
Cancer Detection and Classification by CpG Island Hypermethylation Signatures in Plasma Cell-Free DNA

 , , , , ,
, , , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Acquisition and Clinical Cohort
2.2. Plasma Sample Collection
2.3. cfDNA Extraction
2.4. Filler DNA Generation
2.5. Library Preparation
2.6. cfMBD Methylation Capture
2.7. DNA Sequencing and Alignment
2.8. Quality Control of Methylation Enrichment
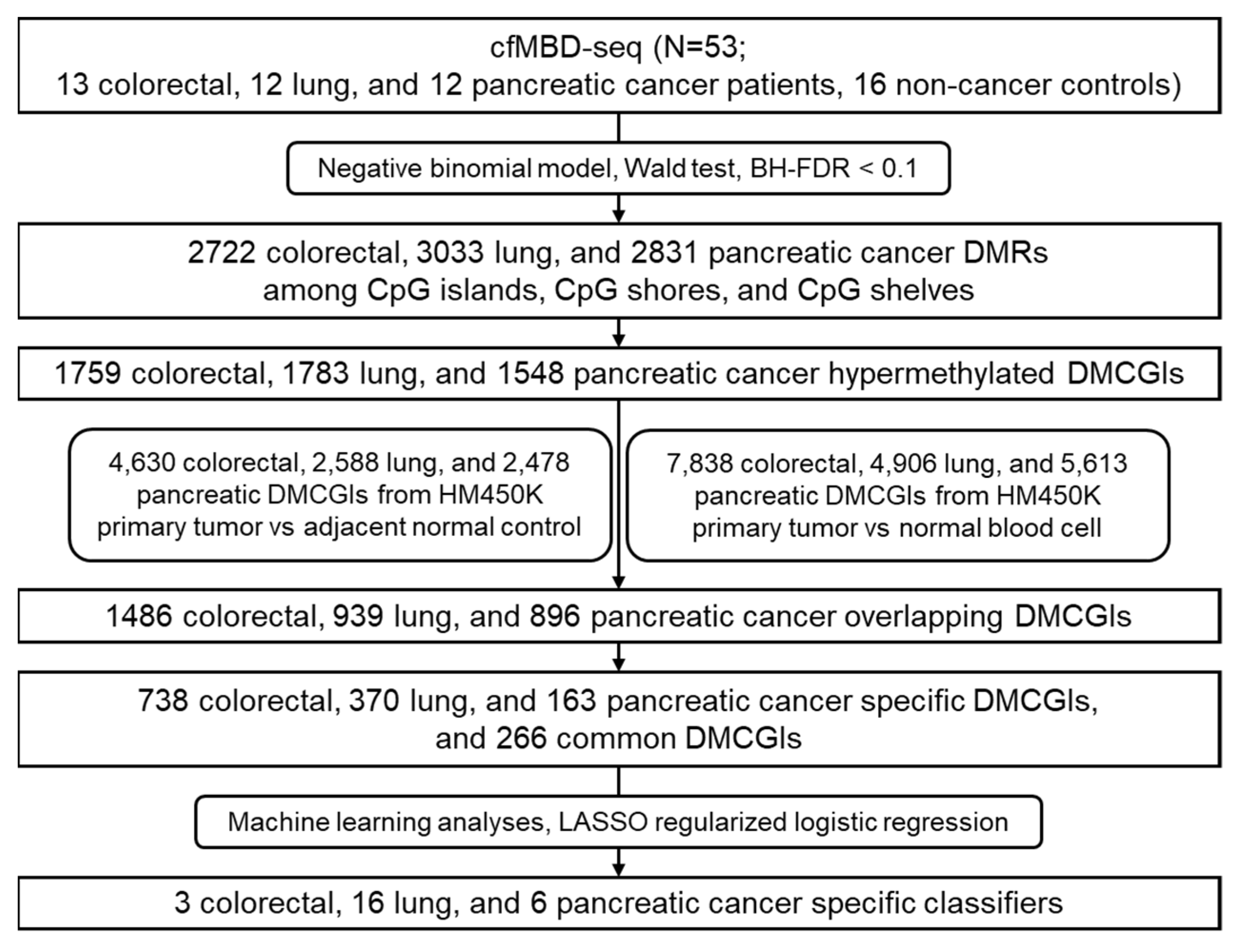
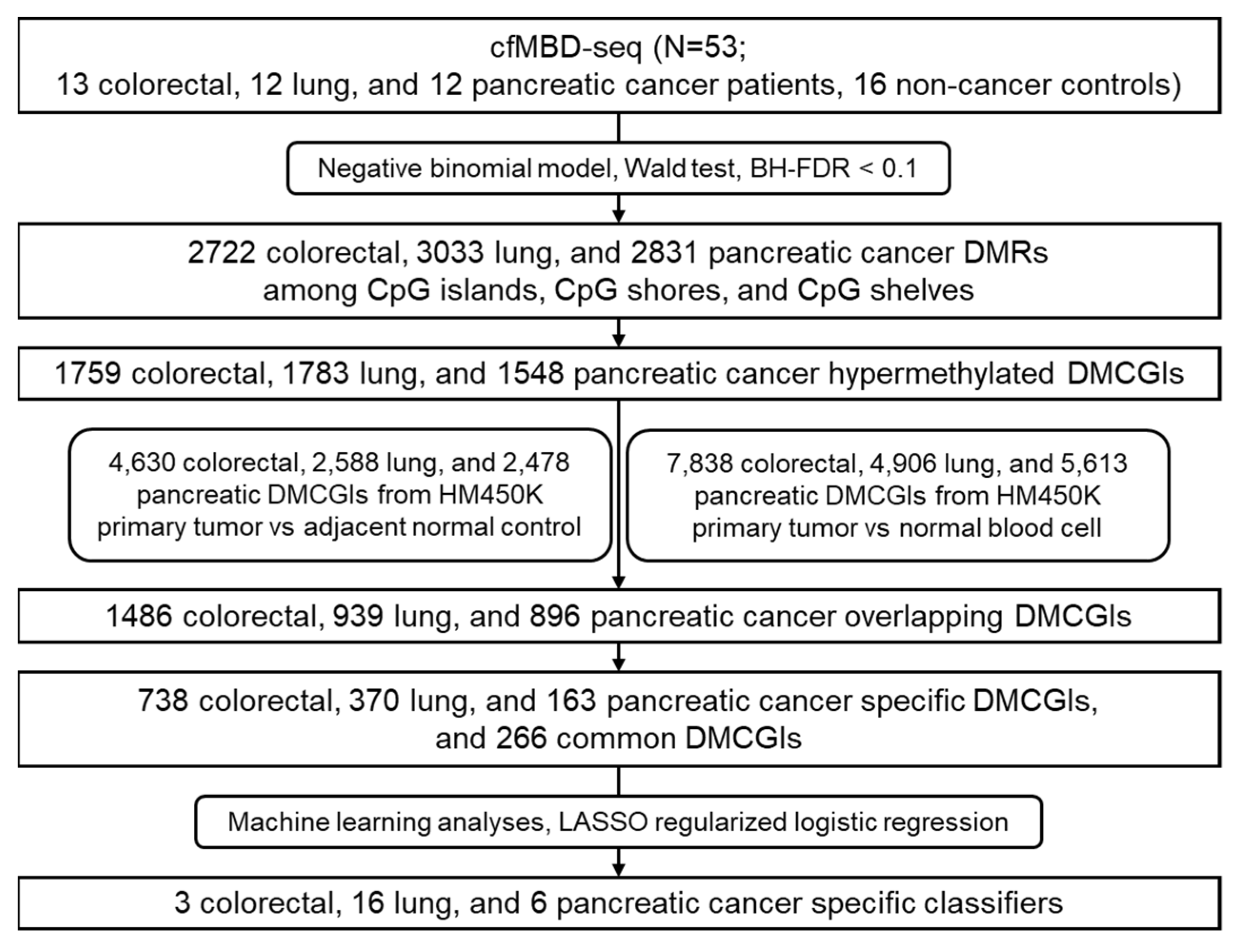
2.9. Differential Methylation Analysis of cfMBD-Seq Data
2.10. Methylation Analyses for Tumor-Tissue-Specific DMCGIs
2.11. Machine Learning Analyses
3. Results
3.1. Significant Enrichment of Methylated CpG Islands in cfDNA
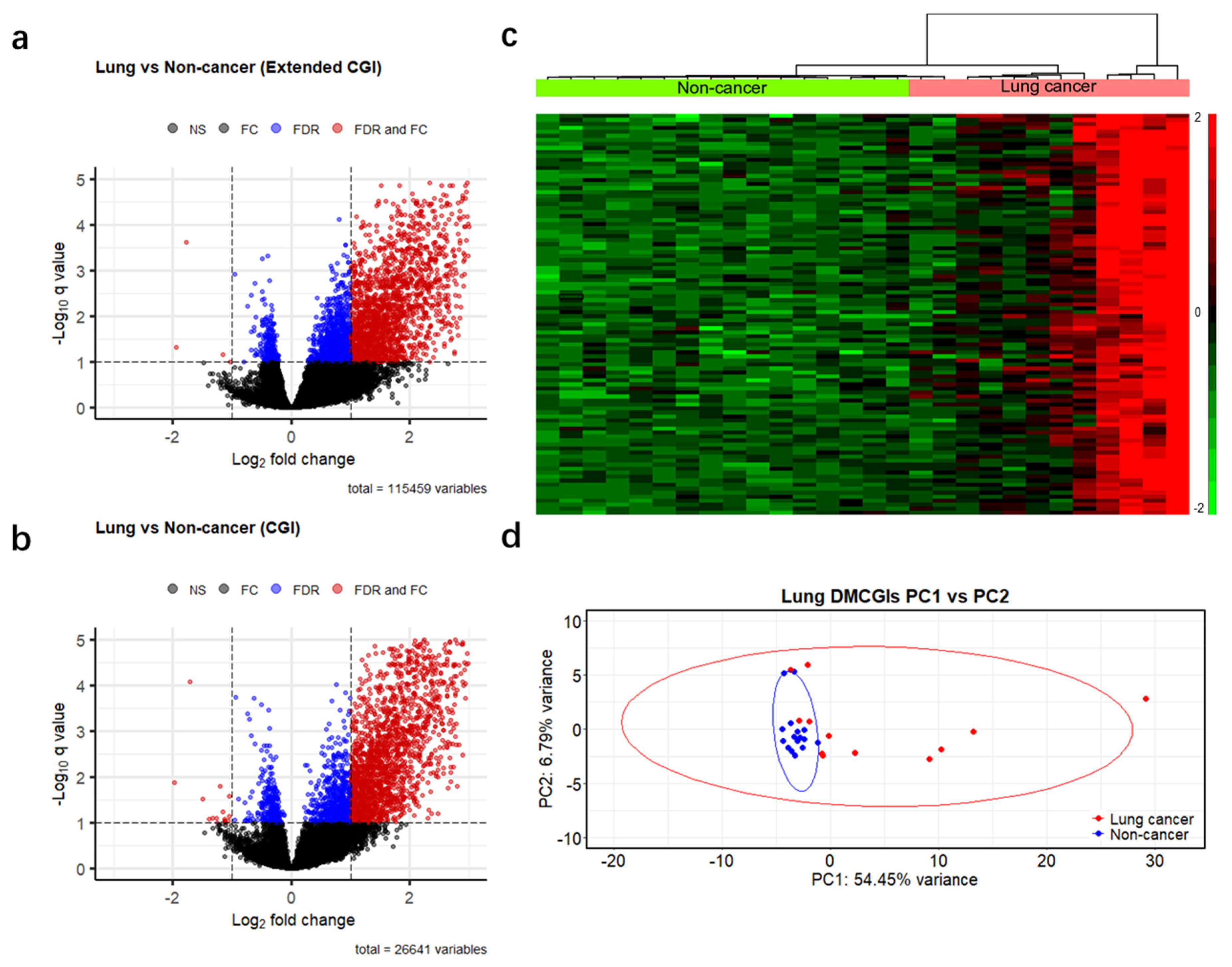
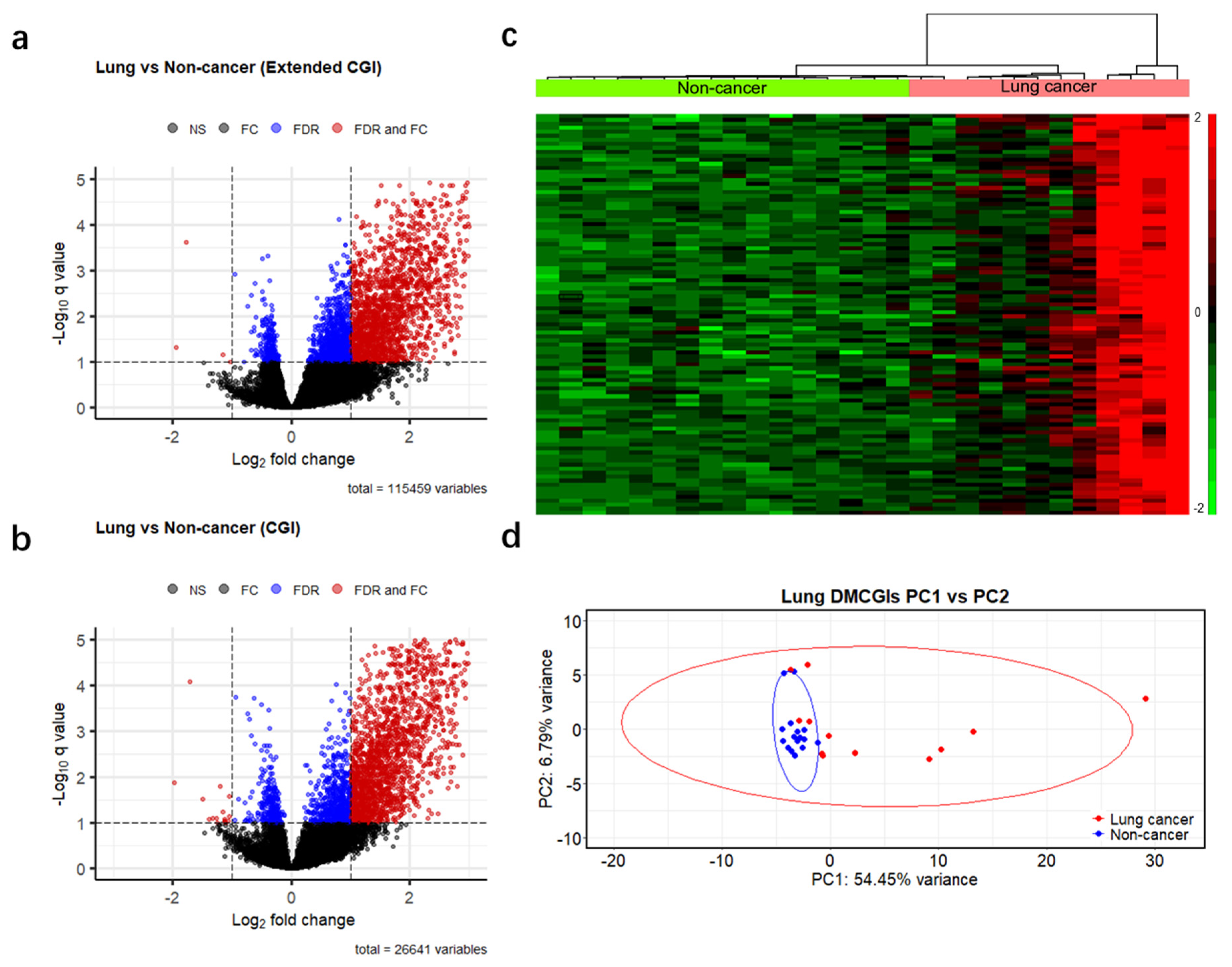
3.2. Differential Methylation Analyses between Cancer Patients and Non-Cancer Controls
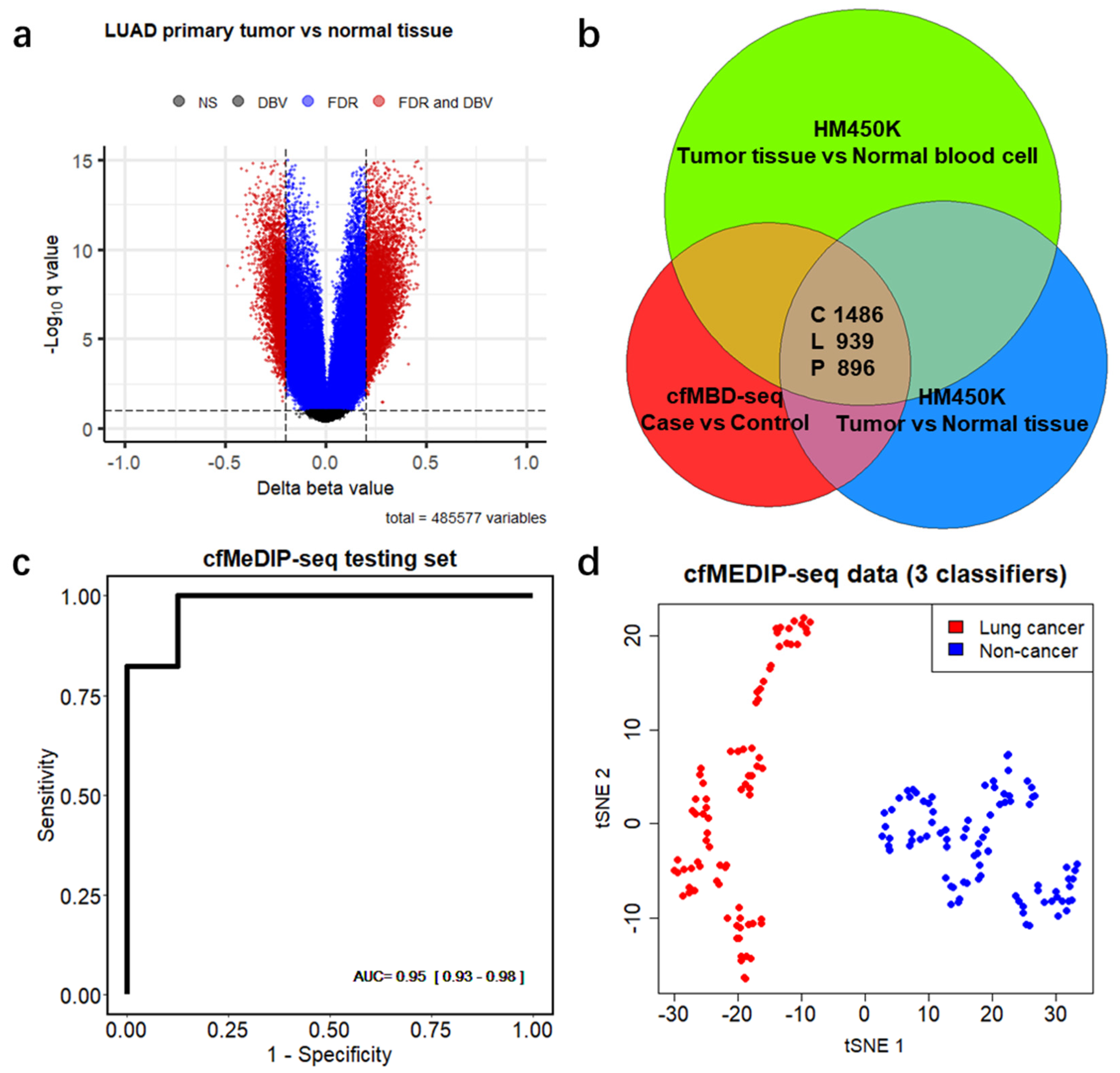
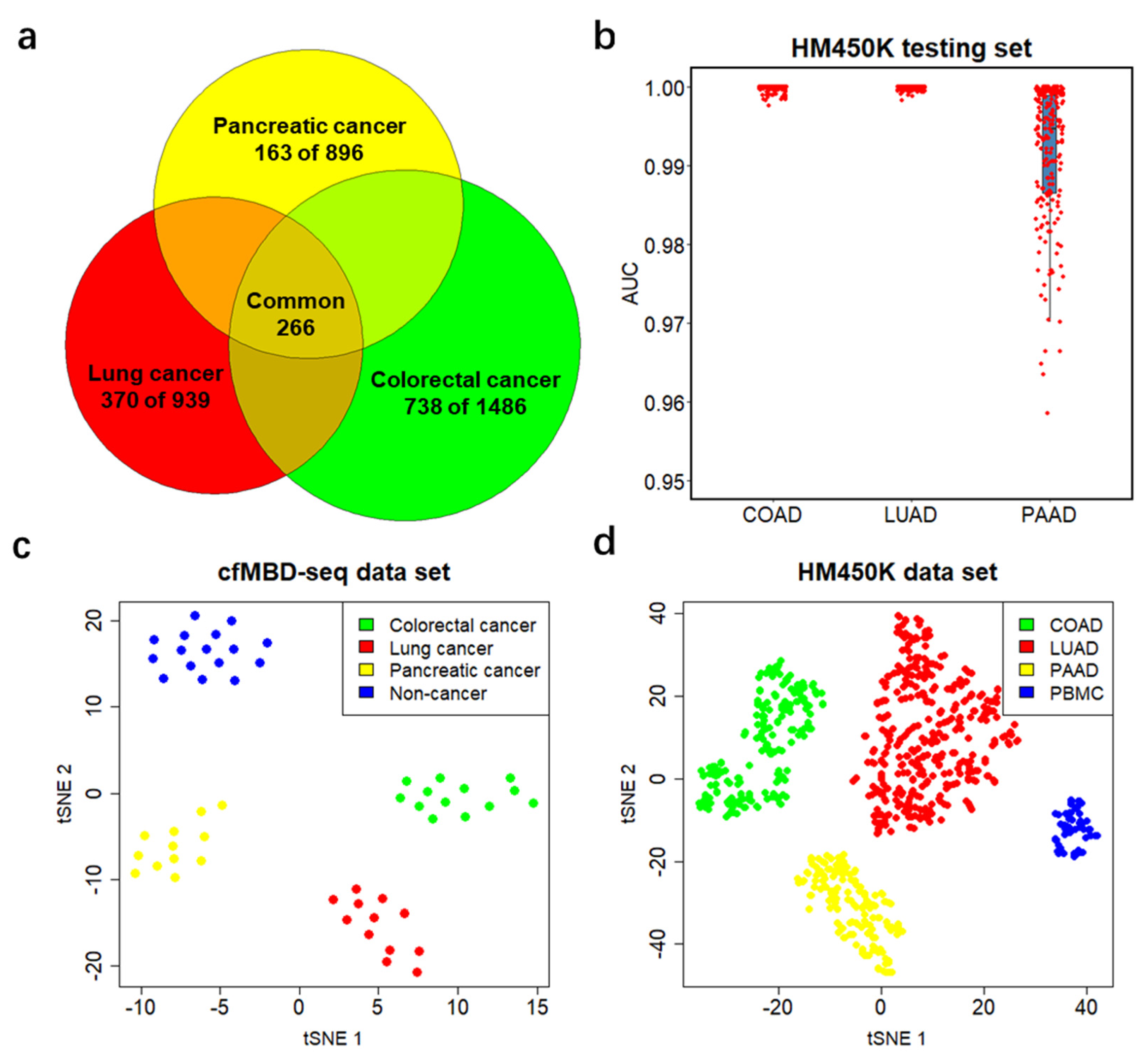
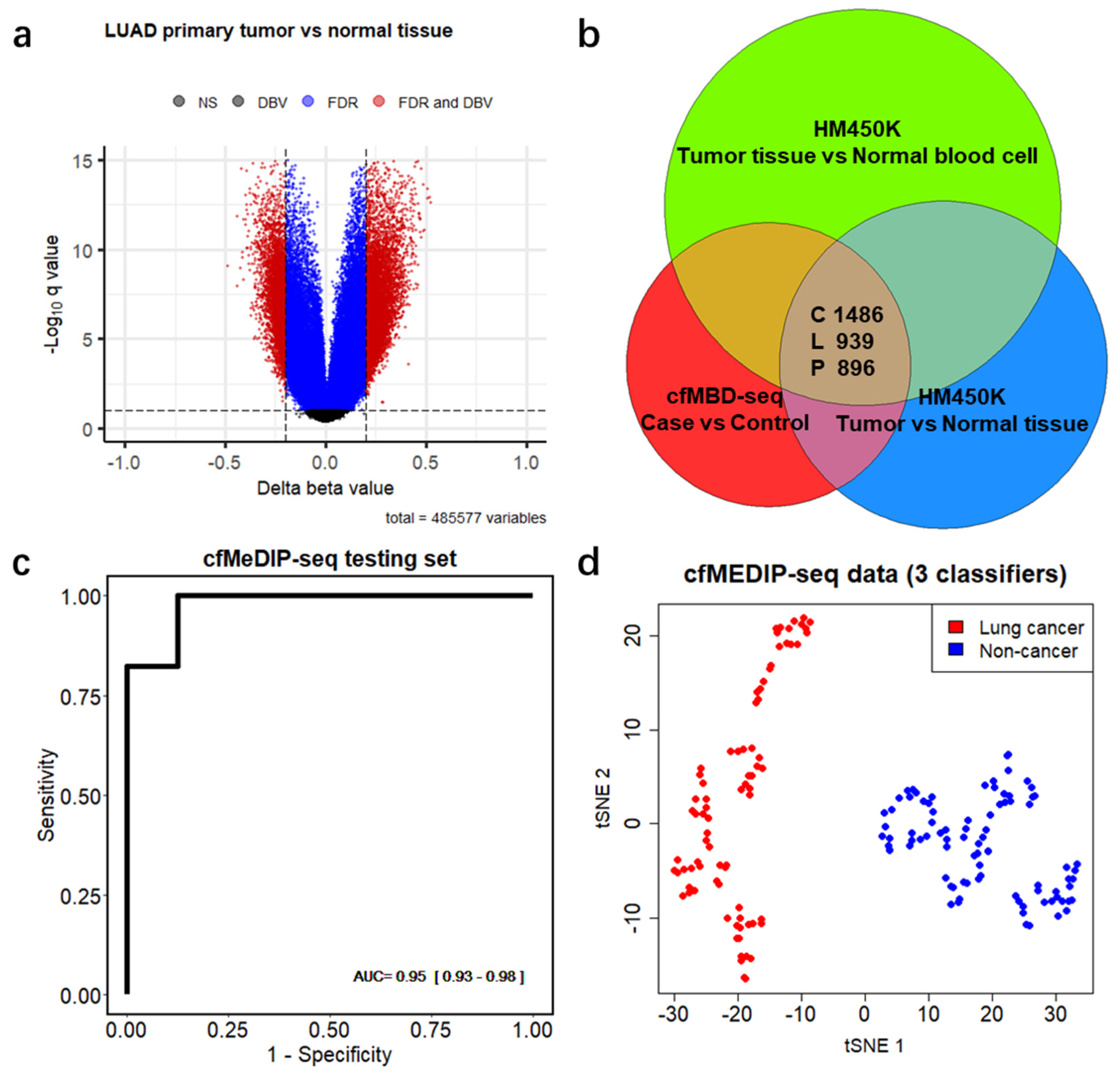
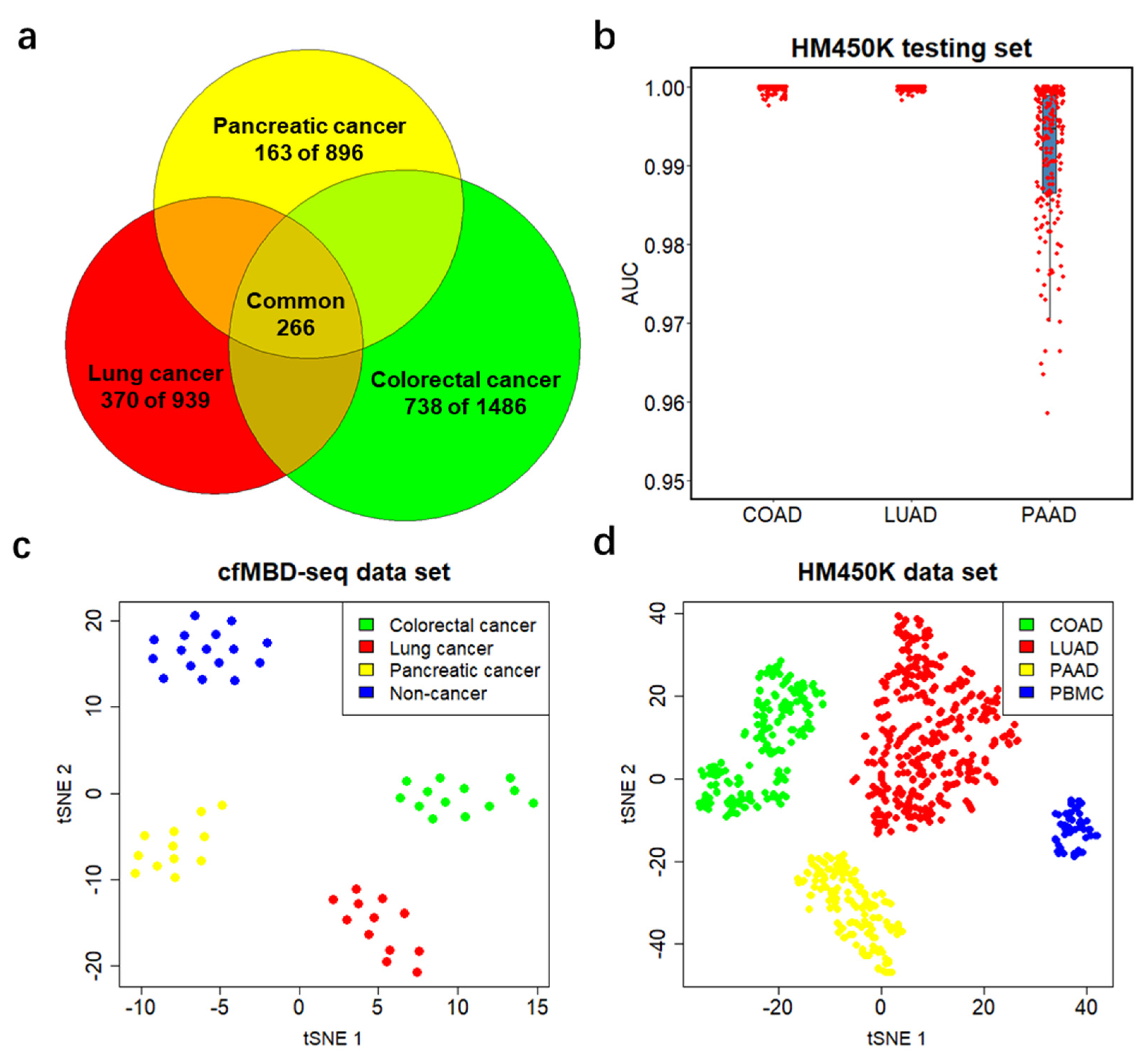
3.3. Significant Overlap between Tumor Tissue-Derived and cfDNA-Derived Differentially Methylated CpG Islands
3.4. Differentially Methylated CpG Islands for Early Lung Cancer Detection
3.5. Differentially Methylated CpG Islands for Cancer Classification
3.6. Gene Annotation of Differentially Methylated CpG Islands
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar] [CrossRef] [Green Version]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Wei, W.; Ye, Z.; Zheng, J.; Xu, R.H. Liquid Biopsy of Methylation Biomarkers in Cell-Free DNA. Trends Mol. Med. 2021, 27, 482–500. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.; Sun, K. Epigenetic Biomarkers in Cell-Free DNA and Applications in Liquid Biopsy. Genes 2019, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Warren, J.D.; Xiong, W.; Bunker, A.M.; Vaughn, C.P.; Furtado, L.V.; Roberts, W.L.; Fang, J.C.; Samowitz, W.S.; Heichman, K.A. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011, 9, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nian, J.; Sun, X.; Ming, S.; Yan, C.; Ma, Y.; Feng, Y.; Yang, L.; Yu, M.; Zhang, G.; Wang, X. Diagnostic Accuracy of Methylated SEPT9 for Blood-based Colorectal Cancer Detection: A Systematic Review and Meta-Analysis. Clin. Transl. Gastroenterol. 2017, 8, e216. [Google Scholar] [CrossRef]
- Sprang, M.; Paret, C.; Faber, J. CpG-Islands as Markers for Liquid Biopsies of Cancer Patients. Cells 2020, 9, 1820. [Google Scholar] [CrossRef]
- Chan, R.F.; Shabalin, A.A.; Xie, L.Y.; Adkins, D.E.; Zhao, M.; Turecki, G.; Clark, S.L.; Aberg, K.A.; van den Oord, E. Enrichment methods provide a feasible approach to comprehensive and adequately powered investigations of the brain methylome. Nucleic Acids Res. 2017, 45, e97. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019, 14, 2749–2780. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, P.V.; Berchuck, J.E.; Korthauer, K.; Spisak, S.; Nassar, A.H.; Abou Alaiwi, S.; Chakravarthy, A.; Shen, S.Y.; Bakouny, Z.; Boccardo, F.; et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 2020, 26, 1041–1043. [Google Scholar] [CrossRef]
- Lasseter, K.; Nassar, A.H.; Hamieh, L.; Berchuck, J.E.; Nuzzo, P.V.; Korthauer, K.; Shinagare, A.B.; Ogorek, B.; McKay, R.; Thorner, A.R.; et al. Plasma cell-free DNA variant analysis compared with methylated DNA analysis in renal cell carcinoma. Genet. Med. 2020, 22, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Soupir, A.C.; Wang, L. Cell-free DNA methylome profiling by MBD-seq with ultra-low input. Epigenetics 2021, 1–14. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabalin, A.A.; Hattab, M.W.; Clark, S.L.; Chan, R.F.; Kumar, G.; Aberg, K.A.; van den Oord, E. RaMWAS: Fast methylome-wide association study pipeline for enrichment platforms. Bioinformatics 2018, 34, 2283–2285. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. GitHub 2021. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 6 November 2021).
- Marini, F.; Binder, H. pcaExplorer: An R/Bioconductor package for interacting with RNA-seq principal components. BMC Bioinform. 2019, 20, 331. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Krijthe, J.H. Rtsne: T-Distributed Stochastic Neighbor Embedding using a Barnes-Hut Implementation. GitHub 2015. Available online: https://github.com/jkrijthe/Rtsne (accessed on 6 November 2021).
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 3164. [Google Scholar] [CrossRef]
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. Role of HOX Genes in Stem Cell Differentiation and Cancer. Stem Cells Int. 2018, 2018, 3569493. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Zhang, C.; Tian, Y.; Du, X.; Wang, Q.; Zhao, H. Expression and Function of WNT6: From Development to Disease. Front. Cell Dev. Biol. 2020, 8, 558155. [Google Scholar] [CrossRef]
- Rolfo, C.; Russo, A. Liquid biopsy for early stage lung cancer moves ever closer. Nat. Rev. Clin. Oncol. 2020, 17, 523–524. [Google Scholar] [CrossRef]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.K.; Janne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [Green Version]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci. Transl. Med. 2012, 4, 162ra154. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wang, L. Cell-Free DNA Methylation Profiling Analysis-Technologies and Bioinformatics. Cancers 2019, 11, 1741. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Jiang, P.; Chan, K.C.; Wong, J.; Cheng, Y.K.; Liang, R.H.; Chan, W.K.; Ma, E.S.; Chan, S.L.; Cheng, S.H.; et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.C.; Jiang, P.; Chan, C.W.; Sun, K.; Wong, J.; Hui, E.P.; Chan, S.L.; Chan, W.C.; Hui, D.S.; Ng, S.S.; et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 18761–18768. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Consortium, C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Fang, H.B.; Jiang, F. An epigenetic classifier for early stage lung cancer. Clin. Epigenet. 2018, 10, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Zhao, Q.; Wei, W.; Zheng, L.; Yi, S.; Li, G.; Wang, W.; Sheng, H.; Pu, H.; Mo, H.; et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci. Transl. Med. 2020, 12, eaax7533. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.H.; Wei, W.; Krawczyk, M.; Wang, W.; Luo, H.; Flagg, K.; Yi, S.; Shi, W.; Quan, Q.; Li, K.; et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater. 2017, 16, 1155–1161. [Google Scholar] [CrossRef]
- Vrba, L.; Futscher, B.W. A suite of DNA methylation markers that can detect most common human cancers. Epigenetics 2018, 13, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.S.; Coolen, M.W.; Stirzaker, C.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Clark, S.J. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 2011, 6, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CpG Islands | Size | Coefficients | Gene | Location | |
|---|---|---|---|---|---|
| Colorectal cancer | |||||
| chr2:29337984-29338909 | 926 | −9.83 | CLIP4 | Promoter | |
| chr2:100937780-100939059 | 1280 | −29.19 | LONRF2 | Promoter | |
| chr6:125283125-125284389 | 1265 | 7.04 | RNF217 | Promoter | |
| Lung cancer | |||||
| chr2:66672432-66673636 | 1205 | −9.04 | MEIS1 | Gene body | |
| chr2:71503548-71504233 | 686 | −5.54 | ZNF638 | Promoter | |
| chr2:219736133-219736592 | 460 | 5.80 | WNT6 | Gene body | |
| chr4:140655963-140657135 | 1173 | 13.48 | MGST2 | Gene body | |
| chr4:174427892-174428192 | 301 | 7.82 | \ | Intergenic | |
| chr5:40679503-40682081 | 2579 | −43.06 | PTGER4 | Promoter | |
| chr7:27265159-27265493 | 335 | −7.47 | \ | Intergenic | |
| chr7:65037625-65037864 | 240 | −14.04 | \ | Intergenic | |
| chr8:124172801-124173541 | 741 | −14.47 | \ | Intergenic | |
| chr9:96108467-96108992 | 526 | 12.82 | C9orf129 | Promoter | |
| chr12:54408427-54408713 | 287 | −5.11 | \ | Intergenic | |
| chr12:58021295-58022037 | 743 | 15.82 | B4GALNT1 | Gene body | |
| chr13:28549840-28550246 | 407 | 5.60 | \ | Intergenic | |
| chr17:46691521-46692097 | 577 | −4.75 | HOXB8 | Gene body | |
| chr17:59539363-59539834 | 472 | −12.18 | TBX4 | Gene body | |
| chr17:70112825-70114271 | 1447 | 9.87 | SOX9 | Promoter | |
| Pancreatic cancer | |||||
| chr1:44883137-44884272 | 1136 | −10.54 | RNF220 | Gene body | |
| chr1:50798668-50799536 | 869 | 8.22 | \ | Intergenic | |
| chr5:92939796-92940216 | 421 | 7.85 | \ | Intergenic | |
| chr10:11059443-11060524 | 1082 | 10.34 | CELF2 | Promoter | |
| chr11:20177609-20178824 | 1216 | 6.63 | DBX1 | Gene body | |
| chr12:114881650-114881937 | 288 | −27.52 | \ | Intergenic | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Soupir, A.C.; Schlick, B.D.; Teng, M.; Sahin, I.H.; Permuth, J.B.; Siegel, E.M.; Manley, B.J.; Pellini, B.; Wang, L. Cancer Detection and Classification by CpG Island Hypermethylation Signatures in Plasma Cell-Free DNA. Cancers 2021, 13, 5611. https://doi.org/10.3390/cancers13225611
Huang J, Soupir AC, Schlick BD, Teng M, Sahin IH, Permuth JB, Siegel EM, Manley BJ, Pellini B, Wang L. Cancer Detection and Classification by CpG Island Hypermethylation Signatures in Plasma Cell-Free DNA. Cancers. 2021; 13(22):5611. https://doi.org/10.3390/cancers13225611
Chicago/Turabian StyleHuang, Jinyong, Alex C. Soupir, Brian D. Schlick, Mingxiang Teng, Ibrahim H. Sahin, Jennifer B. Permuth, Erin M. Siegel, Brandon J. Manley, Bruna Pellini, and Liang Wang. 2021. "Cancer Detection and Classification by CpG Island Hypermethylation Signatures in Plasma Cell-Free DNA" Cancers 13, no. 22: 5611. https://doi.org/10.3390/cancers13225611
APA StyleHuang, J., Soupir, A. C., Schlick, B. D., Teng, M., Sahin, I. H., Permuth, J. B., Siegel, E. M., Manley, B. J., Pellini, B., & Wang, L. (2021). Cancer Detection and Classification by CpG Island Hypermethylation Signatures in Plasma Cell-Free DNA. Cancers, 13(22), 5611. https://doi.org/10.3390/cancers13225611