGPR53 and GPR55: GPR55 is extensively expressed in human brain. Brain Res. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Artmann, A.; Petersen, G.; Hellgren, L.I.; Boberg, J.; Skonberg, C.; Nellemann, C.; Hansen, S.H.; Hansen, H.S. Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim. Biophys. Acta. 2008, 1781, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.; Ramer, R.; Hinz, B. Targeting the endocannabinoid system as a potential anticancer approach. Drug Metab. Rev. 2018, 50, 26–53. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 2009, 60, 77–84. [Google Scholar] [CrossRef]
- Deutsch, D.G.; Chin, S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993, 46, 791–796. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Koutek, B.; Prestwich, G.D.; Howlett, A.C.; Chin, S.A.; Salehani, D.; Akhavan, N.; Deutsch, D.G. Inhibitors of arachidonoyl ethanolamide hydrolysis. J. Biol. Chem. 1994, 269, 22937–22940. [Google Scholar] [CrossRef]
- Street, I.P.; Lin, H.K.; Laliberté, F.; Ghomashchi, F.; Wang, Z.; Perrier, H.; Tremblay, N.M.; Huang, Z.; Weech, P.K.; Gelb, M.H. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry 1993, 32, 5935–5940. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Melck, D.; De Petrocellis, L.; Bobrov, M.Y.; Gretskaya, N.M.; Bezuglov, V.V.; Sitachitta, N.; Gerwick, W.H.; Di Marzo, V. Arachidonoylserotonin and other novel inhibitors of fatty acid amide hydrolase. Biochem. Biophys. Res. Commun. 1998, 248, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, S.; Gaetani, S.; Fegley, D.; Valiño, F.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; La Rana, G.; Calignano, A.; et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003, 9, 76–81. [Google Scholar] [CrossRef]
- Hohmann, A.G.; Suplita, R.L.; Bolton, N.M.; Neely, M.H.; Fegley, D.; Mangieri, R.; Krey, J.F.; Walker, J.M.; Holmes, P.V.; Crystal, J.D.; et al. An endocannabinoid mechanism for stress-induced analgesia. Nature 2005, 435, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Vandevoorde, S.; Jonsson, K.O.; Labar, G.; Persson, E.; Lambert, D.M.; Fowler, C.J. Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br. J. Pharmacol. 2007, 150, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Nomura, D.K.; Cravatt, B.F. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 2009, 16, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Niphakis, M.J.; Cognetta, A.B., 3rd; Chang, J.W.; Buczynski, M.W.; Parsons, L.H.; Byrne, F.; Burston, J.J.; Chapman, V.; Cravatt, B.F. Evaluation of NHS carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2013, 4, 1322–1332. [Google Scholar] [CrossRef]
- Chang, J.W.; Cognetta, A.B., 3rd; Niphakis, M.J.; Cravatt, B.F. Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem. Biol. 2013, 8, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Cisar, J.S.; Weber, O.D.; Clapper, J.R.; Blankman, J.L.; Henry, C.L.; Simon, G.M.; Alexander, J.P.; Jones, T.K.; Ezekowitz, R.A.B.; O’Neill, G.P.; et al. Identification of ABX-1431, a Selective Inhibitor of Monoacylglycerol Lipase and Clinical Candidate for Treatment of Neurological Disorders. J. Med. Chem. 2018, 61, 9062–9084. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/results?recrs=&cond=&term=ABX-1431&cntry=&state=&city=&dist= (accessed on 20 October 2021).
- Chen, A.L.; Lum, K.M.; Lara-Gonzalez, P.; Ogasawara, D.; Cognetta, A.B., 3rd; To, A.; Parsons, W.H.; Simon, G.M.; Desai, A.; Petrascheck, M.; et al. Pharmacological convergence reveals a lipid pathway that regulates C. elegans lifespan. Nat. Chem. Biol. 2019, 15, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Medina-Cleghorn, D.; Bernal-Mizrachi, L.; Bracci, P.M.; Hubbard, A.; Conde, L.; Riby, J.; Nomura, D.K.; Skibola, C.F. The potential relevance of the endocannabinoid, 2-arachidonoylglycerol, in diffuse large B-cell lymphoma. Oncoscience 2016, 3, 31–41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guida, M.; Ligresti, A.; De Filippis, D.; D’Amico, A.; Petrosino, S.; Cipriano, M.; Bifulco, G.; Simonetti, S.; Orlando, P.; Insabato, L.; et al. The levels of the endocannabinoid receptor CB2 and its ligand 2-arachidonoylglycerol are elevated in endometrial carcinoma. Endocrinology 2010, 151, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, H.; Li, Y.; Li, L.; Qiu, Y.; Ren, J. Endocannabinoid and ceramide levels are altered in patients with colorectal cancer. Oncol. Rep. 2015, 34, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Han, L.; Zhang, X.; Li, L.; Jiang, C.; Qiu, Y.; Huang, R.; Xie, B.; Lin, Z.; Ren, J.; et al. Alteration of endocannabinoid system in human gliomas. J. Neurochem. 2012, 120, 842–849. [Google Scholar] [CrossRef]
- Maccarrone, M.; Attinà, M.; Cartoni, A.; Bari, M.; Finazzi-Agrò, A. Gas chromatography-mass spectrometry analysis of endogenous cannabinoids in healthy and tumoral human brain and human cells in culture. J. Neurochem. 2001, 76, 594–601. [Google Scholar] [CrossRef]
- Alberich Jordà, M.; Rayman, N.; Tas, M.; Verbakel, S.E.; Battista, N.; van Lom, K.; Löwenberg, B.; Maccarrone, M.; Delwel, R. The peripheral cannabinoid receptor Cb2, frequently expressed on AML blasts, either induces a neutrophilic differentiation block or confers abnormal migration properties in a ligand-dependent manner. Blood 2004, 104, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, C.; de Ceballos, M.L.; Gomez del Pulgar, T.; Rueda, D.; Corbacho, C.; Velasco, G.; Galve-Roperh, I.; Huffman, J.W.; Ramón y Cajal, S.; Guzmán, M. Inhibition of glioma growth in vivo by selective activation of the CB2 cannabinoid receptor. Cancer Res. 2001, 61, 5784–5789. [Google Scholar]
- Held-Feindt, J.; Dörner, L.; Sahan, G.; Mehdorn, H.M.; Mentlein, R. Cannabinoid receptors in human astroglial tumors. J. Neurochem. 2006, 98, 886–893. [Google Scholar] [CrossRef]
- De Jesús, M.L.; Hostalot, C.; Garibi, J.M.; Sallés, J.; Meana, J.J.; Callado, L.F. Opposite changes in cannabinoid CB1 and CB2 receptor expression in human gliomas. Neurochem. Int. 2010, 56, 829–833. [Google Scholar] [CrossRef]
- Sredni, S.T.; Huang, C.C.; Suzuki, M.; Pundy, T.; Chou, P.; Tomita, T. Spontaneous involution of pediatric low-grade gliomas: High expression of cannabinoid receptor 1 (CNR1) at the time of diagnosis may indicate involvement of the endocannabinoid system. Childs Nerv. Syst. 2016, 32, 2061–2067. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Ciechomska, I.A.; Kaminska, B. Synthetic Cannabinoids Induce Autophagy and Mitochondrial Apoptotic Pathways in Human Glioblastoma Cells Independently of Deficiency in TP53 or PTEN Tumor Suppressors. Cancers 2021, 13, 419. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Sarrió, D.; Palacios, J.; Guzmán, M.; Sánchez, C. Δ9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef] [PubMed]
- Gjerstorff, M.F.; Benoit, V.M.; Laenkholm, A.V.; Nielsen, O.; Johansen, L.E.; Ditzel, H.J. Identification of genes with altered expression in medullary breast cancer vs. ductal breast cancer and normal breast epithelia. Int. J. Oncol. 2006, 28, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.W.; Qamri, Z.; Deol, Y.S.; Smith, D.; Shilo, K.; Zou, X.; Ganju, R.K. Crosstalk between chemokine receptor CXCR4 and cannabinoid receptor CB2 in modulating breast cancer growth and invasion. PLoS ONE 2011, 6, e23901. [Google Scholar] [CrossRef]
- Shubbar, E.; Helou, K.; Kovács, A.; Nemes, S.; Hajizadeh, S.; Enerbäck, C.; Einbeigi, Z. High levels of γ-glutamyl hydrolase (GGH) are associated with poor prognosis and unfavorable clinical outcomes in invasive breast cancer. BMC Cancer 2013, 13, 47. [Google Scholar] [CrossRef]
- Sun, H.; Jiang, L.; Luo, X.; Jin, W.; He, Q.; An, J.; Lui, K.; Shi, J.; Rong, R.; Su, W.; et al. Potential tumor-suppressive role of monoglyceride lipase in human colorectal cancer. Oncogene 2013, 32, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Gómez, E.; Andradas, C.; Blasco-Benito, S.; Caffarel, M.M.; García-Taboada, E.; Villa-Morales, M.; Moreno, E.; Hamann, S.; Martín-Villar, E.; Flores, J.M.; et al. Role of cannabinoid receptor CB2 in HER2 pro-oncogenic signaling in breast cancer. J. Natl. Cancer Inst. 2015, 107, djv077. [Google Scholar] [CrossRef]
- Blasco-Benito, S.; Moreno, E.; Seijo-Vila, M.; Tundidor, I.; Andradas, C.; Caffarel, M.M.; Caro-Villalobos, M.; Urigüen, L.; Diez-Alarcia, R.; Moreno-Bueno, G.; et al. Therapeutic targeting of HER2-CB2R heteromers in HER2-positive breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3863–3872, Erratum in 2019, 116, 6505. [Google Scholar] [CrossRef]
- Yoneten, K.K.; Kasap, M.; Akpinar, G.; Gunes, A.; Gurel, B.; Utkan, N.Z. Comparative Proteome Analysis of Breast Cancer Tissues Highlights the Importance of Glycerol-3-phosphate Dehydrogenase 1 and Monoacylglycerol Lipase in Breast Cancer Metabolism. Cancer Genom. Proteom. 2019, 16, 377–397. [Google Scholar] [CrossRef]
- Ligresti, A.; Bisogno, T.; Matias, I.; De Petrocellis, L.; Cascio, M.G.; Cosenza, V.; D’argenio, G.; Scaglione, G.; Bifulco, M.; Sorrentini, I.; et al. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology 2003, 125, 677–687. [Google Scholar] [CrossRef]
- Cianchi, F.; Papucci, L.; Schiavone, N.; Lulli, M.; Magnelli, L.; Vinci, M.C.; Messerini, L.; Manera, C.; Ronconi, E.; Romagnani, P.; et al. Cannabinoid receptor activation induces apoptosis through tumor necrosis factor α-mediated ceramide de novo synthesis in colon cancer cells. Clin. Cancer Res. 2008, 14, 7691–7700. [Google Scholar] [CrossRef]
- Wang, D.; Wang, H.; Ning, W.; Backlund, M.G.; Dey, S.K.; DuBois, R.N. Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res. 2008, 68, 6468–6476. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, B.; Seviour, E.G.; Tao, K.X.; Liu, X.H.; Ling, Y.; Chen, J.Y.; Wang, G.B. Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett. 2011, 307, 6–17. [Google Scholar] [CrossRef]
- Jung, C.K.; Kang, W.K.; Park, J.M.; Ahn, H.J.; Kim, S.W.; Taek Oh, S.; Choi, K.Y. Expression of the cannabinoid type I receptor and prognosis following surgery in colorectal cancer. Oncol. Lett. 2013, 5, 870–876. [Google Scholar] [CrossRef]
- Martínez-Martínez, E.; Gómez, I.; Martín, P.; Sánchez, A.; Román, L.; Tejerina, E.; Bonilla, F.; Merino, A.G.; de Herreros, A.G.; Provencio, M.; et al. Cannabinoids receptor type 2, CB2, expression correlates with human colon cancer progression and predicts patient survival. Oncoscience 2015, 2, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Hasenoehrl, C.; Feuersinger, D.; Sturm, E.M.; Bärnthaler, T.; Heitzer, E.; Graf, R.; Grill, M.; Pichler, M.; Beck, S.; Butcher, L.; et al. G protein-coupled receptor GPR55 promotes colorectal cancer and has opposing effects to cannabinoid receptor 1. Int. J. Cancer 2018, 142, 121–132. [Google Scholar] [CrossRef]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef] [PubMed]
- Tutino, V.; Caruso, M.G.; De Nunzio, V.; Lorusso, D.; Veronese, N.; Gigante, I.; Notarnicola, M.; Giannelli, G. Down-Regulation of Cannabinoid Type 1 (CB1) Receptor and its Downstream Signaling Pathways in Metastatic Colorectal Cancer. Cancers 2019, 11, 708. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, W.; Shen, K.; Shen, W. ∆9-tetrahydrocannabinol inhibits epithelial-mesenchymal transition and metastasis by targeting matrix metalloproteinase-9 in endometrial cancer. Oncol. Lett. 2018, 15, 8527–8535. [Google Scholar] [CrossRef]
- Ayakannu, T.; Taylor, A.H.; Marczylo, T.H.; Maccarrone, M.; Konje, J.C. Identification of Novel Predictive Biomarkers for Endometrial Malignancies: N-Acylethanolamines. Front. Oncol. 2019, 9, 430. [Google Scholar] [CrossRef]
- Ayakannu, T.; Taylor, A.H.; Bari, M.; Mastrangelo, N.; Maccarrone, M.; Konje, J.C. Expression and Function of the Endocannabinoid Modulating Enzymes Fatty Acid Amide Hydrolase and N-Acylphosphatidylethanolamine-Specific Phospholipase D in Endometrial Carcinoma. Front. Oncol. 2019, 9, 1363. [Google Scholar] [CrossRef]
- Li, X.; Gao, S.; Li, W.; Liu, Z.; Shi, Z.; Qiu, C.; Jiang, J. Effect of monoacylglycerol lipase on the tumor growth in endometrial cancer. J. Obstet. Gynaecol. Res. 2019, 45, 2043–2054. [Google Scholar] [CrossRef]
- Andradas, C.; Byrne, J.; Kuchibhotla, M.; Ancliffe, M.; Jones, A.C.; Carline, B.; Hii, H.; Truong, A.; Storer, L.C.D.; Ritzmann, T.A.; et al. Assessment of Cannabidiol and Δ9-Tetrahydrocannabiol in Mouse Models of Medulloblastoma and Ependymoma. Cancers 2021, 13, 330. [Google Scholar] [CrossRef] [PubMed]
- Hijiya, N.; Shibata, T.; Daa, T.; Hamanaka, R.; Uchida, T.; Matsuura, K.; Tsukamoto, Y.; Nakada, C.; Iha, H.; Inomata, M.; et al. Overexpression of cannabinoid receptor 1 in esophageal squamous cell carcinoma is correlated with metastasis to lymph nodes and distant organs, and poor prognosis. Pathol. Int. 2017, 67, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.; Tang, L.; Wu, C.; Huang, L. mir-23b-3p and mir-130a-5p affect cell growth, migration and invasion by targeting CB1R via the Wnt/β-catenin signaling pathway in gastric carcinoma. Onco Targets Ther. 2018, 11, 7503–7512. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.; Moesgaard, B.; Schmid, P.C.; Schmid, H.H.; Broholm, H.; Kosteljanetz, M.; Hansen, H.S. Endocannabinoid metabolism in human glioblastomas and meningiomas compared to human non-tumour brain tissue. J. Neurochem. 2005, 93, 299–309. [Google Scholar] [CrossRef]
- Schley, M.; Ständer, S.; Kerner, J.; Vajkoczy, P.; Schüpfer, G.; Dusch, M.; Schmelz, M.; Konrad, C. Predominant CB2 receptor expression in endothelial cells of glioblastoma in humans. Brain Res. Bull. 2009, 79, 333–337. [Google Scholar] [CrossRef]
- Klein Nulent, T.J.W.; Van Diest, P.J.; van der Groep, P.; Leusink, F.K.; Kruitwagen, C.L.; Koole, R.; Van Cann, E.M. Cannabinoid receptor-2 immunoreactivity is associated with survival in squamous cell carcinoma of the head and neck. Br. J. Oral Maxillofac. Surg. 2013, 51, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, Y.; Huang, S.; Liu, G.; Xie, C.; Zhou, J.; Fan, W.; Li, Q.; Wang, Q.; Zhong, D.; et al. Overexpression of cannabinoid receptors CB1 and CB2 correlates with improved prognosis of patients with hepatocellular carcinoma. Cancer Genet. Cytogenet. 2006, 171, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, B.; Schuebel, K.; Mukhopadhyay, P.; Cinar, R.; Godlewski, G.; Xiong, K.; Mackie, K.; Lizak, M.; Yuan, Q.; Goldman, D.; et al. Cannabinoid receptor 1 promotes hepatocellular carcinoma initiation and progression through multiple mechanisms. Hepatology 2015, 61, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Z.; Lian, Z.; Liao, R.; Chen, Y.; Qin, Y.; Wang, J.; Jiang, Q.; Wang, X.; Gong, J. Monoacylglycerol Lipase: A Novel Potential Therapeutic Target and Prognostic Indicator for Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 35784. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhao, Y.; Zhou, J.; Wang, X.; Pan, Q.; Zhang, N.; Wang, L.; Wang, M.; Zhan, D.; Liu, Z.; et al. Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-κB-mediated epithelial-mesenchymal transition. J. Hematol. Oncol. 2016, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tian, Y.; Zheng, R.; Li, L.; Qiu, F. Endocannabinoid system and the expression of endogenous ceramides in human hepatocellular carcinoma. Oncol. Lett. 2019, 18, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, X.; Curtiss, C.; Landas, S.; Rong, R.; Sheikh, M.S.; Huang, Y. Monoglyceride lipase gene knockout in mice leads to increased incidence of lung adenocarcinoma. Cell Death Dis. 2018, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ma, H.; Bo, Y.; Shao, M. The oncogenic role of CB2 in the progression of non-small-cell lung cancer. Biomed. Pharmacother. 2019, 117, 109080. [Google Scholar] [CrossRef] [PubMed]
- Milian, L.; Mata, M.; Alcacer, J.; Oliver, M.; Sancho-Tello, M.; Martín de Llano, J.J.; Camps, C.; Galbis, J.; Carretero, J.; Carda, C. Cannabinoid receptor expression in non-small cell lung cancer. Effectiveness of tetrahydrocannabinol and cannabidiol inhibiting cell proliferation and epithelial-mesenchymal transition in vitro. PLoS ONE 2020, 15, e0228909. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Xie, H.; Heier, C.; Huang, J.; Zheng, Q.; Eichmann, T.O.; Schoiswohl, G.; Ni, J.; Zechner, R.; Ni, S.; et al. Enhanced monoacylglycerol lipolysis by ABHD6 promotes NSCLC pathogenesis. eBioMedicine 2020, 53, 102696. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Guo, W.; Zhang, F.; Li, R.; Zhou, Y.; Shao, F.; Feng, X.; Tan, F.; Wang, J.; Gao, S.; et al. Monoacylglycerol Lipase Knockdown Inhibits Cell Proliferation and Metastasis in Lung Adenocarcinoma. Front. Oncol. 2020, 10, 559568. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.C.; Asplund, A.C.; Lindvall, J.M.; Nygren, L.; Liden, J.; Kimby, E.; Christensson, B.; Smith, C.I.; Sander, B. High level of cannabinoid receptor 1, absence of regulator of G protein signalling 13 and differential expression of Cyclin D1 in mantle cell lymphoma. Leukemia 2003, 17, 1880–1890. [Google Scholar] [CrossRef]
- Wasik, A.M.; Nygren, L.; Almestrand, S.; Zong, F.; Flygare, J.; Wennerholm, S.B.; Saft, L.; Andersson, P.; Kimby, E.; Wahlin, B.E.; et al. Perturbations of the endocannabinoid system in mantle cell lymphoma: Correlations to clinical and pathological features. Oncoscience 2014, 1, 550–557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, Z.; Yang, J.; Zhao, H.; Fang, X.; Li, H. Cannabinoid receptor 2 is upregulated in melanoma. J. Cancer Res. Ther. 2012, 8, 549–554. [Google Scholar] [CrossRef]
- Baba, Y.; Funakoshi, T.; Mori, M.; Emoto, K.; Masugi, Y.; Ekmekcioglu, S.; Amagai, M.; Tanese, K. Expression of monoacylglycerol lipase as a marker of tumour invasion and progression in malignant melanoma. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, S.; Giaginis, C.; Alexandrou, P.; Rodriguez, J.; Tasoulas, J.; Danas, E.; Patsouris, E.; Klijanienko, J. Evaluation of cannabinoid CB1 and CB2 receptors expression in mobile tongue squamous cell carcinoma: Associations with clinicopathological parameters and patients’ survival. Tumour Biol. 2016, 37, 3647–3656. [Google Scholar] [CrossRef]
- Hu, W.R.; Lian, Y.F.; Peng, L.X.; Lei, J.J.; Deng, C.C.; Xu, M.; Feng, Q.S.; Chen, L.Z.; Bei, J.X.; Zeng, Y.X. Monoacylglycerol lipase promotes metastases in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3704–3713. [Google Scholar] [PubMed]
- Messalli, E.M.; Grauso, F.; Luise, R.; Angelini, A.; Rossiello, R. Cannabinoid receptor type 1 immunoreactivity and disease severity in human epithelial ovarian tumors. Am. J. Obstet. Gynecol. 2014, 211, 234.e1–234.e6. [Google Scholar] [CrossRef] [PubMed]
- Michalski, C.W.; Oti, F.E.; Erkan, M.; Sauliunaite, D.; Bergmann, F.; Pacher, P.; Batkai, S.; Müller, M.W.; Giese, N.A.; Friess, H.; et al. Cannabinoids in pancreatic cancer: Correlation with survival and pain. Int. J. Cancer 2008, 122, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Pagotto, U.; Marsicano, G.; Fezza, F.; Theodoropoulou, M.; Grübler, Y.; Stalla, J.; Arzberger, T.; Milone, A.; Losa, M.; Di Marzo, V.; et al. Normal human pituitary gland and pituitary adenomas express cannabinoid receptor type 1 and synthesize endogenous cannabinoids: First evidence for a direct role of cannabinoids on hormone modulation at the human pituitary level. J. Clin. Endocrinol. Metab. 2001, 86, 2687–2696. [Google Scholar] [PubMed]
- Endsley, M.P.; Thill, R.; Choudhry, I.; Williams, C.L.; Kajdacsy-Balla, A.; Campbell, W.B.; Nithipatikom, K. Expression and function of fatty acid amide hydrolase in prostate cancer. Int. J. Cancer 2008, 123, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.C.; Hammarsten, P.; Josefsson, A.; Stattin, P.; Granfors, T.; Egevad, L.; Mancini, G.; Lutz, B.; Bergh, A.; Fowler, C.J. A high cannabinoid CB1 receptor immunoreactivity is associated with disease severity and outcome in prostate cancer. Eur. J. Cancer 2009, 45, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Czifra, G.; Varga, A.; Nyeste, K.; Marincsák, R.; Tóth, B.I.; Kovács, I.; Kovács, L.; Bíró, T. Increased expressions of cannabinoid receptor-1 and transient receptor potential vanilloid-1 in human prostate carcinoma. J. Cancer Res. Clin. Oncol. 2009, 135, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Thors, L.; Bergh, A.; Persson, E.; Hammarsten, P.; Stattin, P.; Egevad, L.; Granfors, T.; Fowler, C.J. Fatty acid amide hydrolase in prostate cancer: Association with disease severity and outcome, CB1 receptor expression and regulation by IL-4. PLoS ONE 2010, 5, e12275. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; de Ridder, D.; Bishop, R.T.; Renema, N.; Ponzetti, M.; Sophocleous, A.; Capulli, M.; Aljeffery, A.; Carrasco, G.; Gens, M.D.; et al. Paradoxical effects of JZL184, an inhibitor of monoacylglycerol lipase, on bone remodelling in healthy and cancer-bearing mice. eBioMedicine 2019, 44, 452–466. [Google Scholar] [CrossRef]
- Larrinaga, G.; Sanz, B.; Pérez, I.; Blanco, L.; Cándenas, M.L.; Pinto, F.M.; Gil, J.; López, J.I. Cannabinoid CB₁ receptor is downregulated in clear cell renal cell carcinoma. J. Histochem. Cytochem. 2010, 58, 1129–1134. [Google Scholar] [CrossRef]
- Larrinaga, G.; Sanz, B.; Blanco, L.; Perez, I.; Candenas, M.L.; Pinto, F.M.; Irazusta, A.; Gil, J.; López, J.I. Cannabinoid CB1 receptor is expressed in chromophobe renal cell carcinoma and renal oncocytoma. Clin. Biochem. 2013, 46, 638–641. [Google Scholar] [CrossRef]
- Wang, J.; Xu, Y.; Zhu, L.; Zou, Y.; Kong, W.; Dong, B.; Huang, J.; Chen, Y.; Xue, W.; Huang, Y.; et al. Cannabinoid receptor 2 as a novel target for promotion of renal cell carcinoma prognosis and progression. J. Cancer Res. Clin. Oncol. 2018, 144, 39–52. [Google Scholar] [CrossRef]
- Mergler, S.; Cheng, Y.; Skosyrski, S.; Garreis, F.; Pietrzak, P.; Kociok, N.; Dwarakanath, A.; Reinach, P.S.; Kakkassery, V. Altered calcium regulation by thermosensitive transient receptor potential channels in etoposide-resistant WERI-Rb1 retinoblastoma cells. Exp. Eye Res. 2012, 94, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Oesch, S.; Walter, D.; Wachtel, M.; Pretre, K.; Salazar, M.; Guzmán, M.; Velasco, G.; Schäfer, B.W. Cannabinoid receptor 1 is a potential drug target for treatment of translocation-positive rhabdomyosarcoma. Mol. Cancer Ther. 2009, 8, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Lakiotaki, E.; Giaginis, C.; Tolia, M.; Alexandrou, P.; Delladetsima, I.; Giannopoulou, I.; Kyrgias, G.; Patsouris, E.; Theocharis, S. Clinical Significance of Cannabinoid Receptors CB1 and CB2 Expression in Human Malignant and Benign Thyroid Lesions. Biomed. Res. Int. 2015, 2015, 839403. [Google Scholar] [CrossRef]
- Sailler, S.; Schmitz, K.; Jäger, E.; Ferreiros, N.; Wicker, S.; Zschiebsch, K.; Pickert, G.; Geisslinger, G.; Walter, C.; Tegeder, I.; et al. Regulation of circulating endocannabinoids associated with cancer and metastases in mice and humans. Oncoscience 2014, 1, 272–282. [Google Scholar] [CrossRef]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic activity of cannabinoids. J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Melck, D.; Palmisano, A.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. USA 1998, 95, 8375–8380. [Google Scholar] [CrossRef]
- Ramer, R.; Hinz, B. Antitumorigenic targets of cannabinoids—Current status and implications. Expert Opin. Ther. Targets 2016, 20, 1219–1235. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Sánchez, C.; Cortés, M.L.; Gómez del Pulgar, T.; Izquierdo, M.; Guzmán, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef]
- Ramer, R.; Weinzierl, U.; Schwind, B.; Brune, K.; Hinz, B. Ceramide is involved in R(+)-methanandamide-induced cyclooxygenase-2 expression in human neuroglioma cells. Mol. Pharmacol. 2003, 64, 1189–1198. [Google Scholar] [CrossRef]
- Hinz, B.; Ramer, R.; Eichele, K.; Weinzierl, U.; Brune, K. Up-regulation of cyclooxygenase-2 expression is involved in R(+)-methanandamide-induced apoptotic death of human neuroglioma cells. Mol. Pharmacol. 2004, 66, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Carracedo, A.; Barrado, L.; Real, P.J.; Fernández-Luna, J.L.; Velasco, G.; Malumbres, M.; Guzmán, M. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006, 20, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Caffarel, M.M.; Andradas, C.; Mira, E.; Pérez-Gómez, E.; Cerutti, C.; Moreno-Bueno, G.; Flores, J.M.; García-Real, I.; Palacios, J.; Mañes, S.; et al. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol. Cancer 2010, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Laezza, C.; Pisanti, S.; Crescenzi, E.; Bifulco, M. Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett. 2006, 580, 6076–6082. [Google Scholar] [CrossRef]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Salazar, M.; Olea-Herrero, N.; Guzmán, M.; Velasco, G.; Díaz-Laviada, I. Anti-tumoural action of cannabinoids on hepatocellular carcinoma, role of AMPK-dependent activation of autophagy. Cell Death Differ. 2011, 18, 1099–1111. [Google Scholar] [CrossRef]
- Vara, D.; Morell, C.; Rodríguez-Henche, N.; Diaz-Laviada, I. Involvement of PPARγ in the antitumoural action of cannabinoids on hepatocellular carcinoma. Cell Death Dis. 2013, 4, e618. [Google Scholar] [CrossRef]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Eleni Anagnostou, M.; Babatunde, F.; Corazzari, M.; Redfern, C.P.F.; et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J. Investig. Dermatol. 2015, 135, 1629–1637. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Grabham, P.W.; Wu, C.C.; Hei, T.K. Inhibition of autophagic flux differently modulates cannabidiol-induced death in 2D and 3D glioblastoma cell cultures. Sci. Rep. 2020, 10, 2687. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Santoni, G. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-López, L.; Valle-Reyes, J.S.; Hernández-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell Death Dis. 2019, 10, 779. [Google Scholar] [CrossRef]
- Go, Y.Y.; Kim, S.R.; Kim, D.Y.; Chae, S.W.; Song, J.J. Cannabidiol enhances cytotoxicity of anti-cancer drugs in human head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 20622. [Google Scholar] [CrossRef]
- Bifulco, M.; Laezza, C.; Valenti, M.; Ligresti, A.; Portella, G.; Di Marzo, V. A new strategy to block tumor growth by inhibiting endocannabinoid inactivation. FASEB J. 2004, 18, 1606–1608. [Google Scholar] [CrossRef]
- Izzo, A.A.; Aviello, G.; Petrosino, S.; Orlando, P.; Marsicano, G.; Lutz, B.; Borrelli, F.; Capasso, R.; Nigam, S.; Capasso, F.; et al. Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J. Mol. Med. 2008, 86, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, B.; Romanini, A.; Vanni, A.; Martinotti, E.; Chicca, A.; Fogli, S.; Nieri, P. Anticancer activity of anandamide in human cutaneous melanoma cells. Eur. J. Pharmacol. 2013, 718, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Hamtiaux, L.; Masquelier, J.; Muccioli, G.G.; Bouzin, C.; Feron, O.; Gallez, B.; Lambert, D.M. The association of N-palmitoylethanolamine with the FAAH inhibitor URB597 impairs melanoma growth through a supra-additive action. BMC Cancer 2012, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Ravi, J.; Sneh, A.; Shilo, K.; Nasser, M.W.; Ganju, R.K. FAAH inhibition enhances anandamide mediated anti-tumorigenic effects in non-small cell lung cancer by downregulating the EGF/EGFR pathway. Oncotarget 2014, 5, 2475–2486. [Google Scholar] [CrossRef]
- Winkler, K.; Ramer, R.; Dithmer, S.; Ivanov, I.; Merkord, J.; Hinz, B. Fatty acid amide hydrolase inhibitors confer anti-invasive and antimetastatic effects on lung cancer cells. Oncotarget 2016, 7, 15047–15064. [Google Scholar] [CrossRef]
- Sticht, M.A.; Long, J.Z.; Rock, E.M.; Limebeer, C.L.; Mechoulam, R.; Cravatt, B.F.; Parker, L.A. Inhibition of monoacylglycerol lipase attenuates vomiting in Suncus murinus and 2-arachidonoyl glycerol attenuates nausea in rats. Br. J. Pharmacol. 2012, 165, 2425–2435. [Google Scholar] [CrossRef]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Taïb, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 2019, 9, 19593. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Bai, J.; Ling, Y.; Chang, W.; Xie, G.; Li, R.; Wang, G.; Tao, K. Monoacylglycerol lipase inhibitor JZL184 regulates apoptosis and migration of colorectal cancer cells. Mol. Med. Rep. 2016, 13, 2850–2856. [Google Scholar] [CrossRef]
- Prüser, J.L.; Ramer, R.; Wittig, F.; Ivanov, I.; Merkord, J.; Hinz, B. The Monoacylglycerol Lipase Inhibitor JZL184 Inhibits Lung Cancer Cell Invasion and Metastasis via the CB1 Cannabinoid Receptor. Mol. Cancer Ther. 2021, 20, 787–802. [Google Scholar] [CrossRef]
- Cipriano, M.; Gouveia-Figueira, S.; Persson, E.; Nording, M.; Fowler, C.J. The influence of monoacylglycerol lipase inhibition upon the expression of epidermal growth factor receptor in human PC-3 prostate cancer cells. BMC Res. Notes 2014, 7, 441. [Google Scholar] [CrossRef] [PubMed]
- Matuszak, N.; Hamtiaux, L.; Baldeyroux, B.; Muccioli, G.G.; Poupaert, J.H.; Lansiaux, A.; Lambert, D.M. Dual inhibition of MAGL and type II topoisomerase by N-phenylmaleimides as a potential strategy to reduce neuroblastoma cell growth. Eur. J. Pharm. Sci. 2012, 45, 263–271. [Google Scholar] [CrossRef] [PubMed]
- King, A.R.; Dotsey, E.Y.; Lodola, A.; Jung, K.M.; Ghomian, A.; Qiu, Y.; Fu, J.; Mor, M.; Piomelli, D. Discovery of potent and reversible monoacylglycerol lipase inhibitors. Chem. Biol. 2009, 16, 1045–1052. [Google Scholar] [CrossRef]
- Lei, X.; Zhong, Y.; Huang, L.; Li, S.; Fu, J.; Zhang, L.; Zhang, Y.; Deng, Q.; Yu, X. Identification of a novel tumor angiogenesis inhibitor targeting Shh/Gli1 signaling pathway in Non-small cell lung cancer. Cell Death Dis. 2020, 11, 232. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, Y.; Zhong, J.; Bi, Y.; Liu, Y.; Ren, Z.; Li, X.; Jia, J.; Yu, M.; Yu, X. Pristimerin induces apoptosis and autophagy via activation of ROS/ASK1/JNK pathway in human breast cancer in vitro and in vivo. Cell Death Dis. 2019, 5, 125. [Google Scholar] [CrossRef] [PubMed]
- Nithipatikom, K.; Endsley, M.P.; Isbell, M.A.; Falck, J.R.; Iwamoto, Y.; Hillard, C.J.; Campbell, W.B. 2-arachidonoylglycerol: A novel inhibitor of androgen-independent prostate cancer cell invasion. Cancer Res. 2004, 64, 8826–8830. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wu, T.T.; Jiang, P.C.; Li, Z.Q.; Chen, X.J.; Fu, K.; Wang, W.; Gong, R. Anti-carcinogenic activity of anandamide on human glioma in vitro and in vivo. Mol. Med. Rep. 2016, 13, 1558–1562. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, C.; Pisanti, S.; Laezza, C.; Malfitano, A.M.; Santoro, A.; Vitale, M.; Caruso, M.G.; Notarnicola, M.; Iacuzzo, I.; Portella, G.; et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp. Cell Res. 2006, 312, 363–373. [Google Scholar] [CrossRef]
- Ramer, R.; Hinz, B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J. Natl. Cancer Inst. 2008, 100, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Merkord, J.; Rohde, H.; Hinz, B. Cannabidiol inhibits cancer cell invasion via upregulation of tissue inhibitor of matrix metalloproteinases-1. Biochem. Pharmacol. 2010, 79, 955–966. [Google Scholar] [CrossRef]
- Ramer, R.; Bublitz, K.; Freimuth, N.; Merkord, J.; Rohde, H.; Haustein, M.; Borchert, P.; Schmuhl, E.; Linnebacher, M.; Hinz, B. Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule-1. FASEB J. 2012, 26, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Salazar, M.; Carracedo, A.; Lorente, M.; Egia, A.; González-Feria, L.; Haro, A.; Velasco, G.; Guzmán, M. Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression. Cancer Res. 2008, 68, 1945–1952. [Google Scholar] [CrossRef]
- Pourkhalili, N.; Ghahremani, M.H.; Farsandaj, N.; Tavajohi, S.; Majdzadeh, M.; Parsa, M.; Lavasani, N.J.; Ostad, S.N. Evaluation of anti-invasion effect of cannabinoids on human hepatocarcinoma cells. Toxicol. Mech. Methods 2013, 23, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Rohde, A.; Merkord, J.; Rohde, H.; Hinz, B. Decrease of plasminogen activator inhibitor-1 may contribute to the anti-invasive action of cannabidiol on human lung cancer cells. Pharm. Res. 2010, 27, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Pietrovito, L.; Iozzo, M.; Bacci, M.; Giannoni, E.; Chiarugi, P. Treatment with Cannabinoids as a Promising Approach for Impairing Fibroblast Activation and Prostate Cancer Progression. Int. J. Mol. Sci. 2020, 21, 787. [Google Scholar] [CrossRef]
- McAllister, S.D.; Christian, R.T.; Horowitz, M.P.; Garcia, A.; Desprez, P.Y. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol. Cancer Ther. 2007, 6, 2921–2927. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Murase, R.; Limbad, C.; Singer, E.; Allison, J.; Adrados, I.; Kawamura, R.; Pakdel, A.; Fukuyo, Y.; Nguyen, D.; et al. Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target. Cancer Res. 2013, 73, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.D.; Murase, R.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Allison, J.; Almanza, C.; Pakdel, A.; Lee, J.; Limbad, C.; et al. Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation invasion and metastasis. Breast Cancer Res. Treat. 2011, 129, 37–47, Erratum in 2012, 133, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Murase, R.; Kawamura, R.; Singer, E.; Pakdel, A.; Sarma, P.; Judkins, J.; Elwakeel, E.; Dayal, S.; Martinez-Martinez, E.; Amere, M.; et al. Targeting multiple cannabinoid anti-tumour pathways with a resorcinol derivative leads to inhibition of advanced stages of breast cancer. Br. J. Pharmacol. 2014, 171, 4464–4477. [Google Scholar] [CrossRef]
- Casanova, M.L.; Blázquez, C.; -Palacio, J.; Villanueva, C.; Fernández-Aceñero, M.J.; Huffman, J.W. Inhibition of skin tumour growth and angiogenesis in vivo by activation of cannabinoid receptors. J. Clin. Investig. 2003, 111, 43–50. [Google Scholar] [CrossRef]
- Blázquez, C.; Casanova, M.L.; Planas, A.; Gómez Del Pulgar, T.; Villanuev, C.; Fernández-Aceñero, M.J.; Aragonés, J.; Huffman, J.W.; Jorcano, J.L.; Guzmán, M. Inhibition of tumor angiogenesis by cannabinoids. FASEB J. 2003, 17, 529–531. [Google Scholar] [CrossRef]
- Blázquez, C.; González-Feria, L.; Alvarez, L.; Haro, A.; Casanova, M.L.; Guzmán, M. Cannabinoids inhibit the vascular endothelial growth factor pathway in gliomas. Cancer Res. 2004, 64, 5617–5623. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Fischer, S.; Haustein, M.; Manda, K.; Hinz, B. Cannabinoids inhibit angiogenic capacities of endothelial cells via release of tissue inhibitor of matrix metalloproteinases-1 from lung cancer cells. Biochem. Pharmacol. 2014, 91, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Braile, M.; Cristinziano, L.; Marcella, S.; Varricchi, G.; Marone, G.; Modestino, L.; Ferrara, A.L.; De Ciuceis, A.; Scala, S.; Galdiero, M.R.; et al. LPS-mediated neutrophil VEGF-A release is modulated by cannabinoid receptor activation. J. Leukoc. Biol. 2021, 109, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Picardi, P.; Ciaglia, E.; Proto, M.; Pisanti, S. Anandamide inhibits breast tumor-induced angiogenesis. Transl. Med. UniSa. 2014, 10, 8–12. [Google Scholar] [PubMed]
- Pagano, E.; Borrelli, F.; Orlando, P.; Romano, B.; Monti, M.; Morbidelli, L.; Aviello, G.; Imperatore, R.; Capasso, R.; Piscitelli, F.; et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res. 2017, 119, 227–236. [Google Scholar] [CrossRef]
- Hu, Y.; Ranganathan, M.; Shu, C.; Liang, X.; Ganesh, S.; Osafo-Addo, A.; Yan, C.; Zhang, X.; Aouizerat, B.E.; Krystal, J.H.; et al. Single-cell Transcriptome Mapping Identifies Common and Cell-type Specific Genes Affected by Acute Delta9-tetrahydrocannabinol in Humans. Sci. Rep. 2020, 10, 3450. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huynh, N.; Dumesny, C.; Wang, K.; He, H.; Nikfarjam, M. Cannabinoids Inhibited Pancreatic Cancer via P-21 Activated Kinase 1 Mediated Pathway. Int. J. Mol. Sci. 2020, 21, 8035. [Google Scholar] [CrossRef] [PubMed]
- Glodde, N.; Jakobs, M.; Bald, T.; Tüting, T.; Gaffal, E. Differential role of cannabinoids in the pathogenesis of skin cancer. Life Sci. 2015, 138, 35–40. [Google Scholar] [CrossRef]
- Haustein, M.; Ramer, R.; Linnebacher, M.; Manda, K.; Hinz, B. Cannabinoids increase lung cancer cell lysis by lymphokine-activated killer cells via upregulation of ICAM-1. Biochem. Pharmacol. 2014, 92, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.X.; Sharma, S.; Stolina, M.; Gardner, B.; Roth, M.D.; Tashkin, D.P.; Dubinett, S.M. Δ-9-tetrahydrocannabinol inhibits antitumor immunity by a CB2 receptor-mediated, cytokine-dependent pathway. J. Immunol. 2000, 165, 373–380. [Google Scholar] [CrossRef]
- McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Δ-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J. Immunol. 2005, 174, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Chen, X.; Quan, Y.; Tao, Y.; Li, J. Targeting CYP2J2 to Enhance the Anti-Glioma Efficacy of Cannabinoid Receptor 2 Stimulation by Inhibiting the Pro-Angiogenesis Function of M2 Microglia. Front. Oncol. 2020, 10, 574277. [Google Scholar] [CrossRef]
- Sekiba, K.; Otsuka, M.; Seimiya, T.; Tanaka, E.; Funato, K.; Miyakawa, Y.; Koike, K. The fatty-acid amide hydrolase inhibitor URB597 inhibits MICA/B shedding. Sci. Rep. 2020, 10, 15556. [Google Scholar] [CrossRef]
- Yin, J.; Kim, S.S.; Choi, E.; Oh, Y.T.; Lin, W.; Kim, T.H.; Sa, J.K.; Hong, J.H.; Park, S.H.; Kwon, H.J.; et al. ARS2/MAGL signaling in glioblastoma stem cells promotes self-renewal and M2-like polarization of tumor-associated macrophages. Nat. Commun. 2020, 11, 2978. [Google Scholar] [CrossRef]
- Marino, S.; Carrasco, G.; Li, B.; Shah, K.M.; Lath, D.L.; Sophocleous, A.; Lawson, M.A.; Idris, A.I. JZL184, A Monoacylglycerol Lipase Inhibitor, Induces Bone Loss in a Multiple Myeloma Model of Immunocompetent Mice. Calcif. Tissue Int. 2020, 107, 72–85. [Google Scholar] [CrossRef]
- Hinz, B.; Ramer, R. Anti-tumour actions of cannabinoids. Br. J. Pharmacol. 2019, 176, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Tramèr, M.R.; Carroll, D.; Campbell, F.A.; Reynolds, D.J.; Moore, R.A.; McQuay, H.J. Cannabinoids for control of chemotherapy induced nausea and vomiting: Quantitative systematic review. BMJ 2001, 323, 16–21. [Google Scholar] [CrossRef]
- Khasabova, I.A.; Khasabov, S.; Paz, J.; Harding-Rose, C.; Simone, D.A.; Seybold, V.S. Cannabinoid type-1 receptor reduces pain and neurotoxicity produced by chemotherapy. J. Neurosci. 2012, 32, 7091–7101. [Google Scholar] [CrossRef]
- Long, J.Z.; Nomura, D.K.; Vann, R.E.; Walentiny, D.M.; Booker, L.; Jin, X.; Burston, J.J.; Sim-Selley, L.J.; Lichtman, A.H.; Wiley, J.L.; et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 20270–20275. [Google Scholar] [CrossRef]
- Curry, Z.A.; Wilkerson, J.L.; Bagdas, D.; Kyte, S.L.; Patel, N.; Donvito, G.; Mustafa, M.A.; Poklis, J.L.; Niphakis, M.J.; Hsu, K.L.; et al. Monoacylglycerol Lipase Inhibitors Reverse Paclitaxel-Induced Nociceptive Behavior and Proinflammatory Markers in a Mouse Model of Chemotherapy-Induced Neuropathy. J. Pharmacol. Exp. Ther. 2018, 366, 169–183. [Google Scholar] [CrossRef] [PubMed]
- van Egmond, N.; Straub, V.M.; van der Stelt, M. Targeting Endocannabinoid Signaling: FAAH and MAG Lipase Inhibitors. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 441–463. [Google Scholar] [CrossRef] [PubMed]
- Schlosburg, J.E.; Boger, D.L.; Cravatt, B.F.; Lichtman, A.H. Endocannabinoid modulation of scratching response in an acute allergenic model: A new prospective neural therapeutic target for pruritus. J. Pharmacol. Exp. Ther. 2009, 329, 314–323. [Google Scholar] [CrossRef] [PubMed]
- van Esbroeck, A.C.M.; Janssen, A.P.A.; Cognetta, A.B., 3rd; Ogasawara, D.; Shpak, G.; van der Kroeg, M.; Kantae, V.; Baggelaar, M.P.; de Vrij, F.M.S.; Deng, H.; et al. Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10-2474. Science 2017, 356, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Page, R.L., 2nd; Allen, L.A.; Kloner, R.A.; Carriker, C.R.; Martel, C.; Morris, A.A.; Piano, M.R.; Rana, J.S.; Saucedo, J.F.; American Heart Association Clinical Pharmacology Committee and Heart Failure and Transplantation Committee of the Council on Clinical Cardiology; et al. Medical Marijuana, Recreational Cannabis, and Cardiovascular Health: A Scientific Statement From the American Heart Association. Circulation 2020, 142, e131–e152. [Google Scholar] [CrossRef] [PubMed]
- Teixeira-Clerc, F.; Julien, B.; Grenard, P.; Tran Van Nhieu, J.; Deveaux, V.; Li, L.; Serriere-Lanneau, V.; Ledent, C.; Mallat, A.; Lotersztajn, S. CB1 cannabinoid receptor antagonism: A new strategy for the treatment of liver fibrosis. Nat. Med. 2006, 12, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M.; Duarte, M.J.; Blazquez, C.; Ravina, J.; Rosa, M.C.; Galve-Roperh, I.; Sánchez, C.; Velasco, G.; González-Feria, L. A pilot study of Δ9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br. J. Cancer 2006, 95, 197–203. [Google Scholar] [CrossRef]
- Twelves, C.; Sabel, M.; Checketts, D.; Miller, S.; Tayo, B.; Jove, M.; Brazil, L.; Short, S.C. GWCA1208 study group. A phase 1b randomised, placebo-controlled trial of nabiximols cannabinoid oromucosal spray with temozolomide in patients with recurrent glioblastoma. Br. J. Cancer 2021, 124, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. ClinicalTrials.gov: A Phase I/Ib Study on the Safety of Epidiolex in Patients with Prostate Cancer with Rising PSA after Localized Therapy with Either Surgery or Radiation. Identifier: NCT04428203. Available online: https://clinicaltrials.gov/ct2/show/NCT04428203?term=CBD&cond=cancer&draw=2&rank=2 (accessed on 20 October 2021).
- U.S. National Library of Medicine. ClinicalTrials.gov: Why Antiprogestrone (Mifepristone) and Cyp 26 Inhibitor Must Be Combined with Tamoxifen or (Tamoxifen and Retinoic Acid) for Treating Early Breast Cancer. Identifier: NCT05016349. Available online: https://clinicaltrials.gov/ct2/show/NCT05016349?term=CBD&cond=cancer&draw=1&rank=5 (accessed on 20 October 2021).
- U.S. National Library of Medicine. ClinicalTrials.gov: Phase Ib, Open-Label, Multicenter, Intrapatient Dose-Escalation Clinical Trial to Assess the Safety Profile of the TN-TC11G (THC+CBD) Combination with Temozolomide and Radiotherapy in Patients with Newly-Diagnosed Glioblastoma. Identifier: NCT03529448. Available online: https://clinicaltrials.gov/ct2/show/NCT03529448?term=CBD&cond=cancer&draw=3&rank=42 (accessed on 20 October 2021).
- U.S. Food and Drug Administration. FDA News Release. Available online: https://www.fda.gov/news-events/press-announcements/fda-warns-companies-marketing-unproven-products-derived-marijuana-claim-treat-or-cure-cancer (accessed on 20 October 2021).



{kind=link}
{kind=link}
| Tumour Type | AEA | 2-AG | CB1 | CB2 | FAAH | MAGL | NAPE-PLD | Commentary | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Acute myeloid leukemia | ↑ | [36] | |||||||
| Astrocytomas | ↔ | ↔ | refers to differences between human astrocytomas of low (grades I–II) and high (grade III) malignancy and differences between pure astrocytomas and mixed oligoastrocytomas | [37] | |||||
| ↔ | ↔ | [38] | |||||||
| ↔ | ↑ | [39] | |||||||
| ↓ | ↑ | ↔ | ↑ | ↓ | ↓ | ↓ | [34] | ||
| ↑ | higher expression in pediatric low-grade gliomas of spontaneous involution/stable status than in relapse | [40] | |||||||
| ↔ | ↑ | refers to benign juvenile pilocytic astrocytomas | [41] | ||||||
| B-cell lymphoma | ↑ | refers to analyses of serum from male patients with a body mass index (BMI) ≥ 25 compared to male patients with a BMI < 25 | [31] | ||||||
| Breast cancer | ↑ | [42] | |||||||
| ↑ | ductal breast cancer | [43] | |||||||
| ↑ | immunohistochemical staining revealed 58% CB2 receptor positive samples in 82 patients without comparison to healthy tissue | [44] | |||||||
| ↑ | regulation not associated with disease-specific survival and recurrence-free survival, but with high number of lymph node involvement | [45] | |||||||
| ↓ | [46] | ||||||||
| ↑ | HER2-positive breast cancer | [47] | |||||||
| ↑ | HER2-CB2 heteromers | [48] | |||||||
| ↓ | [49] | ||||||||
| Colorectal cancer | ↑ | ↑ | [50] | ||||||
| ↓ | ↑ | [51] | |||||||
| ↓ | ↔ | [52] | |||||||
| Colorectal cancer | ↑ | [53] | |||||||
| ↓ | proportion of low CB1 expression significantly higher in stage IV than in stage I/II or III cancer | [54] | |||||||
| ↓ | degradation or loss of MAGL expression detected in 60 of 101 cases with colon cancer and in 9 of 18 cases with rectal cancer (from pooled data of different methods) | [46] | |||||||
| ↑ | ↔ | ↑ | ↔ | ↑ | ↑ | [33] | |||
| ↑ | [55] | ||||||||
| ↓ | CB1 mRNA levels significantly reduced in TNM stage I tumours; CB1 mRNA levels, however, increase with greater disease severity | [56] | |||||||
| ↑ | ↓ | refers to tumour-associated macrophages | [57] | ||||||
| ↓ | additional finding: significant downregulation of CB1 in patients with metastases, both in normal mucosa and tumour tissue compared to patients without metastases | [58] | |||||||
| Endometrial cancer | ↑ | ↑ | ↓ | [32] | |||||
| ↓ | [46] | ||||||||
| ↑ | ↑ | [59] | |||||||
| ↑ | [60] | ||||||||
| ↓ | ↑ | [61] | |||||||
| ↑ | [62] | ||||||||
| Ependymoma | ↑ | ↔ | C11orf95 subtype (C11orf95 fusion-positive ependymoma, formerly named EPN_RELA, high-risk brain cancer in children) with higher CB1 receptor expression than the PFA subtype of ependymoma (posterior fossa type A ependymomas); CB2 receptor expression was similar in both subtypes | [63] | |||||
| Esophageal squamous cell carcinoma | ↑ | [64] | |||||||
| Gastric cancer | ↓ | [46] | |||||||
| ↑ | [65] | ||||||||
| Glioblastoma | ↓ | refers to analysis of only one glioblastoma sample | [35] | ||||||
| ↔ | ↑ | refers to differences between glioblastoma (grade IV) and human astrocytomas (grades I–II and III) | [37] | ||||||
| ↑ | ↑ | [66] | |||||||
| ↔ | ↔ | [38] | |||||||
| ↔ | ↑ | [67] | |||||||
| ↓ | ↑ | [39] | |||||||
| ↓ | ↑ | ↑ | ↑ | ↓ | ↓ | ↓ | [34] | ||
| ↔ | ↑ | [41] | |||||||
| Head and neck squamous cell carcinoma | ↑ | [68] | |||||||
| Hepatocellular carcinoma | ↑ | ↑ | early hepatocellular carcinoma only | [69] | |||||
| ↑ | ↑ | ↑ | [70] | ||||||
| ↑ | [71] | ||||||||
| ↑ | [72] | ||||||||
| ↓ | ↑ | ↓ | ↑ | ↑ | ↑ | ↔ | [73] | ||
| Lung cancer | ↓ | [46] | |||||||
| ↓ | [74] | ||||||||
| ↑ | [75] | ||||||||
| ↑ | ↑ | [76] | |||||||
| ↔ | supplementary finding: ABHD6 upregulation | [77] | |||||||
| ↑ | [78] | ||||||||
| Mantle cell lymphoma | ↑ | [79] | |||||||
| ↑ | ↑ | ↓ | ↑ | [80] | |||||
| Medulloblastoma | ↑ | ↔ | SHH subtype (highly aggressive medulloblastoma tumour characterised by activation of the Sonic Hedgehog (SHH) pathway originating from granule cell precursors of the developing cerebellum) with higher CB1 expression levels than other medulloblastoma subtypes; CB2 receptor expression was similar among the subtypes | [63] | |||||
| Melanoma | ↑ | [81] | |||||||
| ↑ | [82] | ||||||||
| Meningioma | ↓ | ↔ | [35] | ||||||
| ↑ | ↑ | [66] | |||||||
| ↔ | ↔ | [39] | |||||||
| Mobile tongue squamous cell carcinoma | ↑ | ↑ | increased CB2 receptor and concomitant CB1 receptor/CB2 receptor expression was observed significantly more frequently in female than in male patients with squamous cell carcinoma of the mobile tongue | [83] | |||||
| Nasopharyngeal carcinoma | ↑ | [84] | |||||||
| Ovary cancer | ↑ | [18] | |||||||
| ↓ | [46] | ||||||||
| ↑ | [85] | ||||||||
| Pancreatic cancer | ↔ | ↔ | ↑ | ↔ | ↓ | ↓ | endocannabinoid regulation compared with healthy controls; indicated cannabinoid receptor, FAAH or MAGL up- or downregulations refer to lower survival time | [86] | |
| Pituitary adenomas | ↑ | ↑ | [87] | ||||||
| Prostate cancer | ↑ | differences in the expression of FAAH depending on the Gleason score of the tumour tissue could not be deduced | [88] | ||||||
| ↑ | [89] | ||||||||
| ↑ | [90] | ||||||||
| ↑ | [91] | ||||||||
| ↑ | [92] | ||||||||
| Renal cell carcinoma | ↓ | refers to analyses of clear renal carcinoma samples | [93] | ||||||
| ↓ | refers to analyses of clear renal carcinoma samples | [94] | |||||||
| ↓ | [46] | ||||||||
| ↑ | [95] | ||||||||
| Retinoblastoma | ↑ | [96] | |||||||
| Rhabdomyosarcoma | ↑ | [97] | |||||||
| Thyroid malignancies | ↔ | [46] | |||||||
| ↑ | ↑ | [98] | |||||||
| Different cancer types | ↓ | ↑ | endocannabinoids measured cross-sectionally in the plasma of age- and sex-matched subgroups of subjects that included 42 control subjects and 44 cancer patients, with no assignment to tumour types at data presentation | [99] |
| Tumour Type | Essential Result of the Studies | CB1 | CB2 | FAAH | MAGL | Reference |
|---|---|---|---|---|---|---|
| Breast cancer | No correlation of FAAH expression with disease-specific survival, but levels of FAAH significantly increased in patients with higher number of axillary lymph node metastases | ↔ | [45] | |||
| Strong association between higher CB2 protein expression in HER2+ breast tumours and lower patient overall, relapse-free and metastasis-free survival | ○ | [47] | ||||
| High HER2–CB2 heteromer expression associated with lower disease-free and overall patient survival | ○ | [48] | ||||
| Colorectal cancer | Higher CB1 expression correlated with poorer overall survival in stage IV; CB1 expression not correlated with patient survival following surgery in stage I/II or III cancer | ○ | [54] | |||
| CB2 mRNA expression as prognostic factor for colon but not for rectal cancer; five-year overall survival for patients without CB2 expression was 76.16% versus 41.94% for patients with CB2 expression | ○ | [55] | ||||
| Higher levels of MAGL or lower levels of CB2 in tumour-associated macrophages of patients with colorectal cancer associated with better survival | ○ | ○ | [57] | |||
| Esophageal squamous cell carcinoma | Overexpression of CB1 in esophageal squamous cell carcinoma correlated with metastasis to lymph nodes and distant organs, and poor prognosis | ○ | [64] | |||
| Head and neck squamous cell carcinoma | Higher CB2 receptor expression associated with reduced disease-specific survival; CB1 receptor immunoreactivity not associated with survival | ↔ | ○ | [68] | ||
| Hepatocellular carcinoma | Disease-free survival in patients with hepatocellular carcinoma with high CB1 and CB2 expression significantly better than in patients with low expression | ○ | ○ | [69] | ||
| MAGL low-expression group with significantly better survival than MAGL high-expression group | ○ | [71] | ||||
| Clinical prognosis for the MAGL high group markedly poorer than that for the MAGL low group in the 1-, 3-, and 5-year overall survival times and recurrence rates | ○ | [72] | ||||
| Lung cancer | Lung adenocarcinoma patients with high CB2 level showed a shorter overall survival | ○ | [75] | |||
| Patients with high expression levels of CB1, CB2 and CB1/CB2 showed increased survival | ○ | ○ | [76] | |||
| Overall survival gradually reduced with increasing ABHD6 levels; no significant association with MAGL expression | ↔ | [77] | ||||
| High MAGL expression associated with worse outcomes | ○ | [78] | ||||
| Mobile tongue squamous cell carcinoma | High CB1 and CB2 expression associated with longer overall and disease-free survival times | ○ | ○ | [83] | ||
| Pancreatic cancer | Correlation between longer survival and low CB1 receptor or high FAAH as well as MAGL levels; no correlation between survival and CB2 immunoreactivity | ○ | ↔ | ○ | ○ | [86] |
| Prostate cancer | High CB1 expression associated with a shorter survival time | ○ | [89] | |||
| High tumour epithelial FAAH associated with a poor disease-specific survival | ○ | [91] | ||||
| Renal cell carcinoma | Higher CB2 expression tending to have poor clinical outcomes in survival analyses | ○ | [95] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramer, R.; Wittig, F.; Hinz, B. The Endocannabinoid System as a Pharmacological Target for New Cancer Therapies. Cancers 2021, 13, 5701. https://doi.org/10.3390/cancers13225701
Ramer R, Wittig F, Hinz B. The Endocannabinoid System as a Pharmacological Target for New Cancer Therapies. Cancers. 2021; 13(22):5701. https://doi.org/10.3390/cancers13225701
Chicago/Turabian StyleRamer, Robert, Felix Wittig, and Burkhard Hinz. 2021. "The Endocannabinoid System as a Pharmacological Target for New Cancer Therapies" Cancers 13, no. 22: 5701. https://doi.org/10.3390/cancers13225701
APA StyleRamer, R., Wittig, F., & Hinz, B. (2021). The Endocannabinoid System as a Pharmacological Target for New Cancer Therapies. Cancers, 13(22), 5701. https://doi.org/10.3390/cancers13225701