
Delineating the Switch between Senescence and Apoptosis in Cervical Cancer Cells under Ciclopirox Treatment

 ,
, 
Abstract
Simple Summary
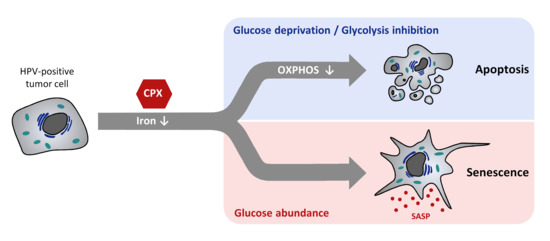
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment Conditions
2.2. Mass-Spectrometric Proteome Analyses and GSEA Analyses
2.3. Protein and RNA Analyses
- 18S rRNA forward: 5’-CATGGCCGTTCTTAGTTGGT-3’ and18S rRNA reverse: 5’ ATGCCAGAGTCTCGTTCGTT-3’;
- Cyclin B1 forward: 5’-GCCTCTACCTTTGCACTTCCT-3’ andCyclin B1 reverse 5’-TGTTGTAGAGTTGGTGTCCATT-3’;
- ID1 forward: 5’-AATCCGAAGTTGGAACCCCC-3’ andID1 reverse: 5’-GAACGCATGCCGCCTCG-3’;
- IL1A forward: 5’-AACCAACGGGAAGGTTCTGA-3 andIL1A reverse: 5‘-AGGCTTGATGATTTCTTCCTCT-3’;
- IL6 forward: 5’-CCACCGGGAACGAAAGAGAA-3‘andIL6 reverse: 5’- CGAAGGCGCTTGTGGAGAA-3’;
- CDKN1A forward: 5’-GACCATGTGGACCTGTCACT-3’ andCDKN1A reverse: 5’-GCGGATTAGGGCTTCCTCTT-3’;
- SERPINE1 forward: 5’-GACCGCAACGTGGTTTTCTC-3’ andSERPINE1 reverse: 5’-GCCATGCCCTTGTCATCAAT-3’.
2.4. Live-Cell Imaging
2.5. Apoptosis Assays
2.6. Colony Formation and Senescence Assays
2.7. Combination Index Analyses
2.8. Statistical Analyses
3. Results
3.1. CPX Regulates Factors Involved in Oxidative Phosphorylation and Glycolysis
3.2. Increased Glucose Availability Protects Cells against CPX-Induced Apoptosis
3.3. Increased Glucose Availability Favors Induction of a Senescent Phenotype under CPX Treatment
3.4. The Glucose-Dependent Apoptosis Induction through CPX Is Shared by Other OXPHOS Inhibitors
3.5. The Pro-Senescent Activity of CPX Is Shared by Other Iron Chelators, but Not by Other OXPHOS Inhibitors
3.6. CPX Synergizes with Glycolysis Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjose, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef]
- Harden, M.E.; Munger, K. Human papillomavirus molecular biology. Mutat. Res. Rev. Mutat. Res. 2017, 772, 3–12. [Google Scholar] [CrossRef]
- Goodwin, E.C.; Yang, E.; Lee, C.J.; Lee, H.W.; DiMaio, D.; Hwang, E.S. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl. Acad. Sci. USA 2000, 97, 10978–10983. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.I.; Francis, D.A.; Karpova, A.Y.; Dowhanick, J.J.; Benson, J.D.; Howley, P.M. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21(CIP)-dependent pathways. EMBO J. 2000, 19, 5762–5771. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Braun, J.A.; Herrmann, A.L.; Blase, J.I.; Frensemeier, K.; Bulkescher, J.; Scheffner, M.; Galy, B.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Effects of the antifungal agent ciclopirox in HPV-positive cancer cells: Repression of viral E6/E7 oncogene expression and induction of senescence and apoptosis. Int. J. Cancer 2020, 146, 461–474. [Google Scholar] [CrossRef]
- Subissi, A.; Monti, D.; Togni, G.; Mailland, F. Ciclopirox: Recent nonclinical and clinical data relevant to its use as a topical antimycotic agent. Drugs 2010, 70, 2133–2152. [Google Scholar] [CrossRef]
- Sehgal, V.N. Ciclopirox: A new topical pyrodonium antimycotic agent. A double-blind study in superficial dermatomycoses. Br. J. Dermatol. 1976, 95, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, S. Reposition of the Fungicide Ciclopirox for Cancer Treatment. Recent. Pat. Anticancer Drug Discov. 2021, 16, 122–135. [Google Scholar] [CrossRef]
- Huang, Y.M.; Cheng, C.H.; Pan, S.L.; Yang, P.M.; Lin, D.Y.; Lee, K.H. Gene Expression Signature-Based Approach Identifies Antifungal Drug Ciclopirox As a Novel Inhibitor of HMGA2 in Colorectal Cancer. Biomolecules 2019, 9, 688. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, L.; Wang, M.; Zhou, L.; Feng, X.; Yu, L.; Lan, J.; Gao, W.; Zhang, C.; Bu, Y.; et al. CPX Targeting DJ-1 Triggers ROS-induced Cell Death and Protective Autophagy in Colorectal Cancer. Theranostics 2019, 9, 5577–5594. [Google Scholar] [CrossRef]
- Mihailidou, C.; Papakotoulas, P.; Papavassiliou, A.G.; Karamouzis, M.V. Superior efficacy of the antifungal agent ciclopirox olamine over gemcitabine in pancreatic cancer models. Oncotarget 2018, 9, 10360–10374. [Google Scholar] [CrossRef][Green Version]
- Zhou, H.; Shen, T.; Luo, Y.; Liu, L.; Chen, W.; Xu, B.; Han, X.; Pang, J.; Rivera, C.A.; Huang, S. The antitumor activity of the fungicide ciclopirox. Int. J. Cancer 2010, 127, 2467–2477. [Google Scholar] [CrossRef]
- Yang, J.; Milasta, S.; Hu, D.; AlTahan, A.M.; Interiano, R.B.; Zhou, J.; Davidson, J.; Low, J.; Lin, W.; Bao, J.; et al. Targeting Histone Demethylases in MYC-Driven Neuroblastomas with Ciclopirox. Cancer Res. 2017, 77, 4626–4638. [Google Scholar] [CrossRef]
- Eberhard, Y.; McDermott, S.P.; Wang, X.; Gronda, M.; Venugopal, A.; Wood, T.E.; Hurren, R.; Datti, A.; Batey, R.A.; Wrana, J.; et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood 2009, 114, 3064–3073. [Google Scholar] [CrossRef] [PubMed]
- Weir, S.J.; Dandawate, P.; Standing, D.; Bhattacharyya, S.; Ramamoorthy, P.; Rangarajan, P.; Wood, R.; Brinker, A.E.; Woolbright, B.L.; Tanol, M.; et al. Fosciclopirox suppresses growth of high-grade urothelial cancer by targeting the gamma-secretase complex. Cell Death Dis. 2021, 12, 562. [Google Scholar] [CrossRef] [PubMed]
- Clement, P.M.; Hanauske-Abel, H.M.; Wolff, E.C.; Kleinman, H.K.; Park, M.H. The antifungal drug ciclopirox inhibits deoxyhypusine and proline hydroxylation, endothelial cell growth and angiogenesis in vitro. Int. J. Cancer 2002, 100, 491–498. [Google Scholar] [CrossRef]
- Sen, S.; Hassane, D.C.; Corbett, C.; Becker, M.W.; Jordan, C.T.; Guzman, M.L. Novel mTOR inhibitory activity of ciclopirox enhances parthenolide antileukemia activity. Exp. Hematol. 2013, 41, 799.e794–807.e794. [Google Scholar] [CrossRef] [PubMed]
- Oexle, H.; Gnaiger, E.; Weiss, G. Iron-dependent changes in cellular energy metabolism: Influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta. 1999, 1413, 99–107. [Google Scholar] [CrossRef]
- Qi, J.; Zhou, N.; Li, L.; Mo, S.; Zhou, Y.; Deng, Y.; Chen, T.; Shan, C.; Chen, Q.; Lu, B. Ciclopirox activates PERK-dependent endoplasmic reticulum stress to drive cell death in colorectal cancer. Cell Death Dis. 2020, 11, 582. [Google Scholar] [CrossRef]
- Shen, T.; Huang, S. Repositioning the Old Fungicide Ciclopirox for New Medical Uses. Curr. Pharm. Des. 2016, 22, 4443–4450. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Bossler, F.; Kuhn, B.J.; Gunther, T.; Kraemer, S.J.; Khalkar, P.; Adrian, S.; Lohrey, C.; Holzer, A.; Shimobayashi, M.; Durst, M.; et al. Repression of Human Papillomavirus Oncogene Expression under Hypoxia Is Mediated by PI3K/mTORC2/AKT Signaling. mBio 2019, 10, e02323-18. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Hoppe-Seyler, K.; Herrmann, A.L.; Daschle, A.; Kuhn, B.J.; Strobel, T.D.; Lohrey, C.; Bulkescher, J.; Krijgsveld, J.; Hoppe-Seyler, F. Effects of Metformin on the virus/host cell crosstalk in human papillomavirus-positive cancer cells. Int. J. Cancer 2021, 149, 1137–1149. [Google Scholar] [CrossRef]
- Honegger, A.; Schilling, D.; Bastian, S.; Sponagel, J.; Kuryshev, V.; Sultmann, H.; Scheffner, M.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Dependence of intracellular and exosomal microRNAs on viral E6/E7 oncogene expression in HPV-positive tumor cells. PLoS Pathog. 2015, 11, e1004712. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Stiban, J.; So, M.; Kaguni, L.S. Iron-Sulfur Clusters in Mitochondrial Metabolism: Multifaceted Roles of a Simple Cofactor. Biochemistry 2016, 81, 1066–1080. [Google Scholar] [CrossRef]
- Rensvold, J.W.; Ong, S.E.; Jeevananthan, A.; Carr, S.A.; Mootha, V.K.; Pagliarini, D.J. Complementary RNA and protein profiling identifies iron as a key regulator of mitochondrial biogenesis. Cell Rep. 2013, 3, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Kyrylkova, K.; Kyryachenko, S.; Leid, M.; Kioussi, C. Detection of apoptosis by TUNEL assay. Methods Mol. Biol. 2012, 887, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Tumour suppression by p53: A role for the DNA damage response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Liu, Y.; Wu, X.; Shell, S.M. Functions of human replication protein A (RPA): From DNA replication to DNA damage and stress responses. J. Cell. Physiol. 2006, 208, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Zhou, H.; Shang, C.; Luo, Y.; Wu, Y.; Huang, S. Ciclopirox activates ATR-Chk1 signaling pathway leading to Cdc25A protein degradation. Genes Cancer 2018, 9, 39–52. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Brown, J.P.; Wei, W.; Sedivy, J.M. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 1997, 277, 831–834. [Google Scholar] [CrossRef]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Alani, R.M.; Young, A.Z.; Shifflett, C.B. Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proc. Natl. Acad. Sci. USA 2001, 98, 7812–7816. [Google Scholar] [CrossRef]
- Gire, V.; Dulic, V. Senescence from G2 arrest, revisited. Cell Cycle 2015, 14, 297–304. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Minden, M.D.; Hogge, D.E.; Weir, S.J.; Kasper, J.; Webster, D.A.; Patton, L.; Jitkova, Y.; Hurren, R.; Gronda, M.; Goard, C.A.; et al. Oral ciclopirox olamine displays biological activity in a phase I study in patients with advanced hematologic malignancies. Am. J. Hematol. 2014, 89, 363–368. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [PubMed]
- Akins, N.S.; Nielson, T.C.; Le, H.V. Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer. Curr. Top. Med. Chem. 2018, 18, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Smola, S. Human Papillomaviruses and Skin Cancer. Adv. Exp. Med. Biol. 2020, 1268, 195–209. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Meyers, J.; Munger, K. Cancer associated human papillomaviruses. Curr. Opin. Virol. 2012, 2, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Patzelt, A.; Lengert, M.; Hutter, K.J.; Kanazawa, K.; Hausen, H.; Rosl, F. Selective down-regulation of human papillomavirus transcription by 2-deoxyglucose. Int. J. Cancer 1998, 76, 639–646. [Google Scholar] [CrossRef]
- Horner, S.M.; DeFilippis, R.A.; Manuelidis, L.; DiMaio, D. Repression of the human papillomavirus E6 gene initiates p53-dependent, telomerase-independent senescence and apoptosis in HeLa cervical carcinoma cells. J. Virol. 2004, 78, 4063–4073. [Google Scholar] [CrossRef] [PubMed]
- Erwig, L.P.; Henson, P.M. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2008, 15, 243–250. [Google Scholar] [CrossRef]
- Angelini, P.D.; Zacarias Fluck, M.F.; Pedersen, K.; Parra-Palau, J.L.; Guiu, M.; Bernado Morales, C.; Vicario, R.; Luque-Garcia, A.; Navalpotro, N.P.; Giralt, J.; et al. Constitutive HER2 signaling promotes breast cancer metastasis through cellular senescence. Cancer Res. 2013, 73, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.H.; Park, T.J. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 2017, 8, 15208. [Google Scholar] [CrossRef]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintas-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrmann, A.L.; Kuhn, B.J.; Holzer, A.; Krijgsveld, J.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Delineating the Switch between Senescence and Apoptosis in Cervical Cancer Cells under Ciclopirox Treatment. Cancers 2021, 13, 4995. https://doi.org/10.3390/cancers13194995
Herrmann AL, Kuhn BJ, Holzer A, Krijgsveld J, Hoppe-Seyler K, Hoppe-Seyler F. Delineating the Switch between Senescence and Apoptosis in Cervical Cancer Cells under Ciclopirox Treatment. Cancers. 2021; 13(19):4995. https://doi.org/10.3390/cancers13194995
Chicago/Turabian StyleHerrmann, Anja L., Bianca J. Kuhn, Angela Holzer, Jeroen Krijgsveld, Karin Hoppe-Seyler, and Felix Hoppe-Seyler. 2021. "Delineating the Switch between Senescence and Apoptosis in Cervical Cancer Cells under Ciclopirox Treatment" Cancers 13, no. 19: 4995. https://doi.org/10.3390/cancers13194995
APA StyleHerrmann, A. L., Kuhn, B. J., Holzer, A., Krijgsveld, J., Hoppe-Seyler, K., & Hoppe-Seyler, F. (2021). Delineating the Switch between Senescence and Apoptosis in Cervical Cancer Cells under Ciclopirox Treatment. Cancers, 13(19), 4995. https://doi.org/10.3390/cancers13194995