Circular RNAs in Hedgehog Signaling Activation and Hedgehog-Mediated Medulloblastoma Tumors

 ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drug Treatments
2.3. Human Normal Brain Cerebellum and Medulloblastoma Tumor Samples
2.4. RNA Isolation
2.5. circRNA Enrichment and RNA-Seq Library Preparations
2.6. RNA-Seq Data Analysis
2.7. Validation of Back-Spliced Junctions by Sanger Sequencing
2.8. cDNA Synthesis, Real-Time qPCR
2.9. Validation of Linear Back-Spliced RNAs by qPCR
2.10. siRNA and Plasmid Transfections
2.11. Cell Proliferation Assays
2.11.1. EdU Incorporation Assay
2.11.2. WST-1 Viability Assay
2.12. Statistical and Bioinformatic Analysis
3. Results
3.1. Detection of Abundantly Expressed circRNAs in the Context of Hedgehog Signaling Upregulation
3.1.1. Hedgehog Signaling Activation in Daoy and HEPM Cells
3.1.2. RNA-Seq: Detection of Linear, Circular, and Linear Back-Spliced Transcripts
3.1.3. Linear RNA-Seq Data Analysis. Confirmation of Hedgehog Pathway Activation in Daoy and HEPM Cells
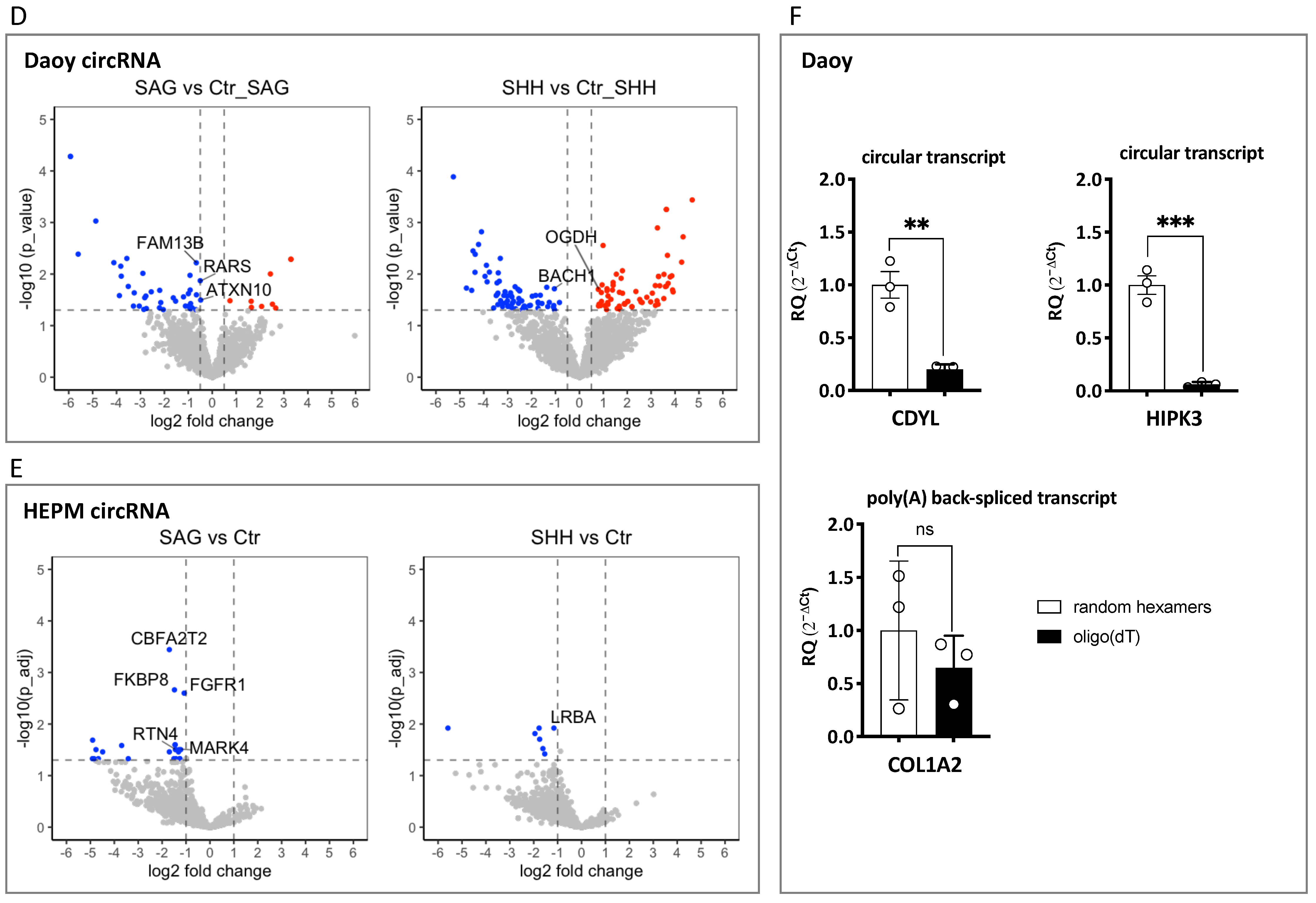
3.1.4. circRNA-Seq Data Analysis. Differentially Expressed circRNAs in SAG and SHH Treated Daoy and HEPM Cells
3.1.5. Linear Back-Spliced RNA-Seq Data Analysis and qPCR Validations
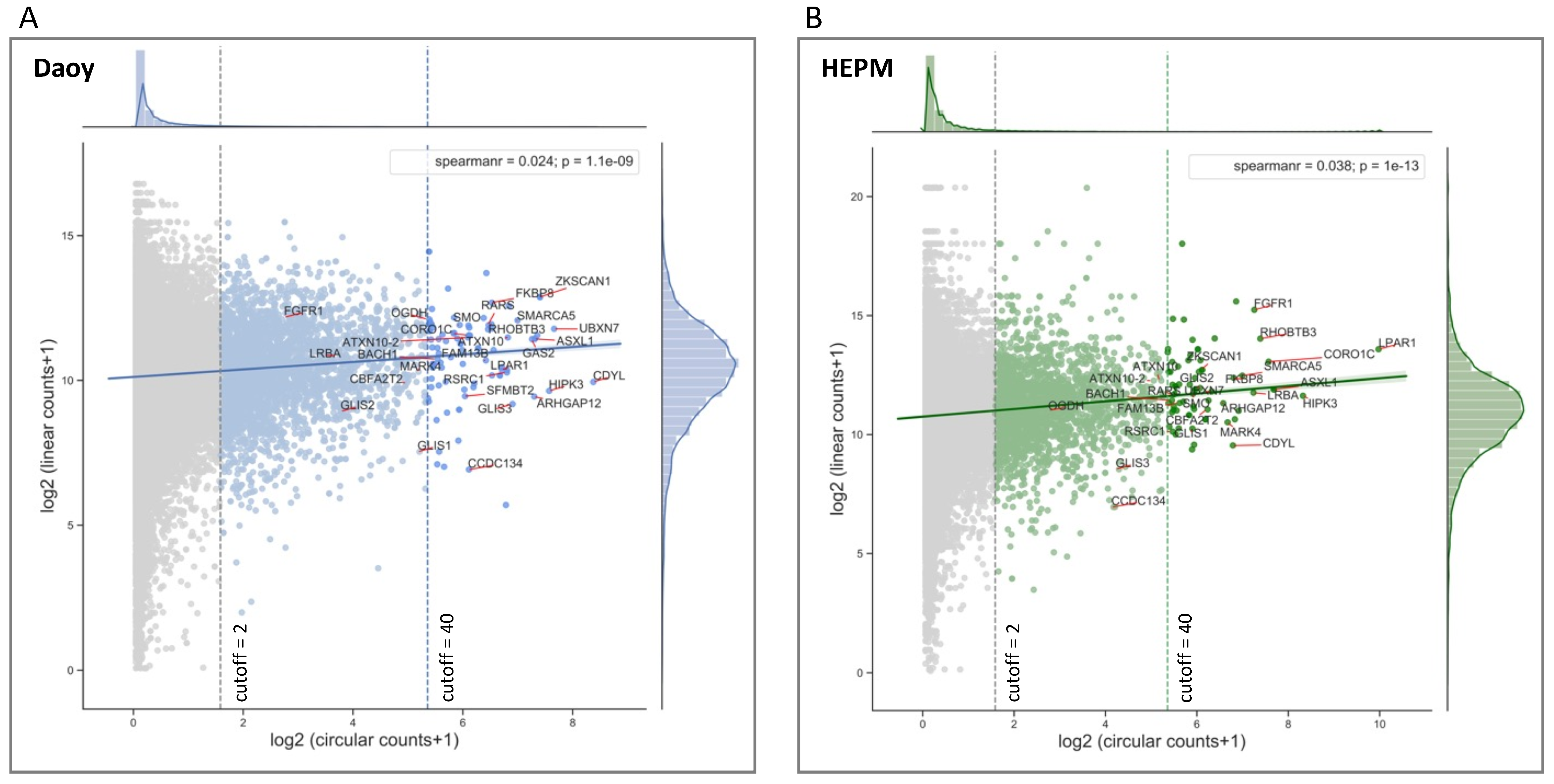
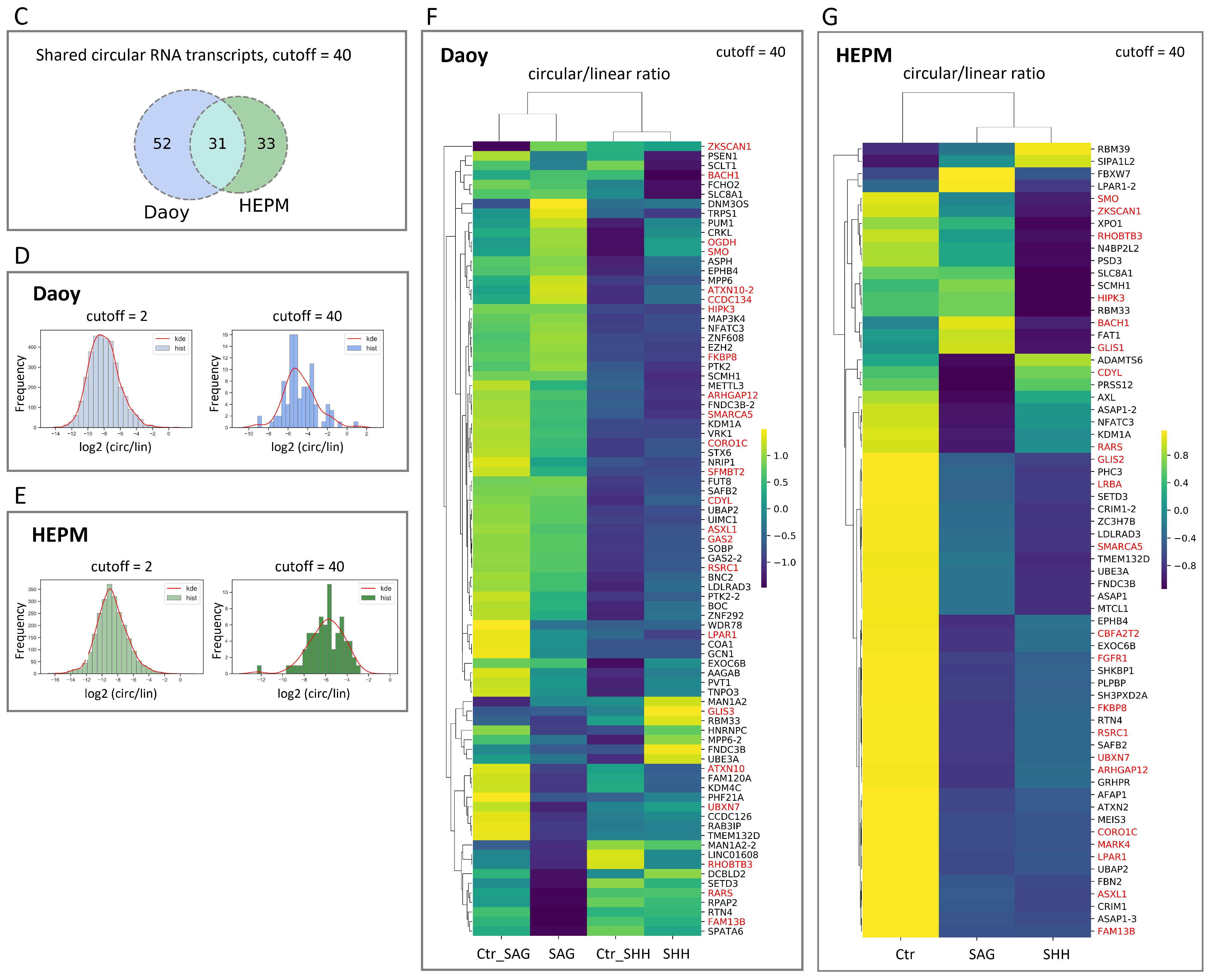
3.2. Circular to Linear Ratio Analysis of RNA-Seq Data
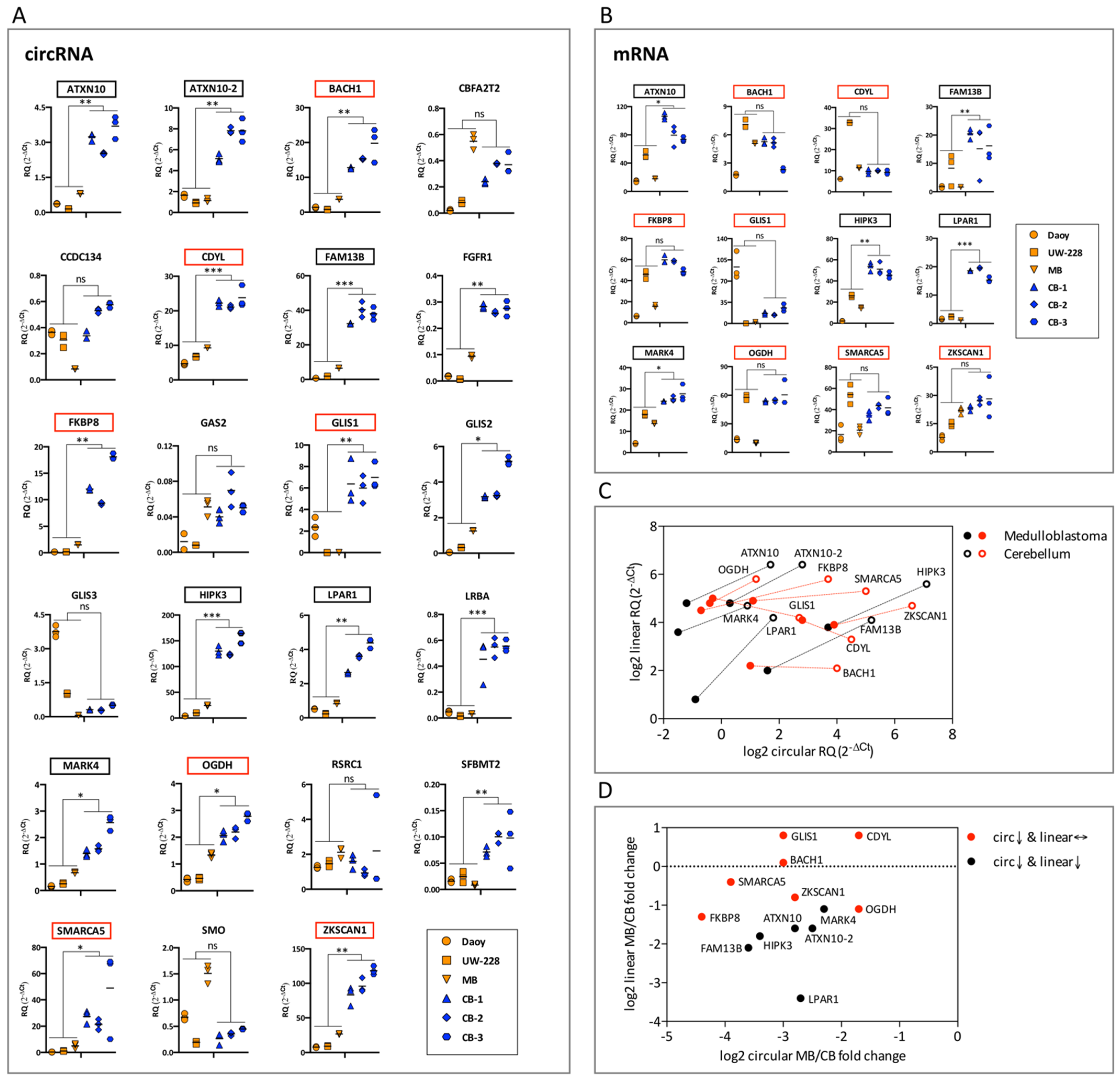
3.3. Differentially Expressed circRNAs in SHH Subtype Medulloblastoma (Daoy, UW-228 Cell Lines and Tumor Sample) and Normal Cerebellum
3.4. Effect of Depletion and Overexpression of the Seven Selected circRNAs on Cell Proliferation in Medulloblastoma Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| circRNA | circular RNA |
| CNS | Central nervous system |
| DE | Differentially expressed |
| GLI1 | Glioma-associated oncogene 1 |
| HHIP | Hedgehog-interacting protein |
| PTCH | Patched |
| SAG | Smoothened agonist |
| SHH | Sonic Hedgehog |
| SMO | Smoothened |
| SUFU | Suppressor of Fused |
| TBP | TATA-binding protein |
References
- Aberger, F.; Ruiz, I.A.A. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Teglund, S.; Toftgård, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, L.; Beachy, P.A. The Hedgehog response network: Sensors, switches, and routers. Science 2004, 304, 1755–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusert, J.M.; Wu, X.; Eberhart, C.G.; Taylor, M.D.; Wechsler-Reya, R.J. SnapShot: Medulloblastoma. Cancer Cell 2014, 26, 940–940.e941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 2019, 24, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Schouten-van Meeteren, A.; Van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H.; Stephen, D.; Joyner, A.; Curran, T. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene 2005, 24, 4026–4036. [Google Scholar] [CrossRef] [Green Version]
- Zurawel, R.H.; Allen, C.; Chiappa, S.; Cato, W.; Biegel, J.; Cogen, P.; de Sauvage, F.; Raffel, C. Analysis ofPTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromosom. Cancer 2000, 27, 44–51. [Google Scholar] [CrossRef]
- Girardi, F.; Allemani, C.; Coleman, M.P. Worldwide Trends in Survival from Common Childhood Brain Tumors: A Systematic Review. J. Glob. Oncol. 2019, 5, 1–25. [Google Scholar] [CrossRef]
- Cocquerelle, C.; Mascrez, B.; Hétuin, D.; Bailleul, B. Mis-splicing yields circular RNA molecules. FASEB J. 1993, 7, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Nigro, J.M.; Cho, K.R.; Fearon, E.R.; Kern, S.E.; Ruppert, J.M.; Oliner, J.D.; Kinzler, K.W.; Vogelstein, B. Scrambled exons. Cell 1991, 64, 607–613. [Google Scholar] [CrossRef]
- Zaphiropoulos, P.G. Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: Correlation with exon skipping. Proc. Natl. Acad. Sci. USA 1996, 93, 6536–6541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwecka, M.; Glažar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Cerda Jara, C.A.; Fenske, P.; et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017, 357, eaam8526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Barrett, S.P.; Wang, P.L.; Salzman, J. Circular RNA biogenesis can proceed through an exon-containing lariat precursor. eLife 2015, 4, e07540. [Google Scholar] [CrossRef] [PubMed]
- Maass, P.G.; Glažar, P.; Memczak, S.; Dittmar, G.; Hollfinger, I.; Schreyer, L.; Sauer, A.V.; Toka, O.; Aiuti, A.; Luft, F.C.; et al. A map of human circular RNAs in clinically relevant tissues. J. Mol. Med. 2017, 95, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Huang, V.; Xu, X.; Livingstone, J.; Soares, F.; Jeon, J.; Zeng, Y.; Hua, J.T.; Petricca, J.; Guo, H.; et al. Widespread and Functional RNA Circularization in Localized Prostate Cancer. Cell 2019, 176, 831–843.e822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.-M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881.e813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Xu, Q.-G.; Wang, Z.-G.; Yang, Y.; Zhang, L.; Ma, J.-Z.; Sun, S.-H.; Yang, F.; Zhou, W.-P. Circular RNA cSMARCA5 inhibits growth and metastasis in hepatocellular carcinoma. J. Hepatol. 2018, 68, 1214–1227. [Google Scholar] [CrossRef]
- Hsiao, K.-Y.; Lin, Y.-C.; Gupta, S.K.; Chang, N.; Yen, L.; Sun, H.S.; Tsai, S.-J. Noncoding Effects of Circular RNA CCDC66 Promote Colon Cancer Growth and Metastasis. Cancer Res. 2017, 77, 2339–2350. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-O.; Dong, R.; Zhang, Y.; Zhang, J.-L.; Luo, Z.; Zhang, J.; Chen, L.-L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Higdon, R.; Kala, J.; Wilkins, D.; Yan, J.F.; Sethi, M.K.; Lin, L.; Liu, S.; Montague, E.; Janko, I.; Choiniere, J.; et al. Integrated Proteomic and Transcriptomic-Based Approaches to Identifying Signature Biomarkers and Pathways for Elucidation of Daoy and UW228 Subtypes. Proteomes 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Triscott, J.; Lee, C.; Foster, C.; Manoranjan, B.; Pambid, M.R.; Berns, R.; Fotovati, A.; Venugopal, C.; O’Halloran, K.; Narendran, A.; et al. Personalizing the Treatment of Pediatric Medulloblastoma: Polo-like Kinase 1 as a Molecular Target in High-Risk Children. Cancer Res. 2013, 73, 6734–6744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finta, C.; Zaphiropoulos, P.G. Intergenic mRNA Molecules Resulting from trans-Splicing. J. Biol. Chem. 2002, 277, 5882–5890. [Google Scholar] [CrossRef] [Green Version]
- Frantz, S.A.; Thiara, A.S.; Lodwick, D.; Ng, L.L.; Eperon, I.C.; Samani, N.J. Exon repetition in mRNA. Proc. Natl. Acad. Sci. USA 1999, 96, 5400–5405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caudevilla, C.; Serra, D.; Miliar, A.; Codony, C.; Asins, G.; Bach, M.; Hegardt, F.G. Natural trans-splicing in carnitine octanoyltransferase pre-mRNAs in rat liver. Proc. Natl. Acad. Sci. USA 1998, 95, 12185–12190. [Google Scholar] [CrossRef] [Green Version]
- Bachmayr-Heyda, A.; Reiner, A.T.; Auer, K.; Sukhbaatar, N.; Aust, S.; Bachleitner-Hofmann, T.; Mesteri, I.; Grunt, T.W.; Zeillinger, R.; Pils, D. Correlation of circular RNA abundance with proliferation—Exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis and normal human tissues. Sci. Rep. 2015, 5, 8057. [Google Scholar] [CrossRef] [PubMed]
- Holdt, L.M.; Kohlmaier, A.; Teupser, D. Circular RNAs as Therapeutic Agents and Targets. Front. Physiol. 2018, 9, 1262. [Google Scholar] [CrossRef] [PubMed]
- Sharova, L.V.; Sharov, A.A.; Nedorezov, T.; Piao, Y.; Shaik, N.; Ko, M.S. Database for mRNA Half-Life of 19 977 Genes Obtained by DNA Microarray Analysis of Pluripotent and Differentiating Mouse Embryonic Stem Cells. DNA Res. 2009, 16, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enuka, Y.; Lauriola, M.; Feldman, M.E.; Sas-Chen, A.; Ulitsky, I.; Yarden, Y. Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res. 2016, 44, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [Green Version]
- Dixon, R.J.; Eperon, I.C.; Samani, N.J. Complementary intron sequence motifs associated with human exon repetition: A role for intragenic, inter-transcript interactions in gene expression. Bioinformatics 2007, 23, 150–155. [Google Scholar] [CrossRef]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [Green Version]
- Ji, P.; Wu, W.; Chen, S.; Zheng, Y.; Zhou, L.; Zhang, J.; Cheng, H.; Yan, J.; Zhang, S.; Yang, P.; et al. Expanded Expression Landscape and Prioritization of Circular RNAs in Mammals. Cell Rep. 2019, 26, 3444–3460.e3445. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Ma, X.K.; Li, G.W.; Yang, L. CIRCpedia v2: An Updated Database for Comprehensive Circular RNA Annotation and Expression Comparison. Genom. Proteom. Bioinform. 2018, 16, 226–233. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, T.; Miao, Y.F.; Jin, K.; Han, S.; Xu, T.Q.; Qiu, Z.L.; Zhang, X.H. Dysregulated circular RNAs in medulloblastoma regulate proliferation and growth of tumor cells via host genes. Cancer Med. 2018, 7, 6147–6157. [Google Scholar] [CrossRef]
- Dodbele, S.; Mutlu, N.; Wilusz, J.E. Best practices to ensure robust investigation of circular RNAs: Pitfalls and tips. EMBO Rep. 2021, 22, e52072. [Google Scholar] [CrossRef]
- Goodall, G.J.; Wickramasinghe, V.O. RNA in cancer. Nat. Rev. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.; Zheng, M.; Xu, Y.; Ma, S.; Zhang, W.; Ju, S. CircRNAs and their regulatory roles in cancers. Mol. Med. 2021, 27, 94. [Google Scholar] [CrossRef]
- Verduci, L.; Tarcitano, E.; Strano, S.; Yarden, Y.; Blandino, G. CircRNAs: Role in human diseases and potential use as biomarkers. Cell Death Dis. 2021, 12, 468. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, C.; Xiong, J.; Ren, H. Emerging important roles of circRNAs in human cancer and other diseases. Genes Dis. 2021, 8, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Zanini, C.; Ercole, E.; Mandili, G.; Salaroli, R.; Poli, A.; Renna, C.; Papa, V.; Cenacchi, G.; Forni, M. Medullospheres from DAOY, UW228 and ONS-76 Cells: Increased Stem Cell Population and Proteomic Modifications. PLoS ONE 2013, 8, e63748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Li, X. hsa_circ_0008285 Facilitates the Progression of Cervical Cancer by Targeting miR-211-5p/SOX4 Axis. Cancer Manag. Res. 2020, 12, 3927–3936. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, J.; Liu, G.; Li, X. Circular RNA hsa_circ_0008285 inhibits colorectal cancer cell proliferation and migration via the miR-382-5p/PTEN axis. Biochem. Biophys. Res. Commun. 2020, 527, 503–510. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| circRNA | circRNA ID | Detected by qPCR | ||
|---|---|---|---|---|
| 1 | ARGHAP12 | hsa_circ_0000231 | ||
| 2 | ASXL1 | * | hsa_circ_0001136 | |
| 3 | ATXN10 | * | hsa_circ_0001247 | √ |
| 4 | ATXN10-2 | hsa_circ_0001246 | √ | |
| 5 | BACH1 | * | hsa_circ_0001181 | √ |
| 6 | CBFA2T2 | * | hsa_circ_0003426 | √ |
| 7 | CCDC134 | hsa_circ_0008806 | √ | |
| 8 | CDYL | * | hsa_circ_0008285 | √ |
| 9 | CORO1C | * | NA | |
| 10 | FAM13B | * | hsa_circ_0001535 | √ |
| 11 | FGFR1 | * | hsa_circ_0008016 | √ |
| 12 | FKBP8 | * | hsa_circ_0000915 | √ |
| 13 | GAS2 | hsa_circ_0021516 | √ | |
| 14 | GLIS1 | * | hsa_circ_0002079 | √ |
| 15 | GLIS2 | * | hsa_circ_0005692 | √ |
| 16 | GLIS3 | * | hsa_circ_0001246 | √ |
| 17 | HIPK3 | * | hsa_circ_0000284 | √ |
| 18 | LPAR1 | * | hsa_circ_0087960 | √ |
| 19 | LRBA | * | hsa_circ_0006867 | √ |
| 20 | MARK4 | * | hsa_circ_0000940 | √ |
| 21 | OGDH | * | hsa_circ_0003340 | √ |
| 22 | RARS | * | NA | |
| 23 | RHOBTB3 | * | hsa_circ_0007444 | |
| 24 | RSRC1 | hsa_circ_0001355 | √ | |
| 25 | SFMBT2 | hsa_circ_0000211 | √ | |
| 26 | SMARCA5 | * | hsa_circ_0001445 | √ |
| 27 | SMO | * | hsa_circ_0001742 | √ |
| 28 | UBXN7 | * | hsa_circ_0001380 | |
| 29 | ZKSCAN1 | * | hsa_circ_0001727 | √ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azatyan, A.; Zhang, S.; Darabi, A.; Siesjö, P.; Wang, T.; Zaphiropoulos, P.G. Circular RNAs in Hedgehog Signaling Activation and Hedgehog-Mediated Medulloblastoma Tumors. Cancers 2021, 13, 5138. https://doi.org/10.3390/cancers13205138
Azatyan A, Zhang S, Darabi A, Siesjö P, Wang T, Zaphiropoulos PG. Circular RNAs in Hedgehog Signaling Activation and Hedgehog-Mediated Medulloblastoma Tumors. Cancers. 2021; 13(20):5138. https://doi.org/10.3390/cancers13205138
Chicago/Turabian StyleAzatyan, Ani, Shasha Zhang, Anna Darabi, Peter Siesjö, Ting Wang, and Peter G. Zaphiropoulos. 2021. "Circular RNAs in Hedgehog Signaling Activation and Hedgehog-Mediated Medulloblastoma Tumors" Cancers 13, no. 20: 5138. https://doi.org/10.3390/cancers13205138
APA StyleAzatyan, A., Zhang, S., Darabi, A., Siesjö, P., Wang, T., & Zaphiropoulos, P. G. (2021). Circular RNAs in Hedgehog Signaling Activation and Hedgehog-Mediated Medulloblastoma Tumors. Cancers, 13(20), 5138. https://doi.org/10.3390/cancers13205138