Simple Summary
Liver cancer is one of the most devastating human malignancies worldwide, especially in Asia, where over 70% of new cases are diagnosed. Most liver cancers are classified as hepatocellular carcinoma (HCC). HCC patients usually present at an advanced stage and have very poor prognosis due to the inaccessibility of curative treatments and ineffective systemic therapies. Fortunately, recent clinical trials using checkpoint inhibitor (ICI) immunotherapy have obtained promising results to significantly prolong the overall survival of patients and improve quality of life. In this review, we summarize the recent efforts of ICI-related clinical trials and also point out the future directions of ICI-related immunotherapy for HCC.
Abstract
HCC usually arises from a chronic inflammation background, driven by several factors including fatty liver, HBV/HCV viral infection and metabolic syndrome. Systemic treatment for advanced HCC remains disappointing due to its strong resistance to chemotherapy and even to tyrosine kinase inhibitors (TKIs). Recently, the use of ICI therapy has revolutionized the systemic treatment of advanced HCC. For the first time, clinical trials testing ICIs, anti-CTLA-4 and anti-PD1/PDL1 reported a survival benefit in patients with sorafenib resistance. However, it took four more years to find the right combination regimen to use ICI in combination with the anti-angiogenic agent bevacizumab to substantially prolong overall survival (OS) of patients with advanced HCC after sorafenib. This review provides a comprehensive history of ICI therapy in HCC, up-to-date information on the latest ICI clinical trials, and discusses the recent development of novel ICIs that would potentially lead to a new checkpoint blockade therapy for advanced HCC.
1. Introduction
Hepatocellular carcinoma is the most prevalent form of liver cancer in the world [1]. Annually, HCC affects approximately 900,000 individuals, and over 70% of new cases are diagnosed in Asia [2]. The etiology of HCC is complicated due to the multiple risk factors involved [3]. HCC usually arises from a background of chronic liver disease caused by alcohol abuse, metabolic syndrome, hepatitis B or C infections and/or aflatoxin exposure that eventually scars the liver parenchyma, leading to the irreversible condition of liver cirrhosis and the subsequent development of HCC [4]. Most HCC patients are diagnosed with advanced disease, and the majority of them are unresectable due to the early dissemination of cancer cells inside the liver. HCC tends to grow near blood vessels such as the portal vein or hepatic vein, which makes surgery an impossible task. α-Fetoprotein (AFP) is commonly used as a serum biomarker for early detection of HCC and also for evaluation of the prognosis and monitoring of response to therapy [5,6]. Systemic chemotherapy for many years is ineffective and therefore not an option for HCC, due to the highly resistant nature of HCC. Locoregional therapies including percutaneous ablation, transarterial chemoembolization (TACE) and radioembolization serve as the main alternative therapies, but largely depend on tumor location, burden and other complications [7]. Ablation is the first line option over surgery for unresectable HCC; however, treatment outcome is still disappointing and recurrence is often seen. In 2008, a tyrosine kinase inhibitor (TKIs), sorafenib, was the first approved targeted therapy to be used as a first-line drug to treat advanced HCC [8]. Subsequently, three additional TKIs, lenvatinib, regorafenib and cabozantinib, have been approved and become available for use in first-line and second-line settings and found to provide beneficial effects and prolonged survival [9,10]. Despite of the application of TKI therapy, median survival with advanced HCC remains unsatisfactory at less than two years [7,11]. Recently, ramucirumab, a therapeutic monoclonal antibody drug that acts against vascular endothelial growth factor (VEGF) receptor 2, has shown significant survival benefits in patients with increased AFP (>400 ng/mL) after sorafenib [12]. A VEGF-neutralizing antibody, bevacinumab, has also proven effective in a single-agent phase II trial in HCC patients. However, serious bleeding complications were observed in 11% [13].
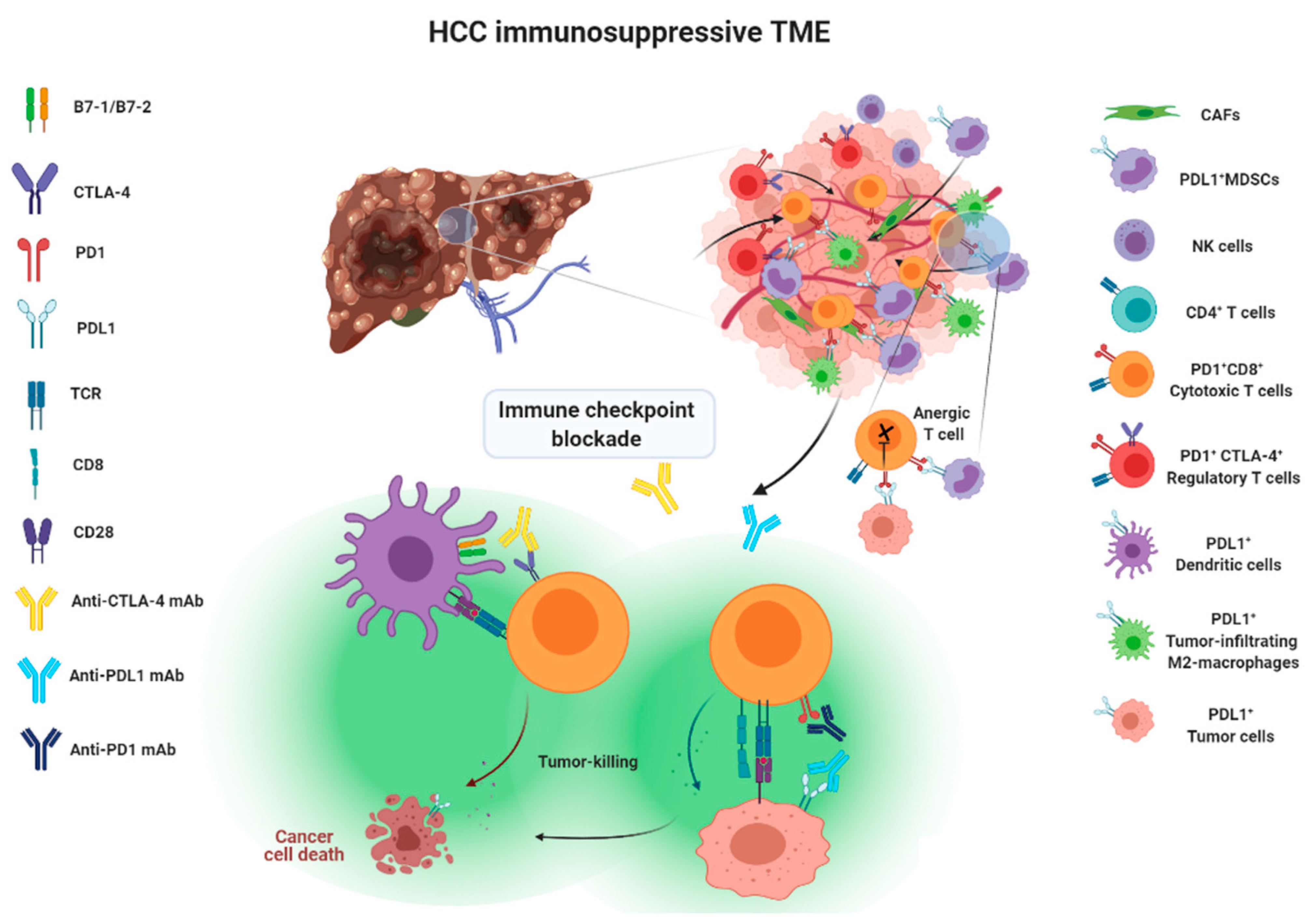
Immune surveillance plays an important role in identifying and eliminating normal cells that become malignant. As when fighting invading pathogens, the innate and adaptive immune systems work together to form an anti-tumor army to destroy cancer cells. However, the interplay between cancer cells, stromal cells, and infiltrating immune cells eventually creates an immunosuppressive tumor microenvironment (TME) that leads to immune evasion, which was previously considered impossible to reverse. There are two main aspects explaining the formation of an immunosuppressive TME. First, as HCC progresses, cancer cells recruit immune cells such as myeloid-derived suppressor cells (MDSCs) or M2 tumor-associated macrophages (TAMs) and FoxP3+ regulatory T cells (Treg), which are known to help the tumor grow better, by secreting chemokines, cytokines and growth factors to form a tumor-promoting niche. As a result, the accumulation of tumor-promoting immunes cells eventually comprises an area of anti-tumor immunity. Second, upregulation of co-inhibitory molecules such as immune checkpoint ligand PDL1 and increased expression of tolerance-related enzymes such as indoleamine 2,3-deoxygenase (IDO) and arginase-1 in cancer cells or tumor-infiltrating immune cells also contribute to the formation of immunosuppressive TME. In addition, downregulation of tumor-associated antigens (TAAs), also known as tumor antigen escape [14,15], and reduced recognition of TAAs by immune cells through alterations in the antigen-processing machinery both play a significant role in the promotion of tumor progression [16]. Therefore, immunotherapies aiming to reverse and overcome the immunosuppressive TME in order to effectively enhance the activity of tumor-killing immune cells point out the future direction of HCC therapy.
2. Tumor Microenvironment of HCC
The liver is the organ responsible for the detoxification of gut-derived blood and systemic circulation. Therefore, the frequent exposure of liver cells to food antigens and to microbial products generated by gut bacteria shapes the dynamic complexities of a liver microenvironment that fosters immune tolerance [17]. This tolerogenic milieu is maintained by liver antigen-presenting cells (APCs), including resident kuffer cells (KCs), sinusoidal endothelial cells (SECs) and stellate cells (SCs), through the secretion of an array of immunosuppressive cytokines, chemokines and growth factors [1]. Kuffer cells are known to express immunosuppressive cytokine IL-10 and IDO, and prostaglandins to promote Treg activation [18,19]. Myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs) and regulatory T cells also produce IL-10 to attenuate the ability of APCs to stimulate T cells and to promote PDL1 expression in monocytes [20]. TGF-β is a well-known soluble factor that attenuates the anti-tumor response by inhibiting the activation of dendritic cells (DCs) [21] and polarizing macrophages towards the M2 phenotype [22], as well as inducing Treg cell activation [23]. In HCC TME, TGF-β is mainly produced by cancer cells, Treg cells and macrophages [17]. High serum TGF-β has been linked to poor prognosis of patients with HCC after sorafenib treatment [24]. SCs-derived hepatocyte growth factor (HGF) also promotes the infiltration and accumulation of MDSCs and Treg cells inside the TME [25,26]. Vascular endothelial growth factor (VEGF) produced by cancer cells or MDSCs plays an essential role in promoting angiogenesis, leading to the formation of an abnormal tumor vasculature that not only serves as a barrier for cytotoxic T lymphocytes (CTLs) but disables them by expressing PDL1 and Fas ligand [27,28,29]. Aside from its role in inducing abnormal tumor vasculature, Courau, et al. showed that VEGF and TGF-β could cooperatively foster immunotolerant TME by blunting the antigen-presenting functions of DC and generating MDSCs [30].
3. Current and Ongoing Strategies of Immune Checkpoint Blockade for HCC
Co-inhibitory molecules expressed by the effector lymphocytes fall into a category of immune checkpoint which serves the purpose of preventing overactivation of lymphocytes upon the engagement of APCs like DCs or macrophages [31]. To evade immune surveillance, tumor cells exploit this mechanism to express the corresponding ligands of co-inhibitory molecules to blunt effector T cells or macrophages. Program death 1 (PD1), cytotoxic T lymphocyte-associated antigen 4 (CTLA4), T cell immunoglobulin and mucin domain containing-3 (TIM3), lymphocyte-activation gene 3 (LAG3), and siglec-10 are among the most extensively studied immune checkpoints [32,33]. PD1 has been found to be expressed by activated T cells, nature killer (NK) cells, MDSCs, monocytes and DCs [34,35,36]. Its corresponding ligand, PDL1, has been found in tumor cells, stromal cells, and myeloid lineage cells like macrophages and DCs [36]. CTLA4 is predominantly expressed by Treg cells and only upregulated in activated T cells [37,38]. In addition, the sialic acid-binding immunoglobulin (Ig)-like lectins 10 (siglec-10) has recently been identified as a new class of co-inhibitory molecule expressed by macrophages to interact with its corresponding ligand, CD24, expressed on the tumor surface [33,39].
Approaches taken to blocking the above-mentioned immune checkpoint receptor/ligand interactions have been considered as a form of immune checkpoint blockade therapy, illustrated in Figure 1.
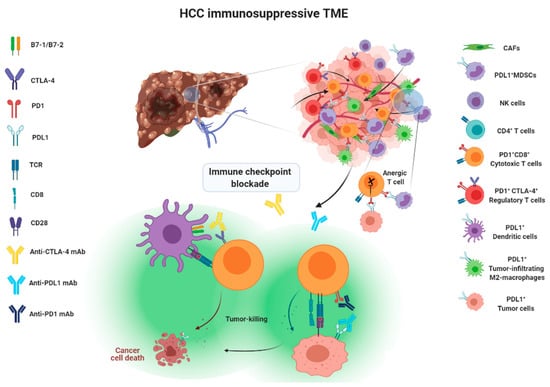
Figure 1.
Action mechanisms of immune checkpoint blockade therapy.
3.1. Monotherapy
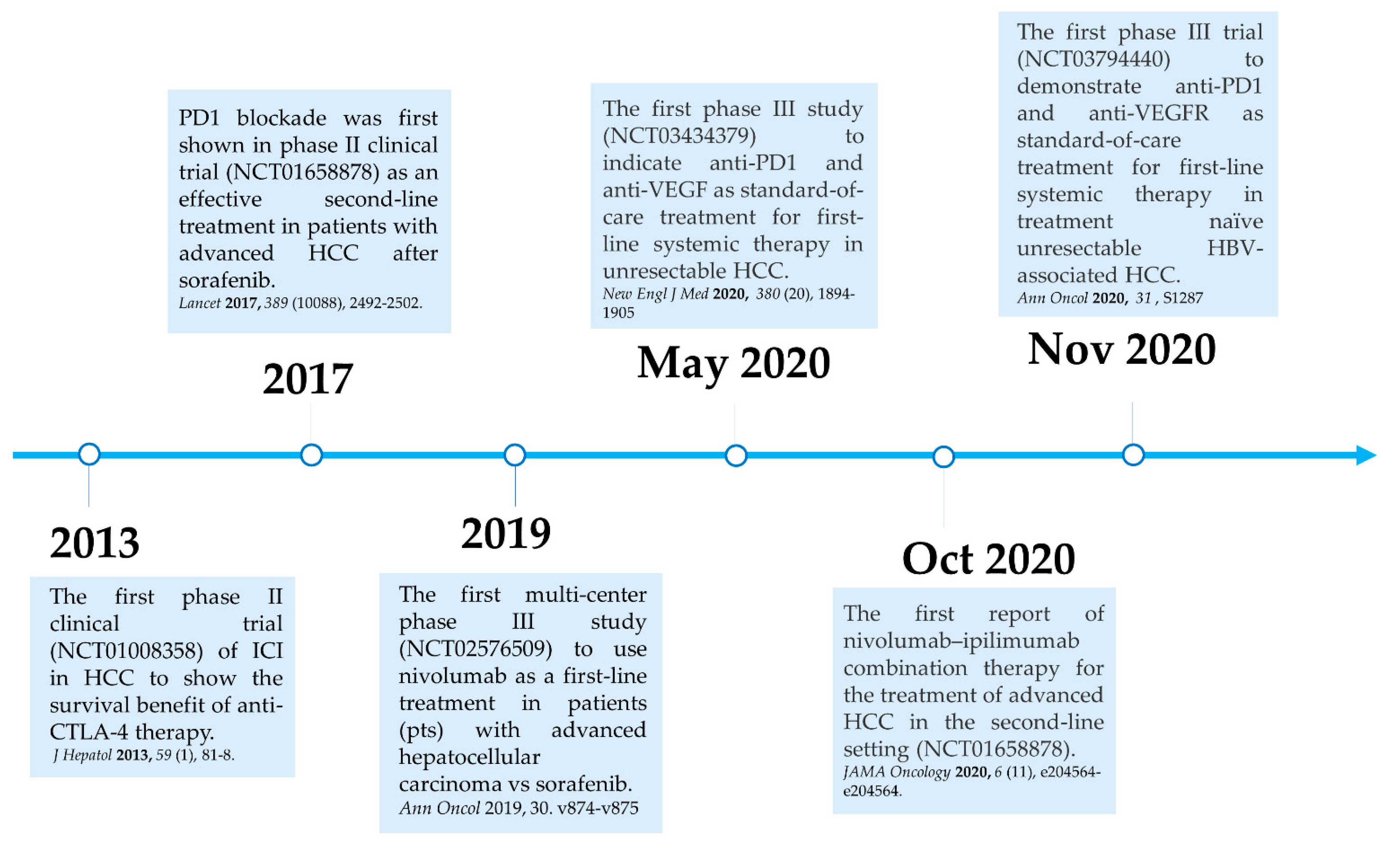
Here, we summarize the current milestones regarding recent clinical trials testing ICIs as a potential systemic treatment for advanced HCC (Figure 2). The first immune checkpoint inhibitor (ICI), ipilimumab, the anti-CTLA-4 monoclonal antibody (mAb), was approved by the U.S. Food and Drug Administration (FDA) in March 2011 for the treatment of patients with advanced melanoma [40]. In 2013, a pilot clinical trial involving 20 patients with advanced HCC and a background of chronic hepatitis C virus (HCV) infection who received tremelimumab treatment, another anti-CTLA-4 mAb, showed promising results in terms of safety, antitumor and antiviral activity [41]. This encouraging result has led to the approval of another ICI, the PD1 inhibitor nivolumab, for the treatment of patients after sorafenib failure in the CheckMate 040 phase I/II clinical trial. In the CheckMate 040 trial (n = 262), durable objective response rate (ORR), defined as the sum of complete (CR) and partial (PR) response rates, was observed in 20% (95% CI 15–26) in patients treated with nivolumab 3 mg/kg in the dose-expansion phase and 15% (95% CI 6–28) in the dose-escalation phase, The median overall survival (OS) in sorafenib-experienced patients was 16.5 months. Further, the two-year survival rate among the responders was over 80% [42]. Soon after, nivolumab was approved by the FDA as a second-line systemic therapy for advanced HCC after sorafenib. Another PD1 inhibitor, pembrolizumab. was also shown effective and tolerable in patients with advanced HCC after sorafenib in a non-randomized, open-label phase II trial. ORR was recorded in 17% 95% CI 11–26) of patients previously treated with sorafenib [43]. Given the consistent results in terms of anti-tumor activity and safety from different PD1 mAb, a randomized, double-blind, phase III trial (KEYNOTE 240) (n = 413) testing pembrolizumab versus placebo after sorafenib failure was launched in a second-line setting. Median OS was 13.9 months (95% CI 11.6–16) in the pembrolizumab treatment group and 10.36 months (95% CI 8.3–13.5) in the placebo group, and a statistically significant survival benefit was observed (Hazard ration HR 0.78; p = 0.0238) in the final analysis [44]. Although, OS and progression-free survival (PFS) did not reach the prespecified criteria, the results were in line with KEYNOTE 224, indicating a favorable risk-to-benefit ratio in the pembrolizumab group. Table 1 summarizes current important phase III trials involving ICI therapy as a major treatment modality.
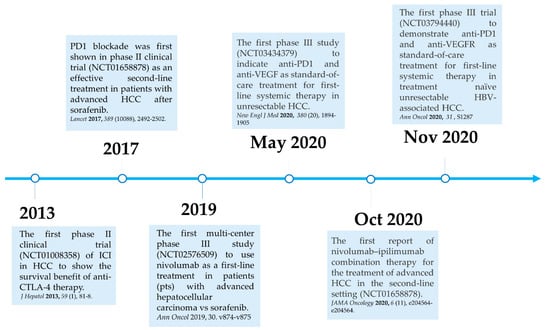
Figure 2.
Milestones for immune checkpoint therapy for patients with advanced HCC.

Table 1.
Current phase III trials involving ICI monotherapy and combinations of ICIs and TKI or VEGF inhibitors.
In 2019, the phase III study CheckMate 459 compared the clinical efficacy and safety of nivolumab with sorafenib in 743 patients with treatment-naïve advanced HCC in a first-line setting [52]. Patients were randomized at a 1:1 ratio to either the nivolumab or sorafenib arm, with a follow-up of 22.8 months. ORR was 15% for patients who received nivolumab and 7% for those who received sorafenib. Prolonged survival was observed in patients after nivolumab, with a median OS of 16.4 months (95% CI 13.9–18.4), compared to patients after sorafenib with median OS of 14.7 months (95% CI 11.9–17.2) (HR 0.85; 95% CI 0.72–1.02; p = 0.0752). Although the predefined threshold of statistical significance for OS was not met (HR 0.84, p = 0.0419) [53], other end point parameters favored nivolumab over sorafenib, with a better disease control (median 7.5 months versus 5.7 months) and safety profile with fewer treatment-related adverse effects (22% versus 49%). Long-term follow-up of CheckMate 459 has also shown that first-line nivolumab monotherapy demonstrated clinical meaningful benefit in patients with advanced HCC at a minimum follow-up of 33.6 months, and that nivolumab had a more favorable safety profile and better preservation of liver function over the course of treatment as compared with sorafenib [53]. Given the promising results from the initial attempts with ICIs in HCC, different mAb targeting PD1/PDL1 have been developed and approved by the U.S. FDA for the treatment of patients with advanced HCC, and a number of combination strategies have been considered and tested in current clinical trials [47,48,49,50,51]. (Table 1).
3.2. Combination Therapy
In the CheckMate 040 randomized, open-label, multicohort phase I/II trial testing nivolumab plus ipilimumab in patients with unresectable HCC after sorafenib, patients were randomized at a 1:1:1 ratio to three treatment arms: (A) nivolumab 1 mg/kg plusipilimumab 3 mg/kg every three weeks (4 doses in total) followed by nivolumab 240 mg; (B) nivolumab 3 mg/kg plus ipilimumab 1 mg/kg followed by nivolumab 240mg; or (C) nivolumab 3 mg/kg every two weeks plus ipilimumab 1 mg/kg every 6 weeks. Although this study reported higher rates of adverse events with combination therapy of nivolumab and ipilimumab than previously reported in nivolumab monotherapy [42], nivolumab plus ipilimumab had manageable safety, promising objective response rate, and durable response with 32% (16 of 50 patients) ORRs in arm A, 31% (15 of 49 patients) in arm B, and 31% (15 of 49) in arm C. Patients in arm A had the best median OS of 22.8 months, and highest 30-month survival rate of 44% with better health improvement compared to that in the other two arms [45]. Four patients in arm A (4 of 49 patients) had a complete response, as did three patients in arm B (3 of 49 patients) [45]. Based on this clinical finding, the arm A regimen received accelerated approval by the U.S. FDA and further investigation of this regimen as first-line treatment for HCC is underway. Moreover, these results showed that the PD1/PDL1 and CTLA-4 pathways have a distinct role in modulating immune activity [54], and also confirmed that CTLA-4 blockade is effective in HCC, as in previous clinical trials [41,55]. Unlike PD1 and PDL1 inhibitors, dosing and timing seem crucial for anti-CTLA-4 therapy.
Another clinical trial attempting to target both PD1 and CTLA-4 with the different mAbs durvalumab (a PD1 inhibitor) and tremelimumab (a CTLA-4 inhibitor) in unresectable HCC also showed promising results; the combination regimen (tremelimumab 300 mg plus durvalumab 1,500 mg followed by durvalumab 1500 mg once every four weeks) had the best benefit-to-risk profile, with one patient having a complete response (1.3%, 1/75 patients), 17 patients a partial response (22.7%, 17/75 patients), and 16 patients stable (21.3%, 16/75 patients) [56]. Based on the phase I/II results, the randomized phase III HIMALAYA is now under way to assess the efficacy and safety of durvalumab plus tremelimumab both in combination and as monotherapy versus sorafenib in treatment-naïve patients with unresectable HCC [47].
Although experiences from ICI clinical trials show that no more than 20% of patients respond to ICI monotherapy, the treatment efficacy and ORRs of ICI therapy are still far better than previous TKI-related targeted therapies [57]. Moreover, combined therapy using nivolumab and ipilimumab has displayed the best ORR (32%) compared to any form of systemic treatment for HCC.
3.3. Combination Therapy of ICIs and VEGF Inhibitor
VEGF secreted by cancer cells is important in fostering an immunosuppressive TME, not only because of its role in recruiting endothelial cells to promote tumor angiogenesis (leading to the formation of abnormal tumor vasculature), but also for its suppressive role in inhibiting the differentiation and maturation of DCs. [29]. In addition, VEGF is also known for its suppressive role in inhibiting the differentiation and maturation of DCs [58]. Therefore, strategies aiming at combining anti-VEGF related signaling with ICI therapy could be the ideal regimen to further overcome the immunosuppressive nature of the TME by inducing tumor vascular normalization as well as enhancing DC maturation, optimizing the treatment efficacy of ICI therapy. Recently, two clinical trials that used anti-VEGF and anti-VEGFR2 antibodies, respectively, as add-on therapies to ICI monotherapy in HCC have shown encouraging results (Table 2). In IMbrave150, a global, randomized, open-label phase III clinical trial, 501 patients were randomized at a 2:1 ratio to receive a standard dose of atezolizumab (1200 mg) followed by a high dose (15 mg/kg) of bevacizumab (anti-VEGF antibody) every three weeks, or sorafenib. This trial did not include atezolizumab or bevacizumab monotherapy [51]. This study met its primary end points (both PFS and OS) at the first interim analysis after a median follow-up of 8.6 months, and therefore stopped. Survival in the patient arm receiving atezolizumab plus bevacizumab still had not been reached at the time the first interim ended; however, improved OS (HR 0.58; 95% CI 0.42–0.79; p = 0.00006) and improved PFS (HR 0.59; 95% CI 0.47–0.76; p < 0.00001) were observed. It was the first time that a phase III trial study had outperformed sorafenib in a first-line setting. Furthermore, updated OS data from IMbrave150 showed even more promising results. The median OS was 19.2 months in the combination therapy arm versus 13.4 months in the sorafenib arm. A survival benefit with combination therapy over sorafenib was observed; 52% of patients who received atezolizumab plus bevacizumab and 40% of patients who received sorafenib had survived after 18 months. The updated ORR in the combination therapy arm was 29.8% (95% CI 24.8–35.0) according to RECIST 1.1 criteria, which was superior to that in the sorafenib arm (11.3%; 95% CI 6.9–17.3), and 18 patients (5.5%, 18/326 patients) in the atezolizumab plus bevacizumab group had a complete response, as compared to no patients in the sorafenib group. Notably, patient health-related quality of life was also significantly improved. A longer median time to deterioration was observed in the combination treatment arm than in the sorafenib arm, 11.2 months versus 3.6 months (HR 0.63, 95% CI 0.46–0.85), respectively [59]. More recently, a similar result was also obtained from a phase II/III trial in China testing a combination of another ICI, sintilimab, and IBI305, a bevacizumab biosimilar, versus sorafenib as a front-line treatment for patients with unresectable HBV-associated HCC [48]. This study also proved that patients with combination treatment (median OS: not reached) also survived much longer than patients with sorafenib (median OS: 10.4 months). Due to the lack of a single-agent arm in the above studies, it is uncertain whether anti-PD1 or anti-VEGF therapy contributed more to the survival benefit of the combination treatment. Nevertheless, the previous phase 1b study showed that the response rates and PFS of the atezolizumab plus bevacizumab arm were significantly better than the bevacizumab treatment arm, suggesting that anti-PD1 may be an indispensable factor in designing future combined regimens for HCC [60].
Table 2.
Safety profiles of immune checkpoint inhibitors for HCC.
3.4. Potential Novel Checkpoint Inhibitors
Aside from PD1/PDL1 and CTLA-4, here we discuss other important co-inhibitory molecules expressed by T cells which have been identified as potential immune checkpoints that modulate T cell activation. T-cell immunoglobulin and mucin-domain containing-3 (TIM3) is a membrane-bound protein that is originally expressed in CD8+ cytotoxic T cells and interferon-γ-producing CD4+ T helper 1 (Th1) cells [65]. Initially, the function of TIM3 was associated with autoimmune disease. Blocking of TIM3 with TIM3-specific antibody or administration of TIM-3 immunoglobulin (Ig) fusion protein resulted in Th1 cell and macrophage hyperactivation in an experimental autoimmune encephalomyelitis (EAE) mouse model [66]. Later, TIM3 expression was found in several immune cells, including Treg cells [67], myeloid cells [68], natural killer (NK) cells [69] and dendritic cells (DCs) [65]. Notably, co-expression of TIM3 and PD1 has been observed in dysfunctional Th1 cells that express low levels of IL-2 and IFN-γ in the preclinical model [70]. A synergistic anti-tumor effect of combined anti-PD1 and anti-TIM3 immunotherapy has been observed in solid tumors in mouse models [70,71]. Therefore, combination treatment of TIM3 and PD1 or CTLA-4 inhibitor could serve as an attractive ICI regimen for HCC. Recently, a phase II trial assessing the efficacy and safety of anti-PD1 and anti-TIM3 combination therapy (NCT03680508) has been launched; the results are still being awaited.
Another potential immune checkpoint is lymphocyte-activation gene 3 (LAG3). The structure of LAG3 is closely related to CD4, and they share the same ligand, MHC-II [72]. LAG3 expression is found on activated CD4+ T cells [73], CD8+ cells [74], Treg cells [75] and plasmacytoid dendritic cells (pDCs) [76]. High levels of LAG3 have been found in tumor-infiltrating lymphocytes (TILs) [77]. Unlike PDL1, high expression of LAG3 has been found in several solid tumors, including HCC [78]. LAG3 also has been shown to have a negative impact on autoimmunity. Loss of LAG3 significantly accelerates disease progression of type 1 diabetes in Non-Obese-Diabetic (NOD) mouse models [79], and is associated with autoimmune disease. Importantly, interplay between LAG3 and other immune checkpoints has been noted. LAG3 could work cooperatively with PD1 or CTLA-4 to suppress MHC-II-mediated T cell receptor (TCR) signaling and subsequent T cell action, making it an ideal target for ICI therapy [78,80]. In an ovarian cancer model, a synergistic anti-tumor effect of combinatory treatment of LAG3 and PD1 mAb shed light on future potential combination therapies [80]. Several clinical trials are ongoing to evaluate the clinical benefits of LAG3 mAb and a soluble LAG3 Ig as well as combination therapy using anti-LAG3 and anti-PD1 mAb in an array of human malignancies [81,82]. Given that upregulation of LAG3 has been found in tumor-infiltrating CD8+ T cells and HCC cells [78,83], potential clinical trials for HCC can be expected in the near future.
Another important aspect to examine is that compensatory upregulation of TIM3 and LAG3 may confer resistance to anti-PD1/PDL1 treatment. In a preclinical mouse model, Oweida et al. demonstrated that TIM3 upregulation was observed in tumor-infiltrating CD4 and CD8 T cells in murine head and neck squamous cell carcinoma (HNSCC) tumors treated with radiotherapy (RT) and anti-PDL1 therapy. Combined treatment with anti-TIM3, anti-PDL1 and RT led to the promotion of T cell cytotoxicity, decreased Treg and significant tumor regression as compared with anti-PDL1 plus RT treatment [84]. In an effort to address the resistance mechanisms of PD1 blockade, Limagne et al. showed that accumulation of galetin-9-expressing monocytic MDSCs (mMDSCs) and TIM3-expressing CD8 T cells was found in lung cancer patients with resistance to anti-PD1 therapy, and may play a crucial role in resistance to PD1 blockade. They demonstrated that anti-TIM3 antibody in vitro could reverse resistance to anti-PD1 in peripheral blood mononuclear cells (PBMCs) isolated from lung cancer patients. Moreover, galetin-9-expressing mMDSCs could impede TIM3+ CD8 T cell activity to reduce anti-PD1 treatment efficacy [85]. In addition, Jikova et al. analyzed fresh HCC biopsies and peripheral blood samples from 21 HCC patients treated with sorafenib or PD1/PDL1 blockade therapy to show that non-responders tended to have TIM3 and LAG3 upregulation on circulating T cells compared with responders [86].
Taken together, blocking the compensatory upregulation of other checkpoint inhibitors such as TIM3 and LAG3 after anti-PD1/PDL1 treatment could be an important pharmaceutical strategy to overcome primary and secondary resistance to PD1/PDL1 blockade in patients with advanced HCC in the future.
4. Challenges and Opportunities in ICI Therapy for HCC
4.1. Management of ICI-Related Immune-Mediated Adverse Effects (IMAEs)
Inhibitory checkpoint molecules expressed by immune cells are to intended to serve as a brake for immune cell overactivation. Therefore, blocking the inhibitory functions of checkpoint molecules will inevitably lead to developing a range of immune-mediated adverse events (IMAEs) including hepatitis, colitis pneumonitis, etc. [87]. The detailed management of autoimmune-specific IMAEs in patients with ICIs therapy has been previously reviewed by Brahmer, et al. [88]. Although HCC usually develops from a background of chronic inflammation induced by HBV/HCV or alcohol, the overall incidence of IMAE in HCC patients receiving ICI therapy is similar to those of patients with other cancer types [89]. Currently, the majority of IMAEs caused by ICI therapy can be relieved by administration of corticosteroids in combination with mycophenolate mofetil or azathioprine [89]. Here, we summarize latest safety profiles of ICIs for HCC (Table 2). As compared to sorafenib treatment, HCC patients receiving nivolumab treatment not only had more durable disease control (median 7.5 months vs. 5.7 months) but also had a better safety profile, with fewer treatment-related adverse effects (TRAES) over grade 3 (22% vs. 49%). However, the use of anti-CTLA-4 agents has often seen more serious IMAEs than that of anti-PD1/PL1 agents. Dose-related adverse effects were frequently observed in HCC patients, causing treatment discontinuation [45]. In the CheckMate040 phase II trial, 50% of patients were treated with corticosteroids in the anti-CTLA-4 high dose group while only 24% in low dose group received corticosteroids [42]. This phenomenon was also observed to a lesser extent in the treatment of patients with other cancer types, such as melanoma and lung cancer [90]. On the other hand, safety profiles between anti-PD1 and anti-PDL1 therapy were comparable [91]. Dual blockade of PD1 and CTLA-4 displayed superior treatment efficacy to each monotherapy alone; however, it came at the cost of an increased rate of hepatic TRAE in the early phase of treatment. Fortunately, the conditions were improved in most of patients after six weeks, suggesting no signs of synergistic toxicities [45]. Overall, PD1/PDL1 inhibitors result in less serious TRAE than CTLA-4 inhibitors. Therefore, dosing and timing for anti-CTLA-4 therapy seems critical in treating patients with HCC.
4.2. Possible Predictive Factors for Response to Anti-PD1/PDL1 Therapy
Given that the action mechanism of PD1/PDL1 inhibitors is to block PD1/PDL interaction between tumor cells and immune cells (Figure 1), PDL1 expression status has served as a biomarker for predicting the treatment outcomes of patients receiving anti-PD1/PDL1 therapy. Recent meta-analysis indicates that a survival benefit from ICI therapy was observed in cancer patients with the up-regulation of PDL1 expression (PDL1 > 1% score index), but not in those with <1% PDL1 [92]. Survival benefit between patients with >1% and ≥50% PDL1 was slightly improved, but not statistically significant. Therefore, PDL1 expression status must be taken into account in the context of ICI therapy.
Interestingly, this seems to not be the case in the recent ICI clinical trials for advanced HCC. As indicated in KEYNOTE-240 phase III trial, HCC patients with PDL1 expression in either tumor cells or stroma cells had a similar response to pembrolizumab treatment as compared to those with no PDL1 expression (median survival 16.1 months versus 16.7 months) [44]. In the CheckMate 040 trial, tumor responses were also independent of PDL1 status [93]. Other cellular factors and immune signatures that could serve as biomarkers to predict ICI immunotherapy outcomes were summarized previously [94].
5. Conclusions
At one time, the introduction of ICIs in advanced HCC was far behind the other human solid tumors. However, with the substantial exciting data produced from recent checkpoint blockade clinical trials in advanced HCC, atezolizumab plus bevacizumab combinatorial treatment is now approved by FDA as the frontline standard of care in advanced HCC [95,96], and the availability of new ICI combination regimens can be expected to significantly increase the treatment efficacy of regional therapy and neoadjuvant therapy in patients with unresectable disease. Although advanced HCC is still among the most difficult-to-treat human cancers, the emergence of immune checkpoint blockade therapies has drastically changed the landscape of clinical treatment of advanced HCC. The potential novel immune checkpoint inhibitors under development will further add to the repertoire of ICI therapy. The challenge of treating patients with cirrhosis remains a difficult task. Nevertheless, treatment of this seemingly incurable disease is now within reach with ICIs, which offer the best hope of reducing the mortality rates of HCC. The quest to eradicate this deadly disease is only beginning.
Author Contributions
Conceptualization, S.-H.C.; P.-Y.C. and S.-H.C.; writing—original draft preparation; writing—review and editing, S.-H.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by China Medical University, grant number CMU110-MF-42 and CMU110-Z-06, and the Ministry of Science and Technology, Taiwan, grant number MOST-109-2320-B-039-062-MY2, MOST 106-2314-B-442-001-MY3, MOST 109-2314-B-442-001, and National Health Research Institutes, Taiwan, grant number, NHRI-109BCCO-MF-202015-01, and Show Chwan Memorial Hospital, Taiwan, grant number SRD-109035, SRD-110008, SRD-110016 and SRD-110017.
Conflicts of Interest
Authors claim that there is no conflict of interest.
References
- Sangro, B.; Sarobe, P.; Hervas-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Suresh, D.; Srinivas, A.N.; Kumar, D.P. Etiology of Hepatocellular Carcinoma: Special Focus on Fatty Liver Disease. Front. Oncol. 2020, 10, 601710. [Google Scholar] [CrossRef]
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (HCC): Epidemiology, etiology and molecular classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [Green Version]
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abecassis, M.M.; Roberts, L.R.; Zhu, A.X.; Murad, M.H.; Marrero, J.A. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef] [Green Version]
- Colagrande, S.; Inghilesi, A.L.; Aburas, S.; Taliani, G.G.; Nardi, C.; Marra, F. Challenges of advanced hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 7645–7659. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Personeni, N.; Pressiani, T.; Santoro, A.; Rimassa, L. Regorafenib in hepatocellular carcinoma: Latest evidence and clinical implications. Drugs Context 2018, 7, 212533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, M. Systemic Therapy for Hepatocellular Carcinoma: Latest Advances. Cancers 2018, 10, 412. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Huitzil-Melendez, F.D.; O’Reilly, E.M.; Saltz, L.B. Current management of advanced hepatocellular carcinoma. Gastrointest. Cancer Res. 2008, 2, 64–70. [Google Scholar] [PubMed]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Siegel, A.B.; Cohen, E.I.; Ocean, A.; Lehrer, D.; Goldenberg, A.; Knox, J.J.; Chen, H.; Clark-Garvey, S.; Weinberg, A.; Mandeli, J.; et al. Phase II trial evaluating the clinical and biologic effects of bevacizumab in unresectable hepatocellular carcinoma. J. Clin. Oncol. 2008, 26, 2992–2998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledererova, A.; Dostalova, L.; Kozlova, V.; Peschelova, H.; Ladungova, A.; Culen, M.; Loja, T.; Verner, J.; Pospisilova, S.; Smida, M.; et al. Hypermethylation of CD19 promoter enables antigen-negative escape to CART-19 in vivo and in vitro. J. Immunother. Cancer 2021, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Michelakos, T.; Yamada, T.; Fan, S.; Wang, X.; Schwab, J.H.; Ferrone, C.R.; Ferrone, S. Defective HLA class I antigen processing machinery in cancer. Cancer Immunol. Immunother. 2018, 67, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Ormandy, L.A.; Hillemann, T.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Korangy, F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005, 65, 2457–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, D.M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Zhong, C.; Cui, W.; Wang, G.; Zheng, G.; Li, L.; Zhang, J.; Ren, R.; Gao, H.; Wang, T.; et al. Induction of tolerogenic dendritic cells by activated TGF-beta/Akt/Smad2 signaling in RIG-I-deficient stemness-high human liver cancer cells. BMC Cancer 2019, 19, 439. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-beta induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [Green Version]
- Yamagiwa, S.; Gray, J.D.; Hashimoto, S.; Horwitz, D.A. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J. Immunol. 2001, 166, 7282–7289. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.H.; Shao, Y.Y.; Chan, S.Y.; Huang, C.Y.; Hsu, C.H.; Cheng, A.L. High Serum Transforming Growth Factor-beta1 Levels Predict Outcome in Hepatocellular Carcinoma Patients Treated with Sorafenib. Clin. Cancer Res. 2015, 21, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Hochst, B.; Schildberg, F.A.; Sauerborn, P.; Gabel, Y.A.; Gevensleben, H.; Goltz, D.; Heukamp, L.C.; Turler, A.; Ballmaier, M.; Gieseke, F.; et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J. Hepatol. 2013, 59, 528–535. [Google Scholar] [CrossRef]
- Dunham, R.M.; Thapa, M.; Velazquez, V.M.; Elrod, E.J.; Denning, T.L.; Pulendran, B.; Grakoui, A. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J. Immunol. 2013, 190, 2009–2016. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Courau, T.; Nehar-Belaid, D.; Florez, L.; Levacher, B.; Vazquez, T.; Brimaud, F.; Bellier, B.; Klatzmann, D. TGF-beta and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI Insight 2016, 1, e85974. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.S.; Gao, F.H. Molecular Mechanism of Tumor Cell Immune Escape Mediated by CD24/Siglec-10. Front. Immunol. 2020, 11, 1324. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3—Potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Hato, T.; Goyal, L.; Greten, T.F.; Duda, D.G.; Zhu, A.X. Immune checkpoint blockade in hepatocellular carcinoma: Current progress and future directions. Hepatology 2014, 60, 1776–1782. [Google Scholar] [CrossRef]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, Z.; Yang, Y.; Jiang, Z.; Gu, Y.; Liu, Y.; Lin, C.; Pan, Z.; Yu, Y.; Jiang, M.; et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 2014, 59, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An anti-CTLA-4 antibody for metastatic melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef] [Green Version]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Inarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Kudo, M.; Lim, H.Y.; Cheng, A.L.; Chao, Y.; Yau, T.; Ogasawara, S.; Kurosaki, M.; Morimoto, N.; Ohkawa, K.; Yamashita, T.; et al. Pembrolizumab as Second-Line Therapy for Advanced Hepatocellular Carcinoma: A Subgroup Analysis of Asian Patients in the Phase 3 KEYNOTE-240 Trial. Liver Cancer 2021, 10, 275–284. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Qin, S.; Finn, R.S.; Kudo, M.; Meyer, T.; Vogel, A.; Ducreux, M.; Mercade, T.M.; Tomasello, G.; Boisserie, F.; Hou, J.; et al. A phase 3, randomized, open-label, multicenter study to compare the efficacy and safety of tislelizumab, an anti-PD-1 antibody, versus sorafenib as first-line treatment in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2018, 36, TPS3110. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Furuse, J.; Galle, P.R.; Kelley, R.K.; Qin, S.; Armstrong, J.; Darilay, A.; Vlahovic, G.; Negro, A.; et al. A randomized, multicenter phase 3 study of durvalumab (D) and tremelimumab (T) as first-line treatment in patients with unresectable hepatocellular carcinoma (HCC): HIMALAYA study. J. Clin. Oncol. 2018, 36, TPS4144. [Google Scholar] [CrossRef]
- Ren, Z.; Xu, J.; Bai, Y.; Xu, A.; Cang, S.; Du, C.; Li, Q.; Lu, Y.; Chen, Y.; Guo, Y.; et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): A randomised, open-label, phase 2–3 study. Lancet Oncol. 2021, 22, 977–990. [Google Scholar] [CrossRef]
- Knox, J.; Cheng, A.; Cleary, S.; Galle, P.; Kokudo, N.; Lencioni, R.; Park, J.; Zhou, J.; Mann, H.; Morgan, S.; et al. A phase 3 study of durvalumab with or without bevacizumab as adjuvant therapy in patients with hepatocellular carcinoma at high risk of recurrence after curative hepatic resection or ablation: EMERALD-2. Ann. Oncol. 2019, 30, iv59–iv60. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kudo, M.; Cheng, A.-L.; Finn, R.S.; Galle, P.R.; Kaneko, S.; Meyer, T.; Qin, S.; Dutcus, C.E.; Chen, E.; et al. Lenvatinib (len) plus pembrolizumab (pembro) for the first-line treatment of patients (pts) with advanced hepatocellular carcinoma (HCC): Phase 3 LEAP-002 study. J. Clin. Oncol. 2019, 37, TPS4152. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Yau, T.; Finn, A.-L.; Cheng, P.; Mathurin, J.; Edeline, M.; Kudo, K.-H.; Han, J.J.; Harding, P.; Merle, O.; Rosmorduc, O.; et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC)[abstract LBA38]. Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Sangro, J.P.; Finn, A.; Cheng, P.; Mathuri, J.; Edeline, M.; Kudo, K.; Han, J.; Harding, P.; Merle, O.; Rosmorduc, O.; et al. CheckMate 459: Long-term (minimum follow-up 33.6 months) survival outcomes with nivolumab versus sorafenib as first-line treatment in patients with advanced hepatocellular carcinoma [abstract LBA-3]. Ann. Oncol. 2020, 31, S241–S242. [Google Scholar] [CrossRef]
- Kyi, C.; Postow, M.A. Immune checkpoint inhibitor combinations in solid tumors: Opportunities and challenges. Immunotherapy 2016, 8, 821–837. [Google Scholar] [CrossRef] [Green Version]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M.; Okusaka, T.; Kang, Y.K.; Qin, S.; Tai, D.W.; Lim, H.Y.; Yau, T.; et al. Safety, Efficacy, and Pharmacodynamics of Tremelimumab Plus Durvalumab for Patients With Unresectable Hepatocellular Carcinoma: Randomized Expansion of a Phase I/II Study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef]
- Merle, P.; Subic, M. Comparison and analysis of the efficacy of drug therapy for liver cancer. Hepatoma Res. 2020, 6, 60. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.V.; Merle, P.; et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J. Clin. Oncol. 2021, 39, 267. [Google Scholar] [CrossRef]
- Lee, M.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Randomised efficacy and safety results for atezolizumab (Atezo) + bevacizumab (Bev) in patients (pts) with previously untreated, unresectable hepatocellular carcinoma (HCC)[abstract LBA39]. Ann. Oncol. 2019, 30, v875. [Google Scholar] [CrossRef]
- Qin, S.; Ren, Z.; Meng, Z.; Chen, Z.; Chai, X.; Xiong, J.; Bai, Y.; Yang, L.; Zhu, H.; Fang, W.; et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: A multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 571–580. [Google Scholar] [CrossRef]
- Kelley, R.K.; Sangro, B.; Harris, W.P.; Ikeda, M.; Okusaka, T.; Kang, Y.-K.; Qin, S.; Tai, W.M.D.; Lim, H.Y.; Yau, T.; et al. Efficacy, tolerability, and biologic activity of a novel regimen of tremelimumab (T) in combination with durvalumab (D) for patients (pts) with advanced hepatocellular carcinoma (aHCC). J. Clin. Oncol. 2020, 38, 4508. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Ikeda, M.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. A phase Ib study of lenvatinib (LEN) plus pembrolizumab (PEMBRO) in unresectable hepatocellular carcinoma (uHCC). J. Clin. Oncol. 2020, 38, 4519. [Google Scholar] [CrossRef]
- Yau, T.; Zagonel, V.; Santoro, A.; Acosta-Rivera, M.; Choo, S.P.; Matilla, A.; He, A.R.; Gracián, A.C.; El-Khoueiry, A.B.; Sangro, B.; et al. Nivolumab (NIVO) + ipilimumab (IPI) + cabozantinib (CABO) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2020, 38, 478. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 2007, 318, 1141–1143. [Google Scholar] [CrossRef]
- Ndhlovu, L.C.; Lopez-Verges, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734. [Google Scholar] [CrossRef] [Green Version]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Chauvin, J.M.; Sander, C.; Janjic, B.; Tarhini, A.A.; Tawbi, H.A.; Kirkwood, J.M.; Moschos, S.; et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8(+) T cells induced by melanoma vaccines. Cancer Res. 2014, 74, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [Green Version]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef] [Green Version]
- Pena, J.; Jones, N.G.; Bousheri, S.; Bangsberg, D.R.; Cao, H. Lymphocyte activation gene-3 expression defines a discrete subset of HIV-specific CD8+ T cells that is associated with lower viral load. AIDS Res. Hum. Retrovir. 2014, 30, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Workman, C.J.; Wang, Y.; El Kasmi, K.C.; Pardoll, D.M.; Murray, P.J.; Drake, C.G.; Vignali, D.A. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J. Immunol. 2009, 182, 1885–1891. [Google Scholar] [CrossRef] [Green Version]
- Demeure, C.E.; Wolfers, J.; Martin-Garcia, N.; Gaulard, P.; Triebel, F. T Lymphocytes infiltrating various tumour types express the MHC class II ligand lymphocyte activation gene-3 (LAG-3): Role of LAG-3/MHC class II interactions in cell-cell contacts. Eur J. Cancer 2001, 37, 1709–1718. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef] [Green Version]
- Bettini, M.; Szymczak-Workman, A.L.; Forbes, K.; Castellaw, A.H.; Selby, M.; Pan, X.; Drake, C.G.; Korman, A.J.; Vignali, D.A. Cutting edge: Accelerated autoimmune diabetes in the absence of LAG-3. J. Immunol. 2011, 187, 3493–3498. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Melero, I.; Bhatia, S.; Bono, P.; Sanborn, R.E.; Lipson, E.J.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S.; et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J. Clin. Oncol. 2017, 35, 9520. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A. Checkpoint inhibitors in melanoma and early phase development in solid tumors: What’s the future? J. Transl. Med. 2017, 15, 173. [Google Scholar] [CrossRef]
- Li, F.J.; Zhang, Y.; Jin, G.X.; Yao, L.; Wu, D.Q. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunol. Lett. 2013, 150, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Oweida, A.; Hararah, M.K.; Phan, A.; Binder, D.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef] [Green Version]
- Limagne, E.; Richard, C.; Thibaudin, M.; Fumet, J.D.; Truntzer, C.; Lagrange, A.; Favier, L.; Coudert, B.; Ghiringhelli, F. Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. Oncoimmunology 2019, 8, e1564505. [Google Scholar] [CrossRef]
- Macek Jilkova, Z.; Aspord, C.; Kurma, K.; Granon, A.; Sengel, C.; Sturm, N.; Marche, P.N.; Decaens, T. Immunologic Features of Patients With Advanced Hepatocellular Carcinoma Before and During Sorafenib or Anti-programmed Death-1/Programmed Death-L1 Treatment. Clin. Transl. Gastroenterol. 2019, 10, e00058. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef]
- Sangro, B.; Chan, S.L.; Meyer, T.; Reig, M.; El-Khoueiry, A.; Galle, P.R. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J. Hepatol. 2020, 72, 320–341. [Google Scholar] [CrossRef] [Green Version]
- Spain, L.; Diem, S.; Larkin, J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat. Rev. 2016, 44, 51–60. [Google Scholar] [CrossRef]
- Duan, J.; Cui, L.; Zhao, X.; Bai, H.; Cai, S.; Wang, G.; Zhao, Z.; Zhao, J.; Chen, S.; Song, J.; et al. Use of Immunotherapy With Programmed Cell Death 1 vs Programmed Cell Death Ligand 1 Inhibitors in Patients With Cancer: A Systematic Review and Meta-analysis. JAMA Oncol. 2020, 6, 375–384. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, L.; Yu, J.; Zhang, Y.; Pang, X.; Ma, C.; Shen, M.; Ruan, S.; Wasan, H.S.; Qiu, S. Clinical efficacy and safety of anti-PD-1/PD-L1 inhibitors for the treatment of advanced or metastatic cancer: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 2083. [Google Scholar] [CrossRef] [Green Version]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.L.; Yau, T.; Furuse, J.; Park, J.W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020, 73, 1460–1469. [Google Scholar] [CrossRef]
- Gok Yavuz, B.; Hasanov, E.; Lee, S.S.; Mohamed, Y.I.; Curran, M.A.; Koay, E.J.; Cristini, V.; Kaseb, A.O. Current Landscape and Future Directions of Biomarkers for Immunotherapy in Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 1195–1207. [Google Scholar] [CrossRef]
- Galle, P.R.; Finn, R.S.; Qin, S.; Ikeda, M.; Zhu, A.X.; Kim, T.-Y.; Kudo, M.; Breder, V.V.; Merle, P.; Kaseb, A.O.; et al. Patient-reported outcomes (PROs) from the Phase III IMbrave150 trial of atezolizumab (atezo) + bevacizumab (bev) vs sorafenib (sor) as first-line treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). J. Clin. Oncol. 2020, 38, 476. [Google Scholar] [CrossRef]
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.; Ducreux, M.; Zhu, A.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. LBA3—IMbrave150: Efficacy and safety results from a ph III study evaluating atezolizumab (atezo) + bevacizumab (bev) vs sorafenib (Sor) as first treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). Ann. Oncol. 2019, 30, ix186–ix187. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).