
Genomic Risk Factors for Cervical Cancer



Abstract
Simple Summary
Abstract
1. Epidemiology and Heritability of Cervical Cancer
1.1. Risk and Prevention
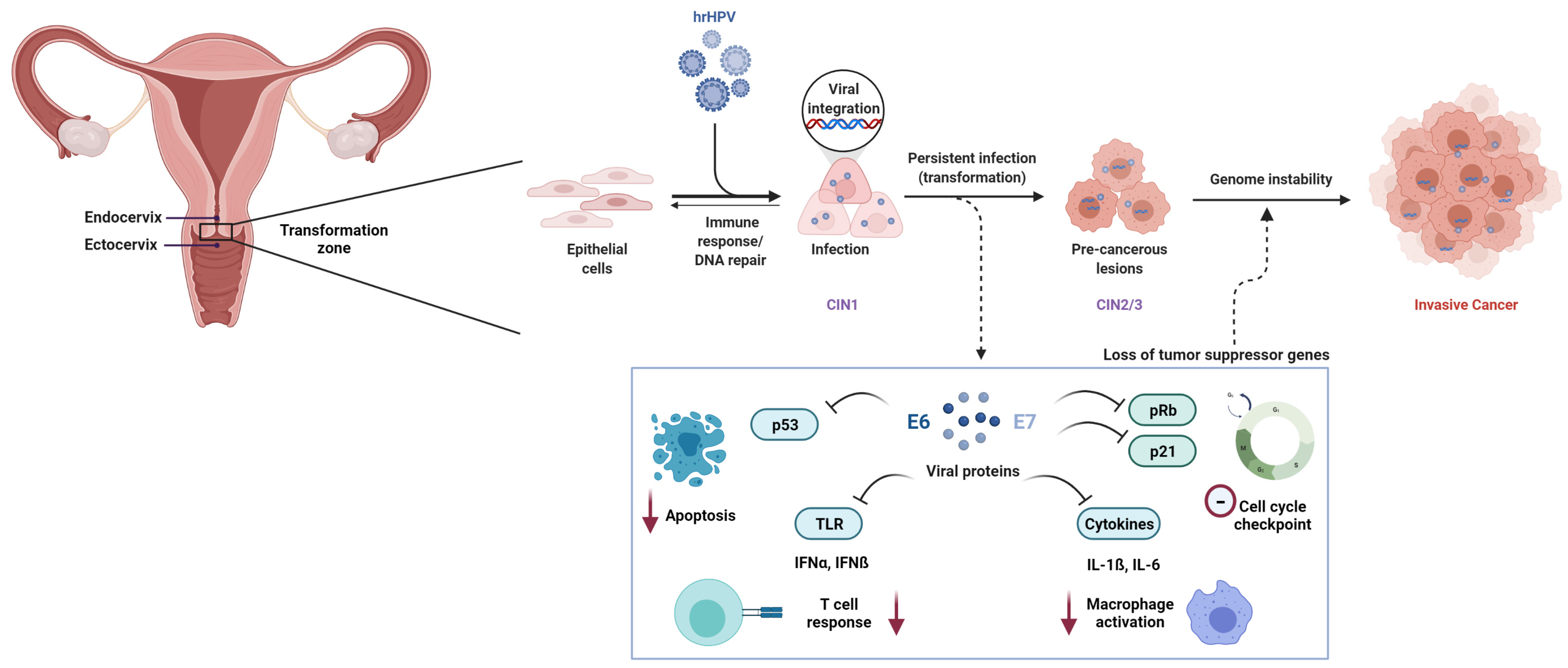
1.2. HPV-Associated Pathogenesis
1.3. Heritability of Cervical Cancer
1.4. Candidate Gene Based Studies
2. Genomic Susceptibility Variants for Cervical Cancer
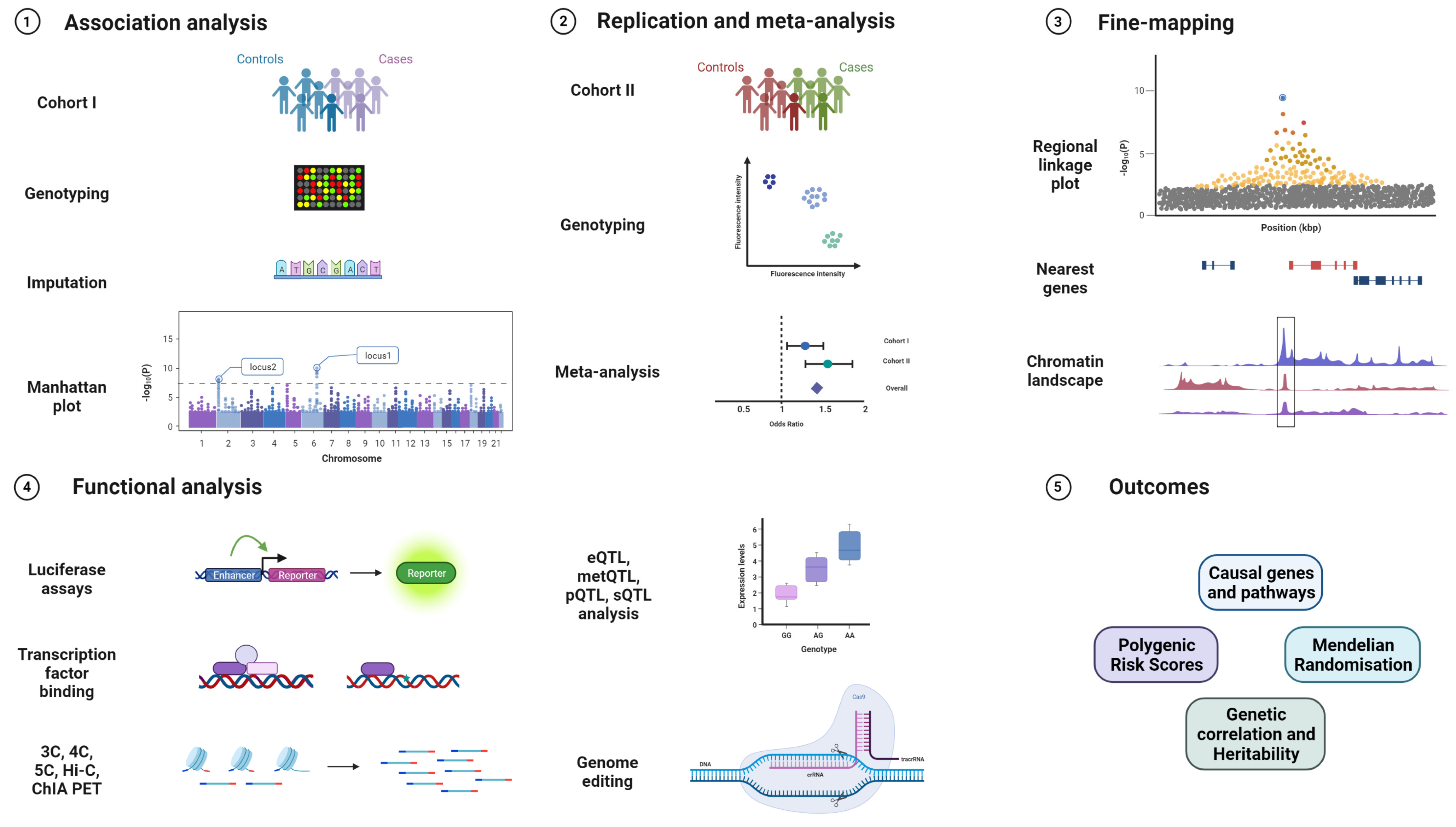
2.1. Genome-Wide Association Studies
2.2. Results from Cervical Cancer GWAS
2.2.1. 6p21.3 (HLA)
2.2.2. 17q12 (GSDMB)
2.2.3. 2q13 (PAX8)
2.2.4. 5p15.33 (CLPTM1L)
2.3. Other GWAS Loci for Cervical Cancer or Dysplasia
2.4. Other GWAS Loci for Viral Infection
3. Follow-Up Studies of Cervical Cancer GWAS Results
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bruni, L.; Albero, G.; Serrano, B.; Mena, M.; Gómez, D.; Muñoz, J.; Bosch, F.; de Sanjosé, S. ICO HPV Information Centre Human Papillomavirus and Related Diseases Report-Germany. Summary Report. ICO/IARC Information Centre HPV Cancer. 2019. Available online: https://hpvcentre.net/statistics/reports/DEU.pdf?t=1575294458729 (accessed on 12 October 2021).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef]
- Burd, E. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 2003, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Burd, E.M.; Dean, C.L. Human Papillomavirus. Microbiol. Spectr. 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, N.; Franceschi, S.; Bosetti, C.; Moreno, V.; Herrero, R.; Smith, J.S.; Shah, K.V.; Meijer, C.J.; Bosch, F.X. Role of parity and human papillomavirus in cervical cancer: The IARC multicentric case-control study. Lancet 2002, 359, 1093–1101. [Google Scholar] [CrossRef]
- Franceschi, S.; Rajkumar, T.; Vaccarella, S.; Gajalakshmi, V.; Sharmila, A.; Snijders, P.J.F.; Muñoz, N.; Meijer, C.J.L.M.; Herrero, R. Human papillomavirus and risk factors for cervical cancer in Chennai, India: A case-control study. Int. J. Cancer 2003, 107, 127–133. [Google Scholar] [CrossRef]
- Brinton, L.A.; Tashima, K.T.; Lehman, H.F.; Levine, R.S.; Mallin, K.; Savitz, D.A.; Stolley, P.D.; Fraumeni, J.F. Epidemiology of cervical cancer by cell type. Cancer Res. 1987, 47, 1706–1711. [Google Scholar]
- Bruni, L.; Albero, G.; Serrano, B.; Mena, M.; Gómez, D.; Muñoz, J.; Bosch, F.; de Sanjosé, S. ICO HPV Information Centre Human Papillomavirus and Related Diseases Report-World Summary Report. ICO/IARC Information Centre HPV Cancer. 2019. Available online: https://hpvcentre.net/statistics/reports/XWX.pdf (accessed on 12 October 2021).
- Bujan Rivera, J.; Klug, S.J. Cervical cancer screening in Germany. Bundesgesundheitsblatt Gesundh. Gesundh. 2018, 61, 1528–1535. [Google Scholar] [CrossRef]
- Hillemanns, P.; Soergel, P.; Hertel, H.; Jentschke, M. Epidemiology and Early Detection of Cervical Cancer. Oncol. Res. Treat. 2016, 39, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Starker, A.; Buttmann-Schweiger, N.; Krause, L.; Barnes, B.; Kraywinkel, K.; Holmberg, C. Cancer screening in Germany: Availability and participation. Bundesgesundheitsblatt Gesundh. Gesundh. 2018, 61, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- de Araujo Souza, P.S.; Sichero, L.; Maciag, P.C. HPV variants and HLA polymorphisms: The role of variability on the risk of cervical cancer. Futur. Oncol. 2009, 5, 359–370. [Google Scholar] [CrossRef] [PubMed]
- De Brot, L.; Pellegrini, B.; Moretti, S.T.; Carraro, D.M.; Soares, F.A.; Rocha, R.M.; Baiocchi, G.; da Cunha, I.W.; de Andrade, V.P. Infections with multiple high-risk HPV types are associated with high-grade and persistent low-grade intraepithelial lesions of the cervix. Cancer Cytopathol. 2017, 125, 138–143. [Google Scholar] [CrossRef]
- Feyrter, F. Über das Oberflächenkarzinom im Bereich des Collum uteri TT-Surface carcinoma of the uterine cervix. Dtsch. Med. Wochenschr. 1955, 80, 1686–1691. [Google Scholar] [CrossRef]
- Gray, L.A.; Barnes, M.L.; Lee, J.J. Carcinoma-in-situ and dysplasia of the cervix. Ann. Surg. 1960, 151, 951–960. [Google Scholar] [CrossRef]
- Cornet, I.; Gheit, T.; Franceschi, S.; Vignat, J.; Burk, R.D.; Sylla, B.S.; Tommasino, M.; Clifford, G.M. Human Papillomavirus Type 16 Genetic Variants: Phylogeny and Classification Based on E6 and LCR. J. Virol. 2012, 86, 6855–6861. [Google Scholar] [CrossRef]
- Chen, A.A.; Heideman, D.A.M.; Boon, D.; Chen, Z.; Burk, R.D.; De Vuyst, H.; Gheit, T.; Snijders, P.J.F.; Tommasino, M.; Franceschi, S.; et al. Human papillomavirus 33 worldwide genetic variation and associated risk of cervical cancer. Virology 2014, 448, 356–362. [Google Scholar] [CrossRef]
- Chen, A.A.; Heideman, D.A.M.; Boon, D.; Gheit, T.; Snijders, P.J.F.; Tommasino, M.; Franceschi, S.; Clifford, G.M. Human Papillomavirus 45 Genetic Variation and Cervical Cancer Risk Worldwide. J. Virol. 2014, 88, 4514–4521. [Google Scholar] [CrossRef]
- Hirose, Y.; Onuki, M.; Tenjimbayashi, Y.; Mori, S.; Ishii, Y.; Takeuchi, T.; Tasaka, N.; Satoh, T.; Morisada, T.; Iwata, T.; et al. Within-Host Variations of Human Papillomavirus Reveal APOBEC Signature Mutagenesis in the Viral Genome. J. Virol. 2018, 92, 1–14. [Google Scholar] [CrossRef]
- Mirabello, L.; Clarke, M.A.; Nelson, C.W.; Dean, M.; Wentzensen, N.; Yeager, M.; Cullen, M.; Boland, J.F.; Alemany, L.; Banks, L.; et al. The intersection of HPV epidemiology, genomics and mechanistic studies of HPV-mediated carcinogenesis. Viruses 2018, 10, 80. [Google Scholar] [CrossRef]
- Chen, A.A.; Gheit, T.; Franceschi, S.; Tommasino, M.; Clifford, G.M. Human Papillomavirus 18 Genetic Variation and Cervical Cancer Risk Worldwide. J. Virol. 2015, 89, 10680–10687. [Google Scholar] [CrossRef]
- Badaracco, G.; Venuti, A.; Sedati, A.; Marcante, M.L. HPV16 and HPV18 in genital tumors: Significantly different levels of viral integration and correlation to tumor invasiveness. J. Med. Virol. 2002, 67, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Pirami, L.; Giache, V.; Becciolini, A. Pre-Invasive and Invasive Lesions of the Uterine Cervix. Obstet. Gynecol. 1997, 600–604. [Google Scholar]
- Burk, R.D.; Chen, Z.Z.; Saller, C.; Tarvin, K.; Carvalho, A.L.; Scapulatempo-Neto, C.; Silveira, H.C.; Fregnani, J.H.; Creighton, C.J.; Anderson, M.L.; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Henken, F.E.; Wilting, S.M.; Overmeer, R.M.; Van Rietschoten, J.G.I.; Nygren, A.O.H.; Errami, A.; Schouten, J.P.; Meijer, C.J.L.M.; Snijders, P.J.F.; Steenbergen, R.D.M. Sequential gene promoter methylation during HPV-induced cervical carcinogenesis. Br. J. Cancer 2007, 97, 1457–1464. [Google Scholar] [CrossRef]
- Dong, S.M.; Kim, H.S.; Rha, S.H.; Sidransky, D. Promoter hypermethylation of multiple genes in carcinoma of the uterine cervix. Clin. Cancer Res. 2001, 7, 1982–1986. [Google Scholar]
- Sartor, M.A.; Dolinoy, D.C.; Jones, T.R.; Colacino, J.A.; Prince, M.E.P.; Carey, T.E.; Rozek, L.S. Genome-wide methylation and expression differences in HPV(+) and HPV(-) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics 2011, 6, 777–787. [Google Scholar] [CrossRef]
- Curty, G.; Menezes, A.N.; Brant, A.C.; de Mulder Rougvie, M.; Moreira, M.Â.M.; Soares, M.A. Expression of Retroelements in Cervical Cancer and Their Interplay with HPV Infection and Host Gene Expression. Cancers 2021, 13, 3513. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; David, P.; Banks, L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 1999, 18, 7690–7700. [Google Scholar] [CrossRef] [PubMed]
- Helt, A.-M.; Funk, J.O.; Galloway, D.A. Inactivation of both the Retinoblastoma Tumor Suppressor and p21 by the Human Papillomavirus Type 16 E7 Oncoprotein Is Necessary To Inhibit Cell Cycle Arrest in Human Epithelial Cells. J. Virol. 2002, 76, 10559–10568. [Google Scholar] [CrossRef]
- Shin, M.K.; Balsitis, S.; Brake, T.; Lambert, P.F. Human papillomavirus E7 oncoprotein overrides the tumor suppressor activity of p21Cip1 in cervical carcinogenesis. Cancer Res. 2009, 69, 5656–5663. [Google Scholar] [CrossRef]
- Jones, D.L.; Alani, R.M.; Münger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21(Cip1)-mediated inhibition of cdk2. Genes Dev. 1997, 11, 2101–2111. [Google Scholar] [CrossRef]
- Sano, T.; Oyama, T.; Kashiwabara, K.; Fukuda, T.; Nakajima, T. Expression Status of p16 Protein Is Associated with Human Papillomavirus Oncogenic Potential in Cervical and Genital Lesions. Am. J. Pathol. 1998, 153, 1741–1748. [Google Scholar] [CrossRef]
- Keating, J.T.; Cviko, A.; Riethdorf, S.; Riethdorf, L.; Quade, B.J.; Sun, D.; Duensing, S.; Sheets, E.E.; Munger, K.; Crum, C.P. Ki-67, Cyclin E, and p16 INK4 Are Complimentary Surrogate Biomarkers for Human Papilloma Virus-Related Cervical Neoplasia. Am. J. Surg. Pathol. 2001, 25, 884–891. [Google Scholar] [CrossRef]
- Von Knebel Doeberitz, M. New markers for cervical dysplasia to visualise the genomic chaos created by aberrant oncogenic papillomavirus infections. Eur. J. Cancer 2002, 38, 2229–2242. [Google Scholar] [CrossRef]
- Nuovo, G.J.; Plaia, T.W.; Belinsky, S.A.; Baylin, S.B.; Herman, J.G. In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 12754–12759. [Google Scholar] [CrossRef]
- Wijetunga, N.A.; Belbin, T.J.; Burk, R.D.; Whitney, K.; Abadi, M.; Greally, J.M.; Einstein, M.H.; Schlecht, N.F. Novel epigenetic changes in CDKN2A are associated with progression of cervical intraepithelial neoplasia. Gynecol. Oncol. 2016, 142, 566–573. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Park, D.; Munger, K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 2013, 110, 16175–16180. [Google Scholar] [CrossRef]
- Le Bon, A.; Tough, D.F. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef]
- Herdman, T.M.; Pett, M.R.; Roberts, I.; Alazawi, W.O.F.; Teschendorff, A.E.; Zhang, X.Y.; Stanley, M.A.; Coleman, N. Interferon-β treatment of cervical keratinocytes naturally infected with human papillomavirus 16 episomes promotes rapid reduction in episome numbers and emergence of latent integrants. Carcinogenesis 2006, 27, 2341–2353. [Google Scholar] [CrossRef] [PubMed]
- Black, A.P.B.; Ardern-Jones, M.R.; Kasprowicz, V.; Bowness, P.; Jones, L.; Bailey, A.S.; Ogg, G.S. Human keratinocyte induction of rapid effector function in antigen-specific memory CD4+ and CD8+ T cells. Eur. J. Immunol. 2007, 37, 1485–1493. [Google Scholar] [CrossRef]
- Karim, R.; Meyers, C.; Backendorf, C.; Ludigs, K.; Offringa, R.; van Ommen, G.J.B.; Melief, C.J.M.; van der Burg, S.H.; Boer, J.M. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS ONE 2011, 6, e17848. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.M.; Raghav, D.; Singh, H.; Raghava, G.P.S. CCDB: A curated database of genes involved in cervix cancer. Nucleic Acids Res. 2011, 39, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Bravo, Á.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Ngo, C.; Samuels, S.; Bagrintseva, K.; Slocker, A.; Hupé, P.; Kenter, G.; Popovic, M.; Samet, N.; Tresca, P.; von der Leyen, H.; et al. From prospective biobanking to precision medicine: BIO-RAIDs—An EU study protocol in cervical cancer. BMC Cancer 2015, 15, 842. [Google Scholar] [CrossRef]
- Way, S.; Hetherington, J.; Galloway, D.C. Simultaneous cytological diagnosis of cervical cancer in three sisters. Lancet 1959, 2, 890–891. [Google Scholar] [CrossRef]
- Ahlbom, A.; Lichtenstein, P.; Malmström, H.; Feychting, M.; Hemminki, K.; Pedersen, N.L. Cancer in twins: Genetic and nongenetic familial risk factors. J. Natl. Cancer Inst. 1997, 89, 287–293. [Google Scholar] [CrossRef]
- Andrews, F.J.; Linehan, J.J.; Melcher, D.H. Cervical carcinoma in both mother and daughter. Acta Cytol. 1981, 25, 3–4. [Google Scholar] [PubMed]
- Magnusson, P.K.E.; Lichtenstein, P.; Gyllensten, U.B. Heritability of cervical tumours. Int. J. Cancer 2000, 88, 698–701. [Google Scholar] [CrossRef]
- Bruinse, H.W.; te Velde, E.R.; de Gast, B.C. Human leukocyte antigen patterns in a family with cervical cancer. Gynecol. Oncol. 1981, 12, 249–252. [Google Scholar] [CrossRef]
- Horn, L.-C.; Raptis, G.; Fischer, U. Familial cancer history in patients with carcinoma of the cervix uteri. Eur. J. Obstet. Gynecol. Reprod. Biol. 2002, 101, 54–57. [Google Scholar] [CrossRef]
- Zoodsma, M.; Sijmons, R.H.; de Vries, E.G.; Zee, A. Familial Cervical Cancer: Case Reports, Review and Clinical Implications. Hered. Cancer Clin. Pract. 2004, 2, 99. [Google Scholar] [CrossRef][Green Version]
- De Oliveira, C.M.; Levi, J.E. The Biological Impact of Genomic Diversity in Cervical Cancer Development. Acta Cytol. 2016, 60, 513–517. [Google Scholar] [CrossRef]
- Rodríguez-Carunchio, L.; Soveral, I.; Steenbergen, R.D.M.; Torné, A.; Martinez, S.; Fusté, P.; Pahisa, J.; Marimon, L.; Ordi, J.; del Pino, M. HPV-negative carcinoma of the uterine cervix: A distinct type of cervical cancer with poor prognosis. BJOG Int. J. Obstet. Gynaecol. 2015, 122, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Tjalma, W. HPV negative cervical cancers and primary HPV screening. Facts Views Vis. ObGyn 2018, 10, 107–113. [Google Scholar]
- Tjalma, W.A.A.; Trinh, X.B.; Rosenlund, M.; Makar, A.P.; Kridelka, F.; Rosillon, D.; Van Dam, P.A.; Collas De Souza, S.; Holl, K.; Simon, P.; et al. A cross-sectional, multicentre, epidemiological study on human papillomavirus (HPV) type distribution in adult women diagnosed with invasive cervical cancer in Belgium. Facts Views Vis. ObGyn 2015, 7, 101–108. [Google Scholar]
- Ruiz, F.J.; Sundaresan, A.; Zhang, J.; Pedamallu, C.S.; Halle, M.K.; Srinivasasainagendra, V.; Zhang, J.; Muhammad, N.; Stanley, J.; Markovina, S.; et al. Genomic Characterization and Therapeutic Targeting of HPV Undetected Cervical Carcinomas. Cancers 2021, 13, 4551. [Google Scholar] [CrossRef] [PubMed]
- Petry, K.U.; Liebrich, C.; Luyten, A.; Zander, M.; Iftner, T. Surgical staging identified false HPV-negative cases in a large series of invasive cervical cancers. Papillomavirus Res. 2017, 4, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Leo, P.J.; Madeleine, M.M.; Wang, S.; Schwartz, S.M.; Newell, F.; Pettersson-Kymmer, U.; Hemminki, K.; Hallmans, G.; Tiews, S.; Steinberg, W.; et al. Defining the genetic susceptibility to cervical neoplasia—A genome-wide association study. PLoS Genet. 2017, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cui, T.; Ek, W.E.; Liu, H.; Wang, H.; Gyllensten, U. Analysis of the genetic architecture of susceptibility to cervical cancer indicates that common SNPs explain a large proportion of the heritability. Carcinogenesis 2015, 36, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Rashkin, S.R.; Graff, R.E.; Kachuri, L.; Thai, K.K.; Alexeeff, S.E.; Blatchins, M.A.; Cavazos, T.B.; Corley, D.A.; Emami, N.C.; Hoffman, J.D.; et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat. Commun. 2020, 11, 4423. [Google Scholar] [CrossRef]
- Von Keyserling, H.; Bergmann, T.; Schuetz, M.; Schiller, U.; Stanke, J.; Hoffmann, C.; Schneider, A.; Lehrach, H.; Dahl, A.; Kaufmann, A.M. Analysis of 4 single-nucleotide polymorphisms in relation to cervical dysplasia and cancer development using a high-throughput ligation-detection reaction procedure. Int. J. Gynecol. Cancer 2011, 21, 1664–1671. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, Z.; Ma, D.; Huettner, P.C.; Massad, L.S.; Nguyen, L.; Borecki, I.; Rader, J.S. TP53, MDM2, NQO1, and susceptibility to cervical cancer. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Kim, J.W.; Roh, J.W.; Choi, J.Y.; Lee, K.M.; Yoo, K.Y.; Song, Y.S.; Kang, D. Genetic polymorphisms of GSTM1, p21, p53 and HPV infection with cervical cancer in Korean women. Gynecol. Oncol. 2004, 93, 14–18. [Google Scholar] [CrossRef]
- Nunobiki, O.; Ueda, M.; Yamamoto, M.; Toji, E.; Sato, N.; Izuma, S.; Okamoto, Y.; Torii, K.; Noda, S. MDM2 SNP 309 human papillomavirus infection in cervical carcinogenesis. Gynecol. Oncol. 2010, 118, 258–261. [Google Scholar] [CrossRef]
- Roszak, A.; Misztal, M.; Sowińska, A.; Jagodziński, P.P. Murine Double-Minute 2 Homolog Single Nucleotide Polymorphisms 285 and 309 in Cervical Carcinogenesis. Mol. Diagnosis Ther. 2015, 19, 235–244. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oliveira, S.; Ribeiro, J.; Sousa, H.; Pinto, D.; Baldaque, I.; Medeiros, R. Genetic polymorphisms and cervical cancer development: ATM G5557A and p53bp1 C1236G. Oncol. Rep. 2012, 27, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.D.; Cai, G.Q.; Zou, W.; Huang, Y.H.; Zhang, J.R.; Wang, D.T.; Chen, B.L. BRIP1 variations analysis reveals their relative importance as genetic susceptibility factor for cervical cancer. Biochem. Biophys. Res. Commun. 2013, 433, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Nava, G.A.; Fernández-Niño, J.A.; Madrid-Marina, V.; Torres-Poveda, K. Cervical cancer genetic susceptibility: A systematic review and meta-analyses of recent evidence. PLoS ONE 2016, 11, e0157344. [Google Scholar] [CrossRef]
- Wang, N.; Wang, S.; Zhang, Q.; Lu, Y.; Wei, H.; Li, W.; Zhang, S.; Yin, D.; Ou, Y. Association of p21 SNPs and risk of cervical cancer among Chinese women. BMC Cancer 2012, 12, 589. [Google Scholar] [CrossRef]
- Lima, G.; Santos, E.; Angelo, H.; Oliveira, M.; Heráclio, S.; Leite, F.; de Melo, C.; Crovella, S.; Maia, M.; Souza, P. Association between p21 Ser31Arg polymorphism and the development of cervical lesion in women infected with high risk HPV. Tumor Biol. 2016, 37, 10935–10941. [Google Scholar] [CrossRef] [PubMed]
- Thakur, N.; Hussain, S.; Nasare, V.; Das, B.C.; Basir, S.F.; Bharadwaj, M. Association analysis of p16 (CDKN2A) and RB1 polymorphisms with susceptibility to cervical cancer in Indian population. Mol. Biol. Rep. 2012, 39, 407–414. [Google Scholar] [CrossRef]
- Juko-Pecirep, I.; Ivansson, E.L.; Gyllensten, U.B. Evaluation of Fanconi anaemia genes FANCA, FANCC and FANCL in cervical cancer susceptibility. Gynecol. Oncol. 2011, 122, 377–381. [Google Scholar] [CrossRef]
- Chung, H.H.; Kim, M.K.; Kim, J.W.; Park, N.H.; Song, Y.S.; Kang, S.B.; Lee, H.P. XRCC1 R399Q polymorphism is associated with response to platinum-based neoadjuvant chemotherapy in bulky cervical cancer. Gynecol. Oncol. 2006, 103, 1031–1037. [Google Scholar] [CrossRef]
- Kim, K.; Kang, S.B.; Chung, H.H.; Kim, J.W.; Park, N.H.; Song, Y.S. XRCC1 Arginine194Tryptophan and GGH-401Cytosine/Thymine polymorphisms are associated with response to platinum-based neoadjuvant chemotherapy in cervical cancer. Gynecol. Oncol. 2008, 111, 509–515. [Google Scholar] [CrossRef]
- Cheng, X.-D.; Lu, W.-G.; Ye, F.; Wan, X.-Y.; Xie, X. The association of XRCC1 gene single nucleotide polymorphisms with response to neoadjuvant chemotherapy in locally advanced cervical carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 91. [Google Scholar] [CrossRef] [PubMed]
- Alsbeih, G.; Al-Harbi, N.; El-Sebaie, M.; Al-Badawi, I. HPV prevalence and genetic predisposition to cervical cancer in Saudi Arabia. Infect. Agent. Cancer 2013, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.J.; Rader, J.S.; Borecki, I.; Zhang, Z.; Hildesheim, A. CD83 polymorphisms and cervical cancer risk. Gynecol. Oncol. 2009, 114, 319–322. [Google Scholar] [CrossRef]
- Zhang, Z.; Borecki, I.; Nguyen, L.; Ma, D.; Smith, K.; Huettner, P.C.; Mutch, D.G.; Herzog, T.J.; Gibb, R.K.; Powell, M.A.; et al. CD83 gene polymorphisms increase susceptibility to human invasive cervical cancer. Cancer Res. 2007, 67, 11202–11208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, L.; Liu, J.; Chen, X.; Zhang, Y.; Liu, L.; Zhu, J.; Chen, J.; Shen, H.; Qiang, F.; Hu, Z. CTLA-4 gene polymorphism +49 A/G contributes to genetic susceptibility to two infection-related cancers-hepatocellular carcinoma and cervical cancer. Hum. Immunol. 2010, 71, 888–891. [Google Scholar] [CrossRef]
- Yin, J.; Wen, J.; Hang, D.; Han, J.; Jiang, J.; Song, C.; Liu, Y.; Liu, J.; Liu, L.; Zhu, L.; et al. Expression quantitative trait loci for CARD8 contributes to risk of two infection-related cancers—Hepatocellular carcinoma and cervical cancer. PLoS ONE 2015, 10, e0132352. [Google Scholar] [CrossRef][Green Version]
- Jin, Y. Association of Single Nucleotide Polymorphisms in Tumor Necrosis Factor-Alpha with Cervical Cancer Susceptibility. Cell Biochem. Biophys. 2015, 71, 77–84. [Google Scholar] [CrossRef]
- Kohaar, I.; Thakur, N.; Salhan, S.; Batra, S.; Singh, V.; Sharma, A.; Sodhani, P.; Das, B.C.; Sarkar, D.P.; Bharadwaj, M. TNF α –308G/A Polymorphism as a Risk Factor for HPV Associated Cervical Cancer in Indian Population. Anal. Cell. Pathol. 2007, 29, 249–256. [Google Scholar] [CrossRef]
- Barbisan, G.; Pérez, L.O.; Contreras, A.; Golijow, C.D. TNF-α and IL-10 promoter polymorphisms, HPV infection, and cervical cancer risk. Tumor Biol. 2012, 33, 1549–1556. [Google Scholar] [CrossRef]
- Badano, I.; Stietz, S.M.; Schurr, T.G.; Picconi, A.M.; Fekete, D.; Quintero, I.M.; Cabrera, M.D.E.; Campos, R.H.; Liotta, J.D. Analysis of TNFα promoter SNPs and the risk of cervical cancer in urban populations of Posadas (Misiones, Argentina). J. Clin. Virol. 2012, 53, 54–59. [Google Scholar] [CrossRef]
- Liu, H.; Lyu, D.; Zhang, Y.; Sheng, L.; Tang, N. Association Between the IL-6 rs1800795 Polymorphism and the Risk of Cervical Cancer: A Meta-Analysis of 1210 Cases and 1525 Controls. Technol. Cancer Res. Treat. 2017, 16, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Gu, Z.; Wang, X. Polymorphisms of the interleukin 6 gene and additional gene–gene interaction contribute to cervical cancer susceptibility in Eastern Chinese women. Arch. Gynecol. Obstet. 2016, 294, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Cho, E.Y.; Lee, T.S.; Kim, J.W.; Park, N.H.; Song, Y.S.; Kim, J.G.; Lee, H.P.; Kang, S.B. Interleukin-12 p40 gene (IL12B) polymorphisms and the risk of cervical caner in Korean women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 140, 71–75. [Google Scholar] [CrossRef]
- Chagas, B.S.; Gurgel, A.P.A.D.; da Cruz, H.L.A.; Amaral, C.M.M.; Cardoso, M.V.; Neto, J.d.S.; Silva, L.A.Ô.F.d.; Albuquerque, E.M.B.d.; Muniz, M.T.C.; Freitas, A.C.d. An interleukin-10 gene polymorphism associated with the development of cervical lesions in women infected with Human Papillomavirus and using oral contraceptives. Infect. Genet. Evol. 2013, 19, 32–37. [Google Scholar] [CrossRef][Green Version]
- Torres-Poveda, K.; Burguete-García, A.I.; Bahena-Román, M.; Méndez-Martínez, R.; Zurita-Díaz, M.A.; López-Estrada, G.; Delgado-Romero, K.; Peralta-Zaragoza, O.; Bermúdez-Morales, V.H.; Cantú, D.; et al. Risk allelic load in Th2 and Th3 cytokines genes as biomarker of susceptibility to HPV-16 positive cervical cancer: A case control study. BMC Cancer 2016, 16, 330. [Google Scholar] [CrossRef]
- Wang, S.S.; Gonzalez, P.; Yu, K.; Porras, C.; Li, Q.; Safaeian, M.; Rodriguez, A.C.; Sherman, M.E.; Bratti, C.; Schiffman, M.; et al. Common genetic variants and risk for HPV persistence and progression to cervical cancer. PLoS ONE 2010, 5, 1–7. [Google Scholar] [CrossRef]
- Klug, S.J.; Ressing, M.; Koenig, J.; Abba, M.C.; Agorastos, T.; Brenna, S.M.; Ciotti, M.; Das, B.; Del Mistro, A.; Dybikowska, A.; et al. TP53 codon 72 polymorphism and cervical cancer: A pooled analysis of individual data from 49 studies. Lancet Oncol. 2009, 10, 772–784. [Google Scholar] [CrossRef]
- Sud, A.; Kinnersley, B.; Houlston, R.S. Genome-wide association studies of cancer: Current insights and future perspectives. Nat. Rev. Cancer 2017, 17, 692–704. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Mbatchou, J.; Barnard, L.; Backman, J.; Marcketta, A.; Kosmicki, J.A.; Ziyatdinov, A.; Benner, C.; O’Dushlaine, C.; Barber, M.; Boutkov, B.; et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 2021, 53, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Cano-Gamez, E.; Trynka, G. From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases. Front. Genet. 2020, 11, 1–21. [Google Scholar] [CrossRef]
- Freedman, M.L.; Monteiro, A.N.A.; Gayther, S.A.; Coetzee, G.A.; Risch, A.; Plass, C.; Casey, G.; De Biasi, M.; Carlson, C.; Duggan, D.; et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet. 2011, 43, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-Wide Association Studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Dayem Ullah, A.Z.; Oscanoa, J.; Wang, J.; Nagano, A.; Lemoine, N.R.; Chelala, C. SNPnexus: Assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res. 2018, 46, W109–W113. [Google Scholar] [CrossRef]
- Arnold, M.; Raffler, J.; Pfeufer, A.; Suhre, K.; Kastenmüller, G. SNiPA: An interactive, genetic variant-centered annotation browser. Bioinformatics 2015, 31, 1334–1336. [Google Scholar] [CrossRef]
- Loos, R.J.F. 15 Years of Genome-Wide Association Studies and No Signs of Slowing Down. Nat. Commun. 2020, 11, 10–12. [Google Scholar] [CrossRef]
- Graff, R.E.; Cavazos, T.B.; Thai, K.K.; Kachuri, L.; Rashkin, S.R.; Hoffman, J.D.; Alexeeff, S.E.; Blatchins, M.; Meyers, T.J.; Leong, L.; et al. Cross-cancer evaluation of polygenic risk scores for 16 cancer types in two large cohorts. Nat. Commun. 2021, 12, 970. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, S.; Hirata, J.; Kanai, M.; Suzuki, K.; Akiyama, M.; Lai Too, C.; Arayssi, T.; Hammoudeh, M.; Al Emadi, S.; Masri, B.K.; et al. Dimensionality reduction reveals fine-scale structure in the Japanese population with consequences for polygenic risk prediction. Nat. Commun. 2020, 11, 1569. [Google Scholar] [CrossRef] [PubMed]
- Reay, W.R.; Cairns, M.J. Advancing the use of genome-wide association studies for drug repurposing. Nat. Rev. Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gyllensten, U. Lessons and implications from association studies and post-GWAS analyses of cervical cancer. Trends Genet. 2015, 31, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Juko-Pecirep, I.; Hammer, J.; Ivansson, E.; Enroth, S.; Gustavsson, I.; Feuk, L.; Magnusson, P.K.E.; McKay, J.D.; Wilander, E.; et al. Genome-wide association study of susceptibility loci for cervical cancer. J. Natl. Cancer Inst. 2013, 105, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, L.; Hu, Z.; Li, S.; Wang, S.S.; Liu, J.J.; Wu, C.; He, L.; Zhou, J.; Li, Z.Z.Z.; et al. A genome-wide association study identifies two new cervical cancer susceptibility loci at 4q12 and 17q12. Nat. Genet. 2013, 45, 918–922. [Google Scholar] [CrossRef]
- Chen, D.; Enroth, S.; Liu, H.; Sun, Y.; Wang, H.; Yu, M.; Deng, L.; Xu, S.; Gyllensten, U. Pooled analysis of genome-wide association studies of cervical intraepithelial neoplasia 3 (CIN3) identifies a new susceptibility locus. Oncotarget 2016, 7, 42216–42224. [Google Scholar] [CrossRef]
- Takeuchi, F.; Kukimoto, I.; Li, Z.; Li, S.; Li, N.; Hu, Z.; Takahashi, A.; Inoue, S.; Yokoi, S.; Chen, J.; et al. Genome-wide association study of cervical cancer suggests a role for ARRDC3 gene in human papillomavirus infection. Hum. Mol. Genet. 2019, 28, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, K.; Akiyama, M.; Kanai, M.; Takahashi, A.; Kawakami, E.; Sugishita, H.; Sakaue, S.; Matoba, N.; Low, S.-K.; Okada, Y.; et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 2020, 52, 669–679. [Google Scholar] [CrossRef]
- Bowden, S.J.; Bodinier, B.; Kalliala, I.; Zuber, V.; Vuckovic, D.; Doulgeraki, T.; Whitaker, M.D.; Wielscher, M.; Cartwright, R.; Tsilidis, K.K.; et al. Genetic variation in cervical preinvasive and invasive disease: A genome-wide association study. Lancet Oncol. 2021, 22, 548–557. [Google Scholar] [CrossRef]
- Koel, M.; Võsa, U.; Lepamets, M.; Laivuori, H.; Lemmelä, S.; Daly, M.; Biobank Research Team, E.; Palta, P.; Mägi, R.; Laisk, T. GWAS meta-analysis and gene expression data link reproductive tract development, immune response and cellular proliferation/apoptosis with cervical cancer and clarify overlap with other cervical phenotypes. medRxiv 2021. [Google Scholar] [CrossRef]
- Olafsdottir, T.; Stacey, S.N.; Sveinbjornsson, G.; Thorleifsson, G.; Norland, K.; Sigurgeirsson, B.; Thorisdottir, K.; Kristjansson, A.K.; Tryggvadottir, L.; Sarin, K.Y.; et al. Loss-of-Function Variants in the Tumor-Suppressor Gene PTPN14 Confer Increased Cancer Risk. Cancer Res. 2021, 81, 1954–1964. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Low, S.K.; Akiyama, M.; Hirata, M.; Ueda, Y.; Matsuda, K.; Kimura, T.; Murakami, Y.; Kubo, M.; Kamatani, Y.; et al. GWAS of five gynecologic diseases and cross-trait analysis in Japanese. Eur. J. Hum. Genet. 2019, 95–107. [Google Scholar] [CrossRef]
- Park, I.; Terasaki, P. Origins of the first HLA specificities. Hum. Immunol. 2000, 61, 185–189. [Google Scholar] [CrossRef]
- Horton, R.; Wilming, L.; Rand, V.; Lovering, R.C.; Bruford, E.A.; Khodiyar, V.K.; Lush, M.J.; Povey, S.; Talbot, C.C.; Wright, M.W.; et al. Gene map of the extended human MHC. Nat. Rev. Genet. 2004, 5, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gyllensten, U. A cis-eQTL of HLA-DRB1 and a frameshift mutation of MICA contribute to the pattern of association of HLA alleles with cervical cancer. Cancer Med. 2014, 3, 445–452. [Google Scholar] [CrossRef]
- Ashiru, O.; Boutet, P.; Fernández-Messina, L.; Agüera-González, S.; Skepper, J.N.; Valés-Gómez, M.; Reyburn, H.T. Natural Killer Cell Cytotoxicity Is Suppressed by Exposure to the Human NKG2D Ligand MICA*008 That Is Shed by Tumor Cells in Exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef]
- Chen, D.; Enroth, S.; Ivansson, E.; Gyllensten, U. Pathway analysis of cervical cancer genome-wide association study highlights the MHC region and pathways involved in response to infection. Hum. Mol. Genet. 2014, 23, 6047–6060. [Google Scholar] [CrossRef]
- McKay, J.; Tenet, V.; Franceschi, S.; Chabrier, A.; Gheit, T.; Gaborieau, V.; Chopin, S.; Avogbe, P.H.; Tommasino, M.; Ainouze, M.; et al. Immuno-related polymorphisms and cervical cancer risk: The IARC multicentric case-control study. PLoS ONE 2017, 12, e0177775. [Google Scholar] [CrossRef]
- Ramachandran, D.; Schürmann, P.; Mao, Q.; Wang, Y.; Bretschneider, L.M.; Speith, L.M.; Hülse, F.; Enßen, J.; Bousset, K.; Jentschke, M.; et al. Association of genomic variants at the human leukocyte antigen locus with cervical cancer risk, HPV status and gene expression levels. Int. J. Cancer 2020, 147, 2458–2468. [Google Scholar] [CrossRef]
- Chen, D.; Gyllensten, U. Systematic investigation of contribution of genetic variation in the HLA-DP region to cervical cancer susceptibility. Carcinogenesis 2014, 35, 1765–1769. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, L.; Lawrence, W.D.; Kukuruga, D.; Eisenbrey, A.B.; Lancaster, W.D. Association between HLA-DQB1 alleles and risk for cervical cancer in African-American women. Int. J. Cancer 1994, 57, 504–507. [Google Scholar] [CrossRef]
- Bao, X.; Hanson, A.L.; Madeleine, M.M.; Wang, S.S.; Schwartz, S.M.; Newell, F.; Pettersson-Kymmer, U.; Hemminki, K.; Tiews, S.; Steinberg, W.; et al. HLA and KIR Associations of Cervical Neoplasia. J. Infect. Dis. 2018, 218, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, G.R.; Olivieri, J.E.; DeBoever, C.; Tanigawa, Y.; Justesen, J.M.; Dilthey, A.; Rivas, M.A. Pervasive additive and non-additive effects within the HLA region contribute to disease risk in the UK Biobank. BioRxiv 2020, 1–19. [Google Scholar] [CrossRef]
- Engelmark, M. Affected sib-pair analysis of the contribution of HLA class I and class II loci to development of cervical cancer. Hum. Mol. Genet. 2004, 13, 1951–1958. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Y.; Bai, Y. Role of GSDMB in Pyroptosis and Cancer. Cancer Manag. Res. 2020, 12, 3033–3043. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef]
- He, H.; Yi, L.; Zhang, B.; Yan, B.; Xiao, M.; Ren, J.; Zi, D.; Zhu, L.; Zhong, Z.; Zhao, X.; et al. USP24-GSDMB complex promotes bladder cancer proliferation via activation of the STAT3 pathway. Int. J. Biol. Sci. 2021, 17, 2417–2429. [Google Scholar] [CrossRef]
- Sun, Q.; Yang, J.; Xing, G.; Sun, Q.; Zhang, L.; He, F. Expression of GSDML Associates with Tumor Progression in Uterine Cervix Cancer. Transl. Oncol. 2008, 1, 73-IN1. [Google Scholar] [CrossRef]
- Ramachandran, D.; Wang, Y.; Schürmann, P.; Hülse, F.; Mao, Q.; Jentschke, M.; Böhmer, G.; Strauß, H.G.; Hirchenhain, C.; Schmidmayr, M.; et al. Association of genomic variants at PAX8 and PBX2 with cervical cancer risk. Int. J. Cancer 2021, 149, 893–900. [Google Scholar] [CrossRef]
- Han, J.; Zhou, W.; Jia, M.; Wen, J.; Jiang, J.; Shi, J.; Zhang, K.; Ma, H.; Liu, J.; Ren, J.; et al. Expression quantitative trait loci in long non-coding RNA PAX8-AS1 are associated with decreased risk of cervical cancer. Mol. Genet. Genom. 2016, 291, 1743–1748. [Google Scholar] [CrossRef]
- Kar, S.P.; Adler, E.; Tyrer, J.; Hazelett, D.; Anton-Culver, H.; Bandera, E.V.; Beckmann, M.W.; Berchuck, A.; Bogdanova, N.; Brinton, L.; et al. Enrichment of putative PAX8 target genes at serous epithelial ovarian cancer susceptibility loci. Br. J. Cancer 2017, 116, 524–535. [Google Scholar] [CrossRef]
- Beesley, J.; Pickett, H.A.; Johnatty, S.E.; Dunning, A.M.; Chen, X.; Li, J.; Michailidou, K.; Lu, Y.; Rider, D.N.; Palmieri, R.T.; et al. Functional polymorphisms in the TERT promoter are associated with risk of serous epithelial ovarian and breast cancers. PLoS ONE 2011, 6, e24987. [Google Scholar] [CrossRef]
- O’Mara, T.A.; Glubb, D.M.; Amant, F.; Annibali, D.; Ashton, K.; Attia, J.; Auer, P.L.; Beckmann, M.W.; Black, A.; Bolla, M.K.; et al. Identification of nine new susceptibility loci for endometrial cancer. Nat. Commun. 2018, 9, 3166. [Google Scholar] [CrossRef]
- Haiman, C.A.; Chen, G.K.; Vachon, C.M.; Canzian, F.; Dunning, A.; Millikan, R.C.; Wang, X.; Ademuyiwa, F.; Ahmed, S.; Ambrosone, C.B.; et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor–negative breast cancer. Nat. Genet. 2011, 43, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Rafnar, T.; Sulem, P.; Stacey, S.N.; Geller, F.; Gudmundsson, J.; Sigurdsson, A.; Jakobsdottir, M.; Helgadottir, H.; Thorlacius, S.; Aben, K.K.H.; et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat. Genet. 2009, 41, 221–227. [Google Scholar] [CrossRef]
- Miura, K.; Mishima, H.; Kinoshita, A.; Hayashida, C.; Abe, S.; Tokunaga, K.; Masuzaki, H.; Yoshiur, K. Genome-Wide Association Study of HPV-Associated Cervical Cancer in Japanese Women. J. Med. Virol. 2014, 86, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Batista, T.M.; Dagdeviren, S.; Carroll, S.H.; Cai, W.; Melnik, V.Y.; Noh, H.L.; Saengnipanthkul, S.; Kim, J.K.; Kahn, C.R.; Lee, R.T. Arrestin domain-containing 3 (Arrdc3) modulates insulin action and glucose metabolism in liver. Proc. Natl. Acad. Sci. USA 2020, 117, 6733–6740. [Google Scholar] [CrossRef]
- Arakaki, A.K.S.; Pan, W.-A.; Wedegaertner, H.; Roca-Mercado, I.; Chinn, L.; Gujral, T.S.; Trejo, J. α-Arrestin ARRDC3 tumor suppressor function is linked to GPCR-induced TAZ activation and breast cancer metastasis. J. Cell Sci. 2021, 134, jcs254888. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Ito, H.; Hirata, J.; Sakaue, S.; Ueda, Y.; Kimura, T.; Takeuchi, F.; Murakami, Y.; Matsuda, K.; Matsuo, K.; et al. Fine Mapping of the Major Histocompatibility Complex Region and Association of the HLA-B*52:01 Allele with Cervical Cancer in Japanese Women. JAMA Netw. Open 2020, 3, 1–9. [Google Scholar] [CrossRef]
- Riancho, J.A.; Vázquez, L.; García-Pérez, M.A.; Sainz, J.; Olmos, J.M.; Hernández, J.L.; Pérez-López, J.; Amado, J.A.; Zarrabeitia, M.T.; Cano, A.; et al. Association of ACACB polymorphisms with obesity and diabetes. Mol. Genet. Metab. 2011, 104, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Huang, K.; Zhang, Q.; Zhou, J.; Sun, H.; Tang, F.; Zhou, H.; Hu, T.; Wang, S.; Jia, Y.; et al. Genome-wide association study identifies four SNPs associated with response to platinum-based neoadjuvant chemotherapy for cervical cancer. Sci. Rep. 2017, 7, 41103. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Pozzoli, U.; Cagliani, R.; Comi, G.P.; Bresolin, N.; Clerici, M.; Sironi, M. Genome-Wide Identification of Susceptibility Alleles for Viral Infections through a Population Genetics Approach. PLoS Genet. 2010, 6, e1000849. [Google Scholar] [CrossRef]
- Chen, D.; McKay, J.D.; Clifford, G.; Gaborieau, V.; Chabrier, A.; Waterboer, T.; Zaridze, D.; Lissowska, J.; Rudnai, P.; Fabianova, E.; et al. Genome-wide association study of HPV seropositivity. Hum. Mol. Genet. 2011, 20, 4714–4723. [Google Scholar] [CrossRef]
- Adebamowo, S.N.; Adeyemo, A.A.; Rotimi, C.N.; Olaniyan, O.; Offiong, R.; Adebamowo, C.A.; Odutola, M.; Dareng, E.O.; Famooto, A.O.; Adebiyi, R. Genome-wide association study of prevalent and persistent cervical high-risk human papillomavirus (HPV) infection. BMC Med. Genet. 2020, 21, 1–10. [Google Scholar] [CrossRef]
- Liu, J.; Tang, W.; Budhu, A.; Forgues, M.; Hernandez, M.O.; Candia, J.; Kim, Y.; Bowman, E.D.; Ambs, S.; Zhao, Y.; et al. A Viral Exposure Signature Defines Early Onset of Hepatocellular Carcinoma. Cell 2020, 182, 317–328. [Google Scholar] [CrossRef]
- Chen, H.; Wang, T.; Huang, S.; Zeng, P. New novel non-MHC genes were identified for cervical cancer with an integrative analysis approach of transcriptome-wide association study. J. Cancer 2021, 12, 840–848. [Google Scholar] [CrossRef]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212. [Google Scholar] [CrossRef] [PubMed]
- Kachuri, L.; Graff, R.E.; Smith-Byrne, K.; Meyers, T.J.; Rashkin, S.R.; Ziv, E.; Witte, J.S.; Johansson, M. Pan-cancer analysis demonstrates that integrating polygenic risk scores with modifiable risk factors improves risk prediction. Nat. Commun. 2020, 11, 6084. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.H.; Wang, Q.; Bolla, M.K.; et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef]
- Franceschi, S. Genomic characterisation of cervical cancer and human papillomavirus: New opportunities for precision medicine. Lancet Oncol. 2021, 22, 419–420. [Google Scholar] [CrossRef]
- Masuda, T.; Ogawa, K.; Kamatani, Y.; Murakami, Y.; Kimura, T.; Okada, Y. A Mendelian randomization study identified obesity as a causal risk factor of uterine endometrial cancer in Japanese. Cancer Sci. 2020, 111, 4646–4651. [Google Scholar] [CrossRef] [PubMed]
- Seddighi, S.; Houck, A.L.; Rowe, J.B.; Pharoah, P.D.P. Evidence of a Causal Association Between Cancer and Alzheimer’s Disease: A Mendelian Randomization Analysis. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Kar, S.; Carter, P.; Vithayathil, M.; Mason, A.M.; Burgess, S.; Larsson, S.C. Is type 2 diabetes causally associated with cancer risk? Evidence from a two-sample mendelian randomization study. Diabetes 2020, 69, 1588–1596. [Google Scholar] [CrossRef]
- Thorsby, E. Invited anniversary review: HLA associated diseases. Hum. Immunol. 1997, 53, 1–11. [Google Scholar] [CrossRef]
- Trowsdale, J.; Knight, J.C. Major Histocompatibility Complex Genomics and Human Disease. Annu. Rev. Genom. Hum. Genet. 2013, 14, 301–323. [Google Scholar] [CrossRef]
- Liu, Z.; Derkach, A.; Yu, K.J.; Yeager, M.; Chang, Y.-S.; Chen, C.-J.; Gyllensten, U.; Lan, Q.; Lee, M.-H.; McKay, J.D.; et al. Patterns of Human Leukocyte Antigen Class I and Class II Associations and Cancer. Cancer Res. 2021, 81, 1148–1152. [Google Scholar] [CrossRef]
- Chen, D.; Hammer, J.; Lindquist, D.; Idahl, A.; Gyllensten, U. A variant upstream of HLA-DRB1 and multiple variants in MICA influence susceptibility to cervical cancer in a Swedish population. Cancer Med. 2014, 3, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Pereyra, F.; Jia, X.; McLaren, P.J.; Telenti, A.; de Bakker, P.I.W.; Walker, B.D.; Ripke, S.; Brumme, C.J.; Pulit, S.L.; Carrington, M.; et al. The Major Genetic Determinants of HIV-1 Control Affect HLA Class I Peptide Presentation. Science 2010, 330, 1551–1557. [Google Scholar] [CrossRef]
- McLaren, P.J.; Fellay, J. HIV-1 and human genetic variation. Nat. Rev. Genet. 2021, 22, 645–657. [Google Scholar] [CrossRef]
- Monteiro, A.N.A.; Freedman, M.L. Lessons from postgenome-wide association studies: Functional analysis of cancer predisposition loci. J. Intern. Med. 2013, 274, 414–424. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| GWAS | Population | GWS Risk Loci | Comment | Ref. | |
|---|---|---|---|---|---|
| Chen et al., 2013 | Swedish | 6p21.33 | rs2516448 (MICA) | [116] | |
| 6p21.32 | rs9272143 (HLA-DRB1/HLA-DQA1) a | ||||
| 6p21.32 | rs3117027 (HLA-DPB2) | ||||
| Shi et al., 2013 | Chinese | 6p21.32 | rs4282438 (HLA-DPB1/HLA-DPB2) | [117] | |
| 4q12 | rs13117307 (EXOC1) | ||||
| 17q12 | rs8067378 (GSDMB) b | ||||
| Chen et al., 2016 | Swedish | 6p21.32 | * rs9271898 (HLA-DQA1) a | Pooled analysis with mainly CIN3 | [118] |
| 6p21.33 | * rs2516448 (MICA) | ||||
| 6p21.32 | * rs3130196 (HLA-DPA2) | ||||
| 6p21.32 | rs73730372 (HLA-DQA1/HLA-DQB1) c | ||||
| 6p21.32–6p21.33 | HLA alleles HLA-B* 07:02, HLA-B* 15:01, HLA-DRB1* 13:01, HLA-DRB1* 15:01, HLA-DQA1* 01:03, HLA-DQB1* 06:03, HLA-DQB1* 06:02, HLA-C* 07:02 | ||||
| Leo et al., 2017 | Caucasian | 6p21.32–6p21.33 | Replication of HLA haplotypes that are determined by the amino-acids carried at positions 13 and 71 of HLA-DRB1 and position 156 in HLA-B | [66] | |
| Rashkin et al., 2020 | UK/US | 2q13 | rs10175462 (PAX8/PAX8-AS1) d | Combined analysis of UK and GERA biobanks | [68] |
| 6p21.32 | rs2856437 (PBX2) | ||||
| Takeuchi et al., 2019 | Japanese | 5q14.3 | rs59661306 (ADGRV1/ARRDC3) | [119] | |
| 7p11.2 | rs7457728 (LINC01445/VSTM2A) | ||||
| Ishigaki et al., 2020 | Japanese | no new loci for cervical cancer | [120] | ||
| Bowden et al., 2021 | UK/Finnish | 2q13 | * rs35724515 (PAX8/PAX8-AS1) d | Analysis of UK Biobank and validation in the FinnGen biobank. | [121] |
| 5p15.3 | rs27069 (CLPTM1L) | ||||
| 6p21.32 | * rs9272050 (HLA-DQA1) a | ||||
| 6p21.33 | rs6938453 (MICA) | ||||
| 6p21.32 | * rs55986091 (HLA-DQB1) c | ||||
| 6p21.33 | rs9266183 (HLA-B) | ||||
| 6p21.32 | * rs9272245 (HLA-DQA1) a | ||||
| 1p36.32 | rs138446575 (TTC34) | ||||
| 12q24.11 | rs117960705 (ACACB) | ||||
| Koel et al., 2021 | Multi- ethnic | 1p36.12 | rs2268177 (LINC00339/CDC42) | Meta-analysis of UK, FinnGen, Japanese RIKEN and Estonian biobanks. | [122] |
| 2q13 | * rs4849177 (PAX8/PAX8-AS1) d | ||||
| 5p15.3 | * rs27069 (CLPTM1L) | ||||
| 17q12 | * rs12603332 (GSDMB) b | ||||
| 2q24.1 | rs12611652 (DAPL1) | ||||
| 6p21.32 | * rs35508382 (HLA-DRB1/HLA-DQA1) c | ||||
| 6p21.33 | rs1053726 (HLA-B) e | ||||
| 6p21.32 | * rs36214159 (HLA-DQA1) c | ||||
| 19p13.3 | rs425787 (CD70) | ||||
| Gene-based analysis | |||||
| Olafsdottir et al., 2021 | Icelandic | 1q32.3–41 | 3 LoF variants (c.-1_2delinsATGG, p.Gln503ArgfsTer12, c.3271 + 1G > A) in PTPN14 | Burden analysis after imputation of rare PVs | [123] |
| Cross-trait analysis | |||||
| Masuda et al., 2019 | Japanese | 11p15.5 | rs150806792 (INS-IGF2) | Combined analysis of cervical cancer with uterine cancer (INS-IGF2, SOX9) or five different cancers | [124] |
| 17q24.3 | rs140991990 (SOX9) | ||||
| 2p16.3 | rs937380553 (LOC730100) | ||||
| 9q22.33 | rs73494486 (GABBR2) | ||||
| 15q25.2 | rs145152209 (SH3GL3/BNC1) | ||||
| 21q22.2 | rs147427629 (LOC107985484) | ||||
| Rashkin et al., 2020 | UK/US | 4q24 | rs10007915 (TET2) | Combined analysis of cervical cancer with 15 different cancers | [68] |
| 6p21.33 | rs17190106 (MUC22/HCG22) | ||||
| 6p21.33 | *rs9266766 (HLA-S/MICA) e | ||||
| 6p21.33 | rs114060326 (MICB/MCCD1) | ||||
| 6p21.33 | rs2763979 (HSPA1B) | ||||
| 6p21.32 | rs34563311 (HLA-DRB1) | ||||
| 6p21.32 | * rs9270747 (HLA-DRB1/HLA-DQA1) a | ||||
| 6p21.32 | rs535777 (HLA-DRB1/HLA-DQA1) | ||||
| 8q24.21 | rs117952826 (CASC8) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramachandran, D.; Dörk, T. Genomic Risk Factors for Cervical Cancer. Cancers 2021, 13, 5137. https://doi.org/10.3390/cancers13205137
Ramachandran D, Dörk T. Genomic Risk Factors for Cervical Cancer. Cancers. 2021; 13(20):5137. https://doi.org/10.3390/cancers13205137
Chicago/Turabian StyleRamachandran, Dhanya, and Thilo Dörk. 2021. "Genomic Risk Factors for Cervical Cancer" Cancers 13, no. 20: 5137. https://doi.org/10.3390/cancers13205137
APA StyleRamachandran, D., & Dörk, T. (2021). Genomic Risk Factors for Cervical Cancer. Cancers, 13(20), 5137. https://doi.org/10.3390/cancers13205137