Hematopoietic versus Solid Cancers and T Cell Dysfunction: Looking for Similarities and Distinctions



Abstract
Simple Summary
Abstract
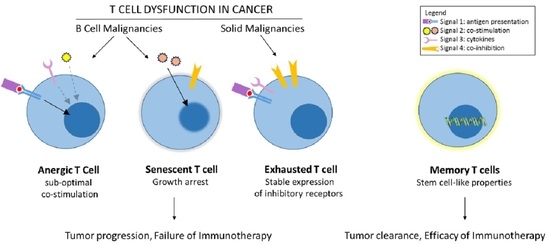
1. Introduction
2. Immunotherapy for Solid Tumors
3. Immunotherapy in Hematopoietic Malignancies
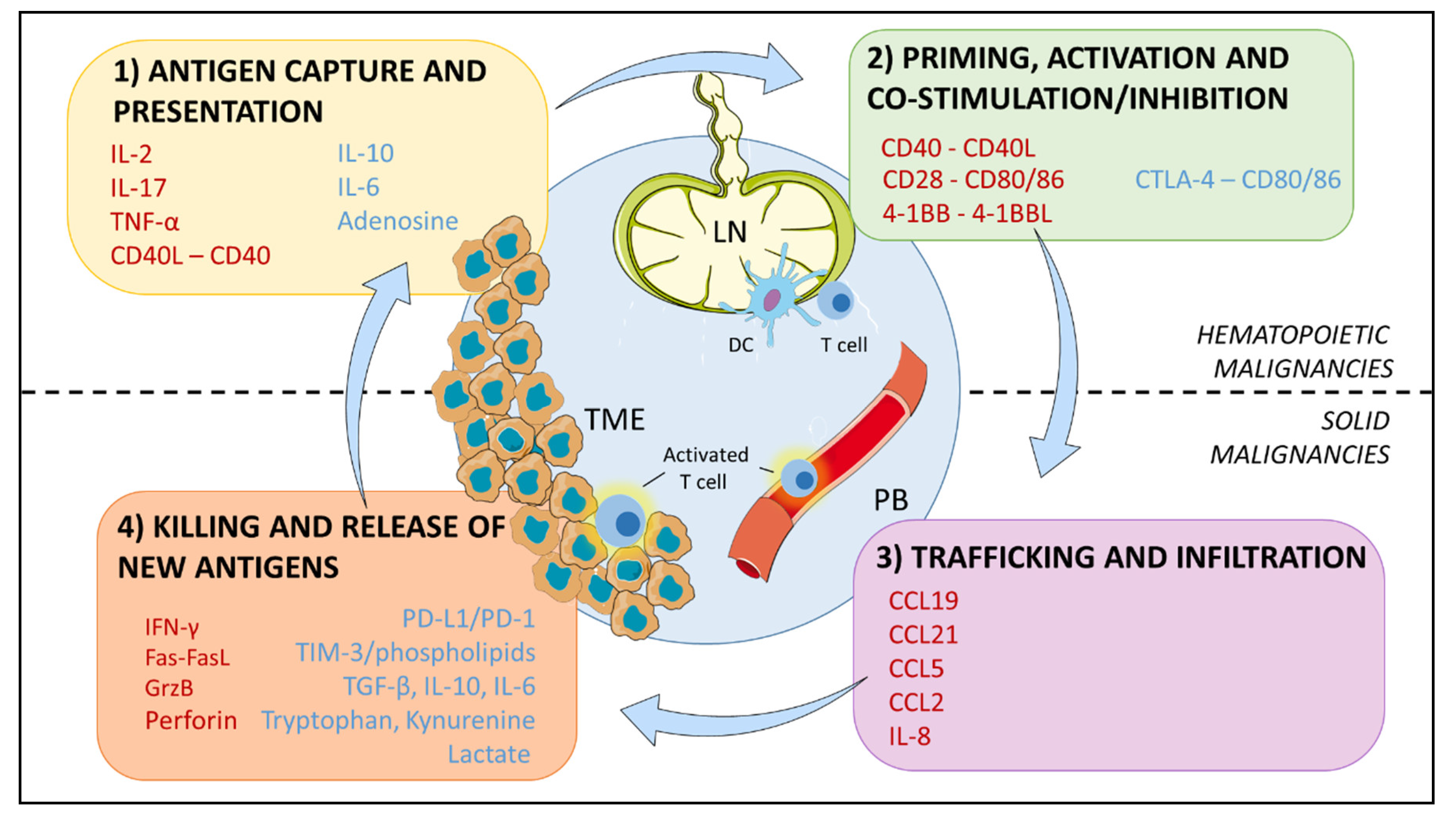
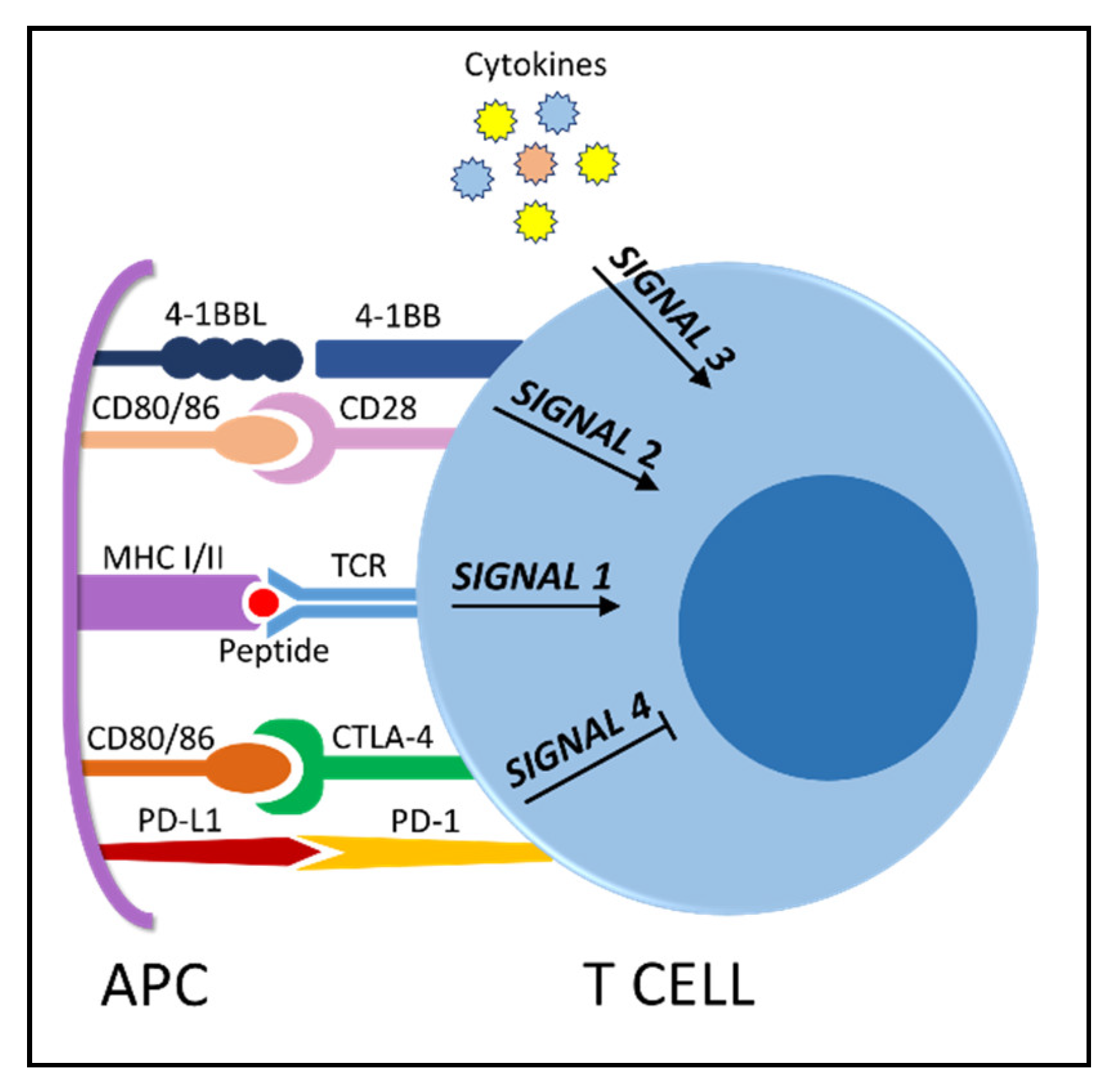
4. The Immunosuppressive Microenvironment: Physical Interactions
5. The Immunosuppressive Microenvironment: Soluble Signals
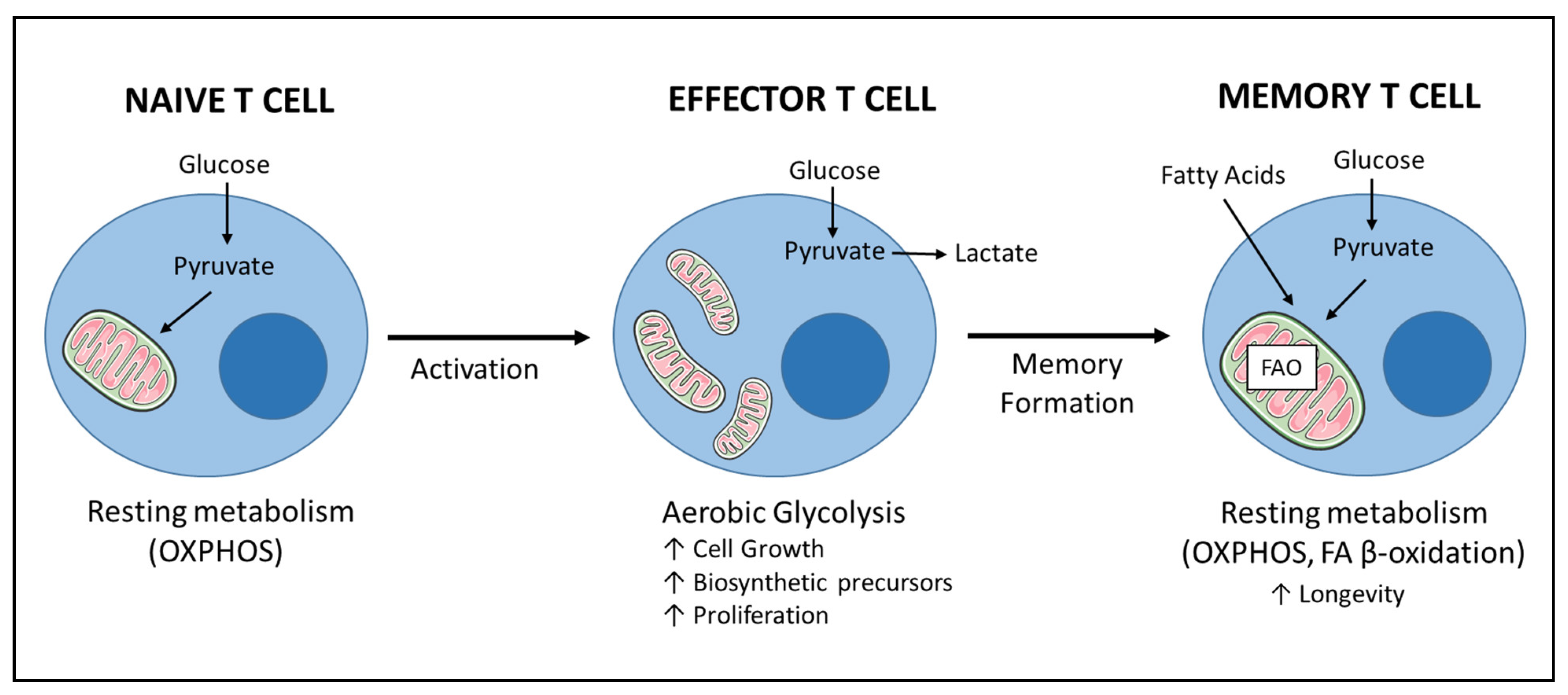
6. Immunotherapy Efficacy Is Linked to T Cell Fitness—The Survival of the Fittest
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pilipow, K.; Darwich, A.; Losurdo, A. T-cell-based breast cancer immunotherapy. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.; Wang, J.; Sahoo, A. T-cell tolerance in cancer. Immunotherapy 2013, 5, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Young, K.H. Checkpoint inhibitors in hematological malignancies. J. Hematol. Oncol. 2017, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Curran, E.K.; Godfrey, J.; Kline, J. Mechanisms of Immune Tolerance in Leukemia and Lymphoma. Trends Immunol. 2017, 38, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Speiser, D.E.; Utzschneider, D.T.; Oberle, S.G.; Münz, C.; Romero, P.; Zehn, D. T cell differentiation in chronic infection and cancer: Functional adaptation or exhaustion? Nat. Rev. Immunol. 2014, 14, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, Z.; Chen, L. Memory T cells: Strategies for optimizing tumor immunotherapy. Protein Cell 2020, 11, 549–564. [Google Scholar] [CrossRef]
- Makkouk, A.; Weiner, G.J. Cancer Immunotherapy and breaking immune tolerance—New approaches to an old challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Aldous, A.R.; Dong, J.Z. Personalized neoantigen vaccines: A new approach to cancer immunotherapy. Bioorg. Med. Chem. 2018, 26, 2842–2849. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, Y.; Ding, Z.Y.; Liu, J.Y. Safety and efficacy of therapeutic cancer vaccines alone or in combination with immune checkpoint inhibitors in cancer treatment. Front. Pharmacol. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kreamer, K.M. Immune Checkpoint Blockade: A New Paradigm in Treating Advanced Cancer. J. Adv. Pract. Oncol. 2014, 5, 418–431. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Vodnala, S.K.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.H.; Lee, P.H.; Shin, M.H.; Patel, S.J.; Yu, Z.; et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363. [Google Scholar] [CrossRef]
- Jafferji, M.S.; Yang, J.C. Adoptive T-Cell Therapy for Solid Malignancies. Surg. Oncol. Clin. N. Am. 2019, 28, 465–479. [Google Scholar] [CrossRef]
- Bansal, R.; Reshef, R. Revving the CAR—Combination strategies to enhance CAR T cell effectiveness. Blood Rev. 2020, 100695. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.; Cetnar, J.P.; Prasad, V. A Timeline of Immune Checkpoint Inhibitor Approvals in Small Cell Lung Cancer. Trends Cancer 2020, 6, 736–738. [Google Scholar] [CrossRef] [PubMed]
- Baretti, M.; Le, D.T. DNA mismatch repair in cancer. Pharmacol. Ther. 2018, 189, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Bañobre, J.; Goel, A. DNA Mismatch Repair Deficiency and Immune Checkpoint Inhibitors in Gastrointestinal Cancers. Gastroenterology 2019, 156, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, S12–S16. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Jones, B.S.; Lamb, L.S.; Goldman, F.; Di Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef]
- Wilkie, S.; Van Schalkwyk, M.C.I.; Hobbs, S.; Davies, D.M.; Van Der Stegen, S.J.C.; Pereira, A.C.P.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying cancers basedon T-cell infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.Y.; Esfahani, M.S.; Moding, E.J.; Hamilton, E.G.; Chabon, J.J.; Rizvi, H.; Steen, C.B.; Chaudhuri, A.A.; Liu, C.L.; Hui, A.B.; et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell 2020, 183, 363–376.e13. [Google Scholar] [CrossRef] [PubMed]
- Holl, E.K.; Frazier, V.N.; Landa, K.; Beasley, G.M.; Hwang, E.S.; Nair, S.K. Examining Peripheral and Tumor Cellular Immunome in Patients With Cancer. Front. Immunol. 2019, 10, 1767. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Wolf, D.; Steurer, M.; Gastl, G.; Gunsilius, E.; Grubeck-Loebenstein, B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin. Cancer Res. 2003, 9, 606–612. [Google Scholar]
- Titov, A.; Valiullina, A.; Zmievskaya, E.; Zaikova, E.; Petukhov, A.; Miftakhova, R.; Bulatov, E.; Rizvanov, A. Advancing CAR T-cell therapy for solid tumors: Lessons learned from lymphoma treatment. Cancers 2020, 12, 125. [Google Scholar] [CrossRef]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 1–12. [Google Scholar] [CrossRef]
- André, T.; Meuleman, N.; Stamatopoulos, B.; De Bruyn, C.; Pieters, K.; Bron, D.; Lagneaux, L. Evidences of Early Senescence in Multiple Myeloma Bone Marrow Mesenchymal Stromal Cells. PLoS ONE 2013, 8, e59756. [Google Scholar] [CrossRef] [PubMed]
- Suen, H.; Brown, R.; Yang, S.; Weatherburn, C.; Ho, P.J.; Woodland, N.; Nassif, N.; Barbaro, P.; Bryant, C.; Hart, D.; et al. Multiple myeloma causes clonal T-cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 2016, 30, 1716–1724. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Armand, P. Immune checkpoint blockade in hematologic malignancies. Blood 2015, 125, 3393–3400. [Google Scholar] [CrossRef]
- Merryman, R.W.; Armand, P.; Wright, K.T.; Rodig, S.J. Checkpoint blockade in Hodgkin and non-Hodgkin lymphoma. Blood Adv. 2017, 1, 2643–2654. [Google Scholar] [CrossRef]
- Westin, J.R.; Chu, F.; Zhang, M.; Fayad, L.E.; Kwak, L.W.; Fowler, N.; Romaguera, J.; Hagemeister, F.; Fanale, M.; Samaniego, F.; et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: A single group, open-label, phase 2 trial. Lancet Oncol. 2014, 15, 69–77. [Google Scholar] [CrossRef]
- Cao, J.; Wang, G.; Cheng, H.; Wei, C.; Qi, K.; Sang, W.; Zhenyu, L.; Shi, M.; Li, H.; Qiao, J.; et al. Potent anti-leukemia activities of humanized CD19-targeted Chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am. J. Hematol. 2018, 93, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Ding, L.; Wang, J.; Hu, Y.; Huang, H. Advances of CD19-directed chimeric antigen receptor-modified T cells in refractory/relapsed acute lymphoblastic leukemia. Exp. Hematol. Oncol. 2017, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10, 204062071984158. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef]
- Younes, A.; Brody, J.; Carpio, C.; Lopez-Guillermo, A.; Ben-Yehuda, D.; Ferhanoglu, B.; Nagler, A.; Ozcan, M.; Avivi, I.; Bosch, F.; et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: A phase 1/2a study. Lancet Haematol. 2019, 6, e67–e78. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-Specific chimeric antigen Receptor-modified T cells after failure of ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Sotillo, E.; Harrington, C.; Wertheim, G.; Paessler, M.; Maude, S.L.; Rheingold, S.R.; Grupp, S.A.; Thomas-Tikhonenko, A.; Pillai, V. Repeated loss of target surface antigen after immunotherapy in primary mediastinal large B cell lymphoma. Am. J. Hematol. 2017, 92, E11–E13. [Google Scholar] [CrossRef] [PubMed]
- Shalabi, H.; Kraft, I.L.; Wang, H.W.; Yuan, C.M.; Yates, B.; Delbrook, C.; Zimbelman, J.D.; Giller, R.; Stetler-Stevenson, M.; Jaffe, E.S.; et al. Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematologica 2018, 103, e215–e218. [Google Scholar] [CrossRef]
- Fan, F.; Zhao, W.; Liu, J.; He, A.; Chen, Y.; Cao, X.; Yang, N.; Wang, B.; Zhang, P.; Zhang, Y.; et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J. Clin. Oncol. 2017, 35, LBA3001. [Google Scholar] [CrossRef]
- Shah, N.N.; Shalabi, H.; Yates, B.; Yuan, C.; Qin, H.; Ombrello, A.; Wang, H.-W.; Hoffman, L.; Tran, M.; Panch, S.; et al. Abstract LB-146: Phase I CD22 CAR T-cell trial updates. In Proceedings of the Cancer Research; American Association for Cancer Research (AACR), Atlanta, GA, USA, 29 March–3 April 2019; Volume 79, p. LB-146-LB-146. [Google Scholar]
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef]
- Yaddanapudi, K.; Putty, K.; Rendon, B.E.; Lamont, G.J.; Faughn, J.D.; Satoskar, A.; Lasnik, A.; Eaton, J.W.; Mitchell, R.A. Control of Tumor-Associated Macrophage Alternative Activation by Macrophage Migration Inhibitory Factor. J. Immunol. 2013, 190, 2984–2993. [Google Scholar] [CrossRef]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef]
- Joshua, D.; Suen, H.; Brown, R.; Bryant, C.; Ho, P.J.; Hart, D.; Gibson, J. The T Cell in Myeloma. Clin. Lymphoma Myeloma Leuk. 2016, 16, 537–542. [Google Scholar] [CrossRef]
- Mills, K.H.G.; Cawley, J.C. Abnormal monoclonal antibody-defined helper/suppressor T-cell subpopulations in multiple myeloma: Relationship to treatment and clinical stage. Br. J. Haematol. 1983, 53, 271–275. [Google Scholar] [CrossRef]
- Kay, N.E.; Leong, T.L.; Bone, N.; Vesole, D.H.; Greipp, P.R.; Van Ness, B.; Oken, M.M.; Kyle, R.A. Blood levels of immune cells predict survival in myeloma patients: Results of an Eastern Cooperative Oncology Group phase 3 trial for newly diagnosed multiple myeloma patients. Blood 2001, 98, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, J.; Brown, R.; Aklilu, E.; Yang, S.; Suen, H.; Hart, D.; Fromm, P.; Gibson, J.; Khoo, L.; Ho, P.J.; et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk. Lymphoma 2014, 55, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Blimark, C.; Holmberg, E.; Mellqvist, U.H.; Landgren, O.; Bjorkholm, M.; Hultcrantz, M.; Kjellander, C.; Turesson, I.; Kristinsson, S.Y. Multiple myeloma and infections: A population-based study on 9253 multiple myeloma patients. Haematologica 2015, 100, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.; Suen, H.; Brown, R.; Yang, S.; Favaloro, J.; Aklilu, E.; Gibson, J.; Ho, P.J.; Iland, H.; Fromm, P.; et al. Long-term survival in multiple myeloma is associated with a distinct immunological profile, which includes proliferative cytotoxic T-cell clones and a favourable Treg/Th17 balance. Blood Cancer J. 2013, 3, e148. [Google Scholar] [CrossRef]
- Forconi, F.; Moss, P. Perturbation of the normal immune system in patients with CLL. Blood 2015, 126, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Pourgheysari, B.; Bruton, R.; Parry, H.; Billingham, L.; Fegan, C.; Murray, J.; Moss, P. The number of cytomegalovirus-specific CD4+ T cells is markedly expanded in patients with B-cell chronic lymphocytic leukemia and determines the total CD4+ T-cell repertoire. Blood 2010, 116, 2968–2974. [Google Scholar] [CrossRef]
- Christopoulos, P.; Pfeifer, D.; Bartholomé, K.; Follo, M.; Timmer, J.; Fisch, P.; Veelken, H. Definition and characterization of the systemic T-cell dysregulation in untreated indolent B-cell lymphoma and very early CLL. Blood 2011, 117, 3836–3846. [Google Scholar] [CrossRef]
- Rossi, D.; Sozzi, E.; Puma, A.; De Paoli, L.; Rasi, S.; Spina, V.; Gozzetti, A.; Tassi, M.; Cencini, E.; Raspadori, D.; et al. The prognosis of clinical monoclonal B cell lymphocytosis differs from prognosis of Rai 0 chronic lymphocytic leukaemia and is recapitulated by biological risk factors. Br. J. Haematol. 2009, 146, 64–75. [Google Scholar] [CrossRef]
- Riches, J.C.; Gribben, J.G. Immunomodulation and Immune Reconstitution in Chronic Lymphocytic Leukemia. Semin. Hematol. 2014, 51, 228–234. [Google Scholar] [CrossRef]
- Chiorazzi, N.; Fu, S.M.; Montazeri, G.; Kunkel, H.G.; Rai, K.; Gee, T. T Cell Helper Defect in Patients with Chronic Lymphocytic Leukemia. J. Immunol. 1979, 122, 1087–1090. [Google Scholar]
- Joshua, D.E.; Brown, R.D.; Ho, P.J.; Gibson, J. Regulatory T cells and multiple myeloma. Clin. Lymphoma Myeloma 2008, 8, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Blaeschke, F.; Willier, S.; Stenger, D.; Lepenies, M.; Horstmann, M.A.; Escherich, G.; Zimmermann, M.; Rojas Ringeling, F.; Canzar, S.; Kaeuferle, T.; et al. Leukemia-induced dysfunctional TIM-3+CD4+ bone marrow T cells increase risk of relapse in pediatric B-precursor ALL patients. Leukemia 2020, 34, 2607–2620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.; Chen, L.; Feng, Z.; Gao, L.; Li, Q. TIGIT expression is upregulated in T cells and causes T cell dysfunction independent of PD-1 and Tim-3 in adult B lineage acute lymphoblastic leukemia. Cell. Immunol. 2019, 344, 103958. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Qing, X.; Wu, C.Y.; Zhu, H.; Zhou, H.Y. Immunophenotype and increased presence of CD4 +CD25 + regulatory T cells in patients with acute lymphoblastic leukemia. Oncol. Lett. 2012, 3, 421–424. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Liu, X.; Kline, D.E.; Teague, R.M.; Gajewski, T.F.; Kline, J. CD40 ligation reverses T cell tolerance in acute myeloid leukemia. J. Clin. Investig. 2013, 123, 1999–2010. [Google Scholar] [CrossRef]
- Rousseau, R.F.; Biagi, E.; Dutour, A.; Yvon, E.S.; Brown, M.P.; Lin, T.; Mei, Z.; Grilley, B.; Popek, E.; Heslop, H.E.; et al. Immunotherapy of high-risk acute leukemia with a recipient (autologous) vaccine expressing transgenic human CD40L and IL-2 after chemotherapy and allogeneic stem cell transplantation. Blood 2006, 107, 1332–1341. [Google Scholar] [CrossRef]
- Castro, J.E.; Melo-Cardenas, J.; Urquiza, M.; Barajas-Gamboa, J.S.; Pakbaz, R.S.; Kipps, T.J. Gene Immunotherapy of Chronic Lymphocytic Leukemia: A Phase I Study of Intranodally Injected Adenovirus Expressing a Chimeric CD154 Molecule. Cancer Res. 2012, 72, 2937–2948. [Google Scholar] [CrossRef]
- Morrison, A.H.; Diamond, M.S.; Hay, C.A.; Byrne, K.T.; Vonderheide, R.H. Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc. Natl. Acad. Sci. USA 2020, 117, 8022–8031. [Google Scholar] [CrossRef]
- Vonderheide, R.H. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018, 33, 563–569. [Google Scholar] [CrossRef]
- Driessens, G.; Kline, J.; Gajewski, T.F. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol. Rev. 2009, 229, 126–144. [Google Scholar] [CrossRef]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef] [PubMed]
- McArthur, H.L.; Diab, A.; Page, D.B.; Yuan, J.; Solomon, S.B.; Sacchini, V.; Comstock, C.; Durack, J.C.; Maybody, M.; Sung, J.; et al. A pilot study of preoperative single-dose ipilimumab and/or cryoablation in women with early-stage breast cancer with comprehensive immune profiling. Clin. Cancer Res. 2016, 22, 5729–5737. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T-cell dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Umansky, V.; Sevko, A.; Gebhardt, C.; Utikal, J. Myeloid-derived suppressor cells in malignant melanoma. JDDG J. Dtsch. Dermatol. Ges. 2014, 12, 1021–1027. [Google Scholar] [CrossRef]
- Jitschin, R.; Braun, M.; Büttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef]
- Topal, G.G.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood J. Am. Soc. Hematol. 2013, 121, 2975–2987. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef]
- De Veirman, K.; Van Valckenborgh, E.; Lahmar, Q.; Geeraerts, X.; De Bruyne, E.; Menu, E.; Van Riet, I.; Vanderkerken, K.; Van Ginderachter, J.A. Myeloid-derived suppressor cells as therapeutic target in hematological malignancies. Front. Oncol. 2014, 4, 349. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Ku, A.W.; Muhitch, J.B.; Powers, C.A.; Diehl, M.; Kim, M.; Fisher, D.T.; Sharda, A.P.; Clements, V.K.; O’Loughlin, K.; Minderman, H.; et al. Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymph nodes. eLife 2016, 5, 1–29. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2015, 41, 49–61. [Google Scholar] [CrossRef]
- Elgert, K.D.; Alleva, D.G.; Mullins, D.W. Tumor-induced immune dysfunction: The macrophage connection. J. Leuk. Biol. 1998, 64, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Jiang, S. Immune Cell-Derived Exosomes in the Cancer-Immunity Cycle. Trends Cancer 2020, 6, 506–517. [Google Scholar] [CrossRef]
- Semionatto, I.F.; Palameta, S.; Toscaro, J.M.; Manrique-Rincón, A.J.; Ruas, L.P.; Paes Leme, A.F.; Bajgelman, M.C. Extracellular vesicles produced by immunomodulatory cells harboring OX40 ligand and 4-1BB ligand enhance antitumor immunity. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Landskron, G.; De La Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Busse, A.; Keilholz, U. Role of TGF-β in Melanoma. Curr. Pharm. Biotechnol. 2011, 12, 2165–2175. [Google Scholar] [CrossRef]
- Moses, H.; Barcellos-Hoff, M.H. TGF-β Biology in mammary development and breast cancer. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Pang, M.F.; Georgoudaki, A.M.; Lambut, L.; Johansson, J.; Tabor, V.; Hagikura, K.; Jin, Y.; Jansson, M.; Alexander, J.S.; Nelson, C.M.; et al. TGF-β1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene 2016, 35, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Horie, M.; Nagase, T. TGF-β signaling in lung health and disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed]
- Gastl, G.A.; Abrams, J.S.; Nanus, D.M.; Oosterkamp, R.; Silver, J.; Liu, F.; Chen, M.; Albino, A.P.; Bander, N.H. Interleukin-10 production by human carcinoma cell lines and its relationship to interleukin-6 expression. Int. J. Cancer 1993, 55, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Grütz, G.; Warszawska, K.; Kirsch, S.; Witte, E.; Wolk, K.; Geginat, J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010, 21, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The Master Regulator of Immunity to Infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Hamidullah; Changkija, B.; Konwar, R. Role of interleukin-10 in breast cancer. Breast Cancer Res. Treat. 2012, 133, 11–21. [Google Scholar] [CrossRef]
- Alhakeem, S.S.; McKenna, M.K.; Oben, K.Z.; Noothi, S.K.; Rivas, J.R.; Hildebrandt, G.C.; Fleischman, R.A.; Rangnekar, V.M.; Muthusamy, N.; Bondada, S. Chronic Lymphocytic Leukemia–Derived IL-10 Suppresses Antitumor Immunity. J. Immunol. 2018, 200, 4180–4189. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Prado-Garcia, H.; Aguilar-Cazares, D.; Flores-Vergara, H.; Mandoki, J.J.; Lopez-Gonzalez, J.S. Effector, memory and naïve CD8+ T cells in peripheral blood and pleural effusion from lung adenocarcinoma patients. Lung Cancer 2005, 47, 361–371. [Google Scholar] [CrossRef]
- De Chaisemartin, L.; Goc, J.; Damotte, D.; Validire, P.; Magdeleinat, P.; Alifano, M.; Cremer, I.; Fridman, W.H.; Sautès-Fridman, C.; Dieu-Nosjean, M.C. Characterization of chemokines and adhesion molecules associated with T cell presence in tertiary lymphoid structures in human lung cancer. Cancer Res. 2011, 71, 6391–6399. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef]
- López-González, J.S.; Aguilar-Cázares, D.; Prado-García, H.; Nieto-Rodríguez, A.; Mandoki, J.J.; Avila-Moreno, F.; Rivera, R.M.; Chavarría-Garcés, J. Lack of correlation between growth inhibition by TGF-β and the percentage of cells expressing type II TGF-β receptor in human non-small cell lung carcinoma cell lines. Lung Cancer 2002, 38, 149–158. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Hildeman, D.A.; Mitchell, T.; Kappler, J.; Marrack, P. T cell apoptosis and reactive oxygen species. J. Clin. Investig. 2003, 111, 575–581. [Google Scholar] [CrossRef]
- Linnemann, C.; Schildberg, F.A.; Schurich, A.; Diehl, L.; Hegenbarth, S.I.; Endl, E.; Lacher, S.; Müller, C.E.; Frey, J.; Simeoni, L.; et al. Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology 2009, 128, e728. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.; Cekic, C. Regulation of lymphocyte function by adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2097–2103. [Google Scholar] [CrossRef]
- Rad Pour, S.; Morikawa, H.; Kiani, N.A.; Yang, M.; Azimi, A.; Shafi, G.; Shang, M.; Baumgartner, R.; Ketelhuth, D.F.J.; Kamleh, M.A.; et al. Exhaustion of CD4+ T-cells mediated by the Kynurenine Pathway in Melanoma. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Klein Geltink, R.I.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4 + T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Carmen, F.C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.C.; Van Der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. XPosttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239. [Google Scholar] [CrossRef] [PubMed]
- Cham, C.M.; Driessens, G.; O’Keefe, J.P.; Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 2008, 38, 2438–2450. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Smolková, K.; Plecitá-Hlavatá, L.; Bellance, N.; Benard, G.; Rossignol, R.; Ježek, P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 2011, 43, 950–968. [Google Scholar] [CrossRef]
- Terness, P.; Bauer, T.M.; Röse, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of Allogeneic T Cell Proliferation by Indoleamine 2,3-Dioxygenase-expressing Dendritic Cells: Mediation of Suppression by Tryptophan Metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Musella, M.; Galassi, C.; Vitale, I.; De Maria, R. Tuning Cancer Fate: Tumor Microenvironment’s Role in Cancer Stem Cell Quiescence and Reawakening. Front. Immunol. 2020, 11, 2166. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Danhier, P.; Bański, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Singer, K.; Evert, K.; Renner, K.; Kreutz, M. The immunological Warburg effect: Can a metabolic-tumor-stroma score (MeTS) guide cancer immunotherapy? Immunol. Rev. 2020, 295, 187–202. [Google Scholar] [CrossRef]
- Puschel, F.; Favaro, F.; Redondo-Pedraza, J.; Lucendo, E.; Iurlaro, R.; Marchetti, S.; Majem, B.; Eldering, E.; Nadal, E.; Ricci, J.E.; et al. Starvation and antimetabolic therapy promote cytokine release and recruitment of immune cells. Proc. Natl. Acad. Sci. USA 2020, 117, 9932–9941. [Google Scholar] [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; MacIntyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Siska, P.J.; van der Windt, G.J.W.; Kishton, R.J.; Cohen, S.; Eisner, W.; MacIver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef]
- Van Bruggen, J.A.C.; Martens, A.W.J.; Fraietta, J.A.; Hofland, T.; Tonino, S.H.; Eldering, E.; Levin, M.D.; Siska, P.J.; Endstra, S.; Rathmell, J.C.; et al. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8+ T cells and impede CAR T-cell efficacy. Blood 2019, 134, 44–58. [Google Scholar] [CrossRef]
- Galicia-Vázquez, G.; Aloyz, R. Metabolic rewiring beyond Warburg in chronic lymphocytic leukemia: How much do we actually know? Crit. Rev. Oncol. Hematol. 2019, 134, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Vardhana, S.A.; Hwee, M.A.; Berisa, M.; Wells, D.K.; Yost, K.E.; King, B.; Smith, M.; Herrera, P.S.; Chang, H.Y.; Satpathy, A.T.; et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 2020, 21, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, G.J.W.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8 + T Cell Memory Development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.-C.; et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef]
- Buck, M.D.D.; O’Sullivan, D.; Klein Geltink, R.I.I.; Curtis, J.D.D.; Chang, C.H.; Sanin, D.E.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.J.W.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e9. [Google Scholar] [CrossRef]
- McLellan, A.D.; Ali Hosseini Rad, S.M. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019, 97, 664–674. [Google Scholar] [CrossRef]
- Enamorado, M.; Iborra, S.; Priego, E.; Cueto, F.J.; Quintana, J.A.; Martýnez-Cano, S.; Mejyás-Perez, E.; Esteban, M.; Melero, I.; Hidalgo, A.; et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Similarities | Solid Malignancies | B-Cell Malignancies |
|---|---|---|
| T cell dysfunction mechanism | Trafficking/infiltration, Exhaustion in TME | Priming and activation, Anergy, Senescence |
| Hurdles to adoptive (CAR) T cell therapy | Antigen choice, tumor infiltration | T cell state pre-expansion |
| Immunosuppression by soluble factors | By cancer cells, cancer associated fibroblasts and innate immune cells | By B cancer cells or by-stander cells |
| Immunosuppression by physical interactions | Co-inhibition; killing; interaction with by-stander cells | Priming, co-stimulation; killing; interaction with by-stander cells |
| T cell fitness and metabolism | Metabolic competition restricts T cells | Unknown mechanism(s) alter T cell metabolism |
| Therapy | Intervention in the Cancer- Immunity Cycle | Strategy |
|---|---|---|
| Cancer Vaccines | Promoting T cell Priming | Not discussed |
| Transfer of immune effectors | Enhance or generate T cell response | TILs |
| CAR T cells | ||
| bispecific antibodies | ||
| Immunomodulation | Counteract immunosuppression | Immune Checkpoint Inhibitors (ICIs) |
| Cytokines and cytokine blockade |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montironi, C.; Muñoz-Pinedo, C.; Eldering, E. Hematopoietic versus Solid Cancers and T Cell Dysfunction: Looking for Similarities and Distinctions. Cancers 2021, 13, 284. https://doi.org/10.3390/cancers13020284
Montironi C, Muñoz-Pinedo C, Eldering E. Hematopoietic versus Solid Cancers and T Cell Dysfunction: Looking for Similarities and Distinctions. Cancers. 2021; 13(2):284. https://doi.org/10.3390/cancers13020284
Chicago/Turabian StyleMontironi, Chiara, Cristina Muñoz-Pinedo, and Eric Eldering. 2021. "Hematopoietic versus Solid Cancers and T Cell Dysfunction: Looking for Similarities and Distinctions" Cancers 13, no. 2: 284. https://doi.org/10.3390/cancers13020284
APA StyleMontironi, C., Muñoz-Pinedo, C., & Eldering, E. (2021). Hematopoietic versus Solid Cancers and T Cell Dysfunction: Looking for Similarities and Distinctions. Cancers, 13(2), 284. https://doi.org/10.3390/cancers13020284