Simple Summary
The tumor suppressor gene TP53 is found mutated in around half of human cancers. The accumulation of the mutant p53 protein in the form of aggregates in cancer cells have led to the emergence of the “prion p53” hypothesis which states that mutant p53 is able to drive the wild-type form of the protein into an alternate conformation, thereby contributing to tumor progression. This report challenges the “prion p53” hypothesis by reviewing evidence of p53 behavior in light of our current knowledge regarding amyloid proteins, prionoids and prions.
Abstract
Identified in the late 1970s as an oncogene, a driving force leading to tumor development, p53 turned out to be a key tumor suppressor gene. Now p53 is considered a master gene regulating the transcription of over 3000 target genes and controlling a remarkable number of cellular functions. The elevated prevalence of p53 mutations in human cancers has led to a recurring questioning about the roles of mutant p53 proteins and their functional consequences. Both mutants and isoforms of p53 have been attributed dominant-negative and gain of function properties among which is the ability to form amyloid aggregates and behave in a prion-like manner. This report challenges the ongoing “prion p53” hypothesis by reviewing evidence of p53 behavior in light of our current knowledge regarding amyloid proteins, prionoids and prions.
1. Introduction
p53 is a 393-amino-acids-long transcription factor composed of a globular DNA binding domain flanked by a transcription activation domain in N-terminal and a tetramerization domain in the C-terminal part of the protein (Figure 1a) [1] and is active in a homo-tetrameric state [2]. p53’s best-described functions mainly revolve around genome maintenance. p53 is usually kept at a low basal protein level by the E3-ubiquitin ligase MDM2 (murine double minute 2) and its paralog MDM4 [3]. Following DNA damages or other cellular stresses, p53 level rises as the protein is phosphorylated and stabilized, which in turn facilitates DNA repair by pausing the cell cycle (via p21) or guides damaged cells toward apoptosis (via BAX) [4,5,6].
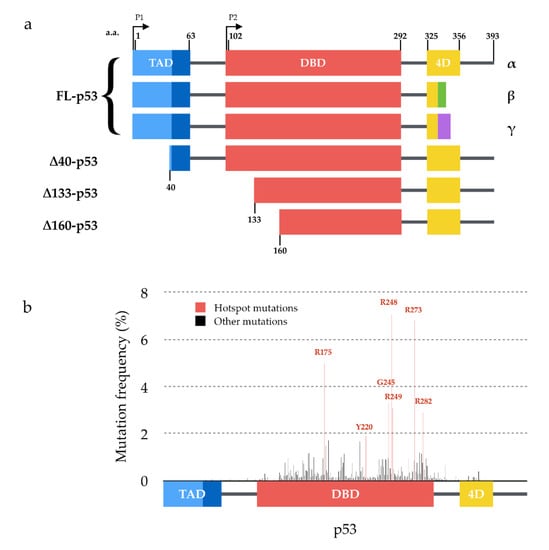
Figure 1.
Structure of p53 isoforms and distribution of mutations within p53. (a) p53 isoforms are generated from the use of two alternative promoters (P1 and P2), secondary initiation codons (∆40, ∆133 or ∆160) and/or alternate splicing sites (α, β or γ C-termini) leading to 12 combinations described to date. (b) Distribution and frequency of p53 mutations described in human tumors (data from the International Agency for Research on Cancer). Hotspot mutations are shown in red and are all located within the DNA binding domain of p53.
However, the progressive accumulation of data regarding p53 target genes has highlighted the fact that the p53 response is remarkably flexible and that it regulates different sets of genes depending on the cell type, stress conditions and microenvironment signaling. p53 indeed participates in autophagy, senescence, metabolism, proliferation, differentiation, immune response and inflammation [7,8,9,10,11,12]. The complexity of the characterization of p53 functions is also increased by the fact that the TP53 gene encodes at least 12 isoforms (Figure 1a), whose roles remain to be fully unraveled [13,14,15]. In addition, the identification of two p53 paralogues, p63 [16] and p73 [17,18], which also encode multiple isoforms [19], adds further complexity to an already dense network. Both p63 and p73 display a modular structure similar to that of p53 with which they also share a high degree of similarity at the protein level [20,21]. This leads to the regulation by these three genes of a common set of target genes involved in apoptosis and cell cycle arrest, and highlights p63 and p73 as candidate tumor suppressor genes [22].
Given the broad spectrum of p53’s functions, its inactivation obviously places a heavy burden on the cell. Mutations of p53 are indeed found in about half of human tumors on average [23,24]. The vast majority of these mutations are amino acid substitutions, primarily located in the DNA binding domain (Figure 1b). The functional consequences of p53 mutations are manifold. In particular, mutant p53 proteins have been shown to be less efficiently ubiquitinated [25] and can thus be stabilized by evading proteasomal degradation through the MDM2 degradation loop, thereby increasing their cellular levels [26]. Several mutations are considered to confer gain-of-function properties to p53 (e.g., increased protein half-life, activation of additional downstream pathways) and have been associated with poorer prognosis for the patients [26,27,28,29,30]. When the mutations are heterozygous, p53 mutant proteins have notably been shown to interfere by a dominant-negative effect with the transcriptional activity of the wild-type p53 proteins encoded by the remaining wild-type allele [14,31,32,33,34]. Among the mechanisms proposed to explain the dominant-negative effect of mutant p53 over wild-type p53, the “prion-like” hypothesis has been steadily gaining momentum during the past decade. Twenty-five years after this hypothesis was first introduced, there is now enough data to review this theory. We have scrutinized the current research dedicated to deciphering the ability of p53 dominant-negative mutants to actually behave in a prion-like manner. To begin with, we will define the terms amyloid, prion and prion-like in order to clearly lay the foundations of our line of argument.
2. Amyloid, Prion and Prionoids
2.1. Members Only, the Defining Traits of Amyloid Proteins
One of the defining traits of prion and prion-like proteins is their amyloid structure. The term amyloid was coined in 1838 by botanist Matthias Schleiden (from the Latin amylum (starch)) to describe an amylaceous constituent of plants. In 1854, Rudolph Virchow first used the term amyloid because of the peculiar reaction of iodine with the corpora amylacea of the nervous system, which he first mistakenly took for starch [35].
In vivo amyloids share three main hallmarks [36]. Firstly, they display a fibrillary morphology. As the amyloid architecture is a consequence of the physicochemical properties of a polypeptide chain, a given peptide sequence can be incorporated in fibrils in multiple ways and give rise to various fibril morphologies. Secondly, amyloids are formed by proteins enriched in β-sheet secondary structure as a result of conformation change. These proteins then adopt a cross-β structure in which β-strands are oriented perpendicularly to the fibril axis and are assembled into β-sheets that run the length of the fibrils with a parallel or antiparallel in-register arrangement. Only a fraction of the polypeptide chain is incorporated in the cross-β core of the fibrils. Thirdly, amyloids specifically bind dyes, such as thioflavin (ThT or ThS) or Congo Red [36,37]. Any given protein aggregate needs to display these three features to be classified as amyloid in vivo. Remarkably, said fibrils are formed by polypeptide chains which have no similarity in sequence, structure or function whatsoever, nevertheless sharing morphological and structural properties. The term amyloid has thus evolved into a polysemous term as it refers to protein-based structures associated with protein misfolding diseases (PMDs), as well as to a wide panel of essential functions performed by proteins called functional amyloids [38].
So far, among the large panel of amyloid-forming proteins, 36 proteins and peptides (e.g., PrP, α-syn, IAPP) have been formally proven to form pathological amyloid in vivo [36]. Among the pathological amyloids, prion and prionoid have the property of being able to self-replicate; but the prion protein PrP presents the seemingly unique property of being inter-individually transmittable.
2.2. PrP, Still the Only One in the Prion Category
The term prion refers to the proteinaceous agent causing prion diseases, now known to be PrPSc, the pathological isoform of the cellular prion protein PrPC, which is encoded by the PRNP gene [39,40]. Prions are thought to multiply by a nucleation and fragmentation process during which PrPSc oligomers grow in size through incorporation of endogenous PrPC [41]. Large PrPSc aggregates may then be taken apart into smaller fragments, called propagons, able to singlehandedly initiate a new nucleation–fragmentation cycle [42,43]. PrPC protein can adopt various PrPSc self-propagating conformations, each being able to propagate its biochemical signature to naive PrPC proteins. The existence of these structurally distinct assemblies is referred to as prion strains and leads to different molecular, histopathological and clinical phenotypes (reviewed in [44]).
Prion diseases are also called transmissible spongiform encephalopathies (TSE) as they have all the characteristics of infectious diseases, such as transmissibility, species barriers, as well as the existence of strains. Prion diseases are found in several mammalian species, among which cattle, where the mad cow disease pandemic originated in the late 1990s [45,46], and cervids whose wild population is increasingly affected by the chronic wasting disease (CWD) in North America wooded areas and more recently in Europe [47]. Prion diseases also affect humans with an incidence of 1–2 cases per million. Human prion diseases can be either genetic, sporadic or acquired. Genetic prion diseases like genetic Creutzfeldt–Jakob disease (gCJD), fatal familial insomnia (FFI) and Gerstmann–Straussler–Scheinker syndrome (GSS) account for 5% of cases and are all caused by mutations in the PRNP gene (for review see [48]). Sporadic prion diseases like sCJD and sporadic FFI represent 85% of cases and have yet unknown etiology. Acquired prion diseases are due to the transmission of prions within the human species (through medical procedure, growth hormone supplementation, cannibalism) or from animal to human (for example by the consumption of products derived from cattle suffering from “mad cow” disease, leading to vCJD). All these prion diseases are characterized by the accumulation of PrPSc amyloid aggregates in the central nervous system.
2.3. Prionoids, the New Challengers
An increasing number of neurodegenerative disorders, including Alzheimer disease (AD), Parkinson disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and also metabolic diseases like type 2 diabetes (T2D) and AA amyloidosis A, have been linked to protein misfolding and aggregation [41,49,50,51,52]. In these PMDs, also often termed “prion-like diseases,” specific proteins are detected under the form of amyloid fibers in patients’ tissues, just as PrPSc (e.g., Aβ and tau in AD, α-Synucelin in PD, TDP-43 in ALS, HTT in HD, IAPP in T2D). The clinical consequences of their misfolding vary between these diseases and can occur in different organs, among which the central nervous system, liver, spleen, pancreas or the peripheral nervous system. Yet, all PMD-associated proteins are found in extracellular and/or intracellular amyloid aggregate deposits and share critical features with prion proteins (for review, [50,53,54,55]). Firstly, alternatively folded or partially unfolded isoforms of a disease-associated protein interact with each other to form cross-β spines [56] that assemble into propagons able to self-propagate [42]. Propagons grow into protofilaments that interact with each other to form higher-order fibrillary aggregates. Fibrils can also be fragmented, which frees up additional seeds that possess templating (and therefore self-perpetuating) activity of their own [41]. Secondly, PMD-associated proteins are capable of cell-to-cell propagation in vitro, and aggregates have also been shown to seed and spread upon inoculation and initiate disease in vivo [55,57]. Thirdly, PMD-associated proteins also display strains and species barriers [55,58,59,60,61,62].
Because PMD-associated amyloids resemble prions in their self-templating propagation, cell-to-cell transmissibility and existence of species barrier and strains, PMD-associated amyloids are often referred to as prion or prion-like proteins. However, inter-individual transmissibility of PMD-associated proteins, which is a defining trait of prions, has not been definitively proven yet. The term “prionoid” thus emerged as a way to classify non-infectious self-propagating amyloid aggregates capable of cell-to-cell propagation within individuals [41,49,50].
A given prionoid might however change classification if its infectivity happens to be conclusively demonstrated. Notably, some evidence of the experimental transmission of protein aggregates at cellular and organism levels are emerging regarding Aβ [63,64,65], α-Synuclein [66], tau [67], and amyloid A seeds [68,69,70,71]. Further studies exploring the natural donor/host transmission are eagerly awaited as they should assess whether or not certain prionoids are also transmissible or if prions are still the only protein agents capable of actually transmitting diseases in a natural environment.
2.4. Functional Amyloids, Cellular Multitools
The term amyloid is essentially associated with neurodegenerative diseases and systemic amyloidosis, and is thus perceived as negative and deleterious for the cell. However, knowledge about functional amyloids is emerging and certain amyloid assemblies have now been described as performing a variety of essential life processes in many organisms [38]. In mammals, functional amyloids are, for instance, involved in the chemical storage of peptide hormones [72] and of the blood clotting proteins fibrin [73], the scaffolding avoiding melanin toxicity (Pmel17, [74]), the long-term memory (CPEB3, [75]), mouse fertility (CRES genes, Cornwall 2019 Andrology) and the necroptosis signaling in virus-infected cells (RIP1/RIP3, [76]). But essential functions are also driven by amyloids or super-assemblies in other organisms, like biofilm formation in Escherichia coli (Curli) and Bacillus subtilis (TasA), plasmid replication control in Bacteria (RepA, [77]), long-term memory in Aplysia (CPEB, [78]) and Drosophila (Orb2), memory of past unsuccessful mating encounters leading to aging in Saccharomyces cerevisiae Whi3, [79,80]), protection of germline components in dormant oocyte in Xenopus laevis (Xvelo, [81,82]), Drosophila (Oskar), Zebrafish (Bucky ball) and Caenorhabditis elegans (P granules) or immune response in Drosophila (Imd, [83]). HET-s, which is involved in heterokaryon incompatibility in Podospora anserina, has a special place in the amyloid world as it is a functional amyloid with prion characteristics [84]. It is now accepted that protein aggregation actually plays a native role in cellular functions. The high stability of the amyloid structures indeed provides structural organizing scaffolds which can be easily and quickly mobilized in response to environmental and physiological conditions.
Despite their structural similarities, the kinetics of aggregation of pathological and functional amyloids diverge, the formation of pathological amyloids being much slower than the fibrillation of functional ones. Moreover, a specificity of functional amyloids is that their formation and disappearance are tightly controlled as they can be disintegrated and release functional monomers on demand. Amyloid formation is thus not necessarily an irreversible process as it is thought to be for prions and prionoids. It is important to note that the pathological entities in prions and prionoid-based diseases are most probably the small oligomers and not the amyloid fibers per se [85,86]. Large fibrils can have a buffering effect by sequestering toxic oligomers within more inert structures [87], which makes them comparable to certain functional amyloids, the quick aggregation kinetics of which may spare cells from oligomer toxicity.
Prion, prionoid and amyloid entities are now more clearly defined in the literature and although putting p53 in a box is not the point of this review, it is high time to reassess its actual prion-like properties.
3. Prion p53, the Origin Story
The first mention of a “prion p53” actually dates back to 1995 [88]. As a basis to explain the dominant-negative effect of mutant p53 over the wild-type protein, Jo Milner and Elizabeth Medcalf evaluated the in vitro effect of several mutants of p53 (p. R151S, p. R247I, p. R273P, p. R273L) on wild-type p53. They showed that these mutants drive a wild-type p53 bearing a “wild-type” conformation [p53WT] toward a mutant conformation [p53MUT] when co-translated, thereby discriminating two in vitro allosteric variants of wild-type p53 [89].
The conformational flexibility of p53 has also provided grounds to the prion p53 hypothesis. Changes in p53 conformation have been monitored by their reactivity to conformation-specific antibodies [90]. For example, pAb1620 antibody recognizes the [p53WT] conformation while pAb240 [91] recognizes the [p53MUT] conformation. From there on, p53 mutants were classified as having wild-type [p53WT] or mutant [p53MUT] conformations based on their reactivity with either pAb1620 or pAb240 antibody. Mutants harboring a [p53WT] conformation were, however, not systematically transcriptionally active. These observations later gave rise to the subdivision of p53 mutants into two categories: (i) “DNA contact” mutants (R273H, R248Q, R248W), which have a decreased ability to bind DNA, and (ii) “conformation or structural” mutants (R175H, G245S, R249S, R282H), in which mutations induce a local to global destabilization of the protein structure [88,92] that exposes specific motives at the surface of the protein, thereby allowing reactivity with the pAb240 antibody. However, the relevance of these two classes of mutants is being challenged [93,94]. Some mutations such as p.V135A lead to a [p53WT] conformation when translated at 30 °C, but the protein undergoes a conversion to a [p53MUT] conformation within two minutes when the temperature shifts from 30 °C to 37 °C [95], also illustrating p53 conformational flexibility. Hence, the apparent ability of some p53 mutants to induce a conformational shift of a wild-type p53 protein bearing a [p53WT] conformation towards a [p53MUT] conformation, together with the existence of alternative conformations of p53 gave ground to the “prion p53” hypothesis [88,96], at a time when the prion protein PrPSc was in the limelight (Figure 2a).
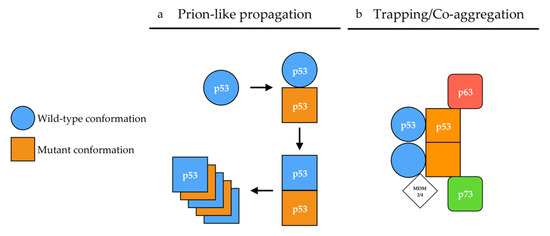
Figure 2.
Proposed mechanisms of p53 aggregation. (a) According to the prion-like hypothesis, mutant p53 (orange) in a [p53MUT] conformation (square) induces a conformational shift of wild-type p53 (blue) from a [p53WT] (circle) conformation toward a [p53MUT] conformation in a prion-like manner. In a [p53MUT] conformation, wild-type and mutant p53 form amyloid fibers. (b) According to the co-aggregation/trapping hypothesis, mutant p53 hetero-tetramerizes with wild-type p53. The unfolded core of mutant p53 allows new interactions with p63/p73 isoforms and MDM2/4 in a trapping/co-aggregation mechanism.
3.1. p53 Aggregation in Tissue Samples and Tumor Cell Lines
p53 mutants have early on been described as forming aggregates in tissue samples and tumor cell lines. p53 has indeed been shown to form protein aggregates that abnormally accumulate in the nucleus and sometimes in the cytoplasm of cells from several types of tumor samples [97,98,99,100,101,102]. The aggregative features of p53 have been monitored for a time by immunohistochemistry (anti-p53 antibodies DO-1 and others) as a surrogate for the identification of tumors carrying p53 mutation [98,103,104,105], which is associated with worsened cancer prognosis (reviewed in [106]). However, immunohistochemistry often fails to deliver consistent results and is thus not the expected gold standard [28,104].
The amyloid nature of p53 aggregates has been mostly explored by co-localization experiments relying on anti-p53 antibodies, anti-fibrils antibodies (OC, A11 [107,108,109]) or amyloid dyes (thioflavin T, thioflavin S or Congo red). However, major variations are observed in the quantity and features of p53 aggregation, depending on the cell type, the mutation of p53 or the method used to quantify said aggregation [106,110,111,112,113,114], thereby rendering arduous any extensive comparison. In addition, the nature of the proteins forming the A11/OC-positive or amyloid dye-positive protein aggregates was not determined, thus the sometimes-partial co-localization of p53 with these amyloid aggregates precludes, for now, p53 from being classified as a genuine amyloid by the International Society of Amyloidosis nomenclature committee [36,115].
Studies concerning p53 aggregation have been limited to the canonical (full-length FL-p53α) isoform of p53 and its mutant forms. However, recent reports focused on the potential aggregation of p53 isoforms ∆40p53 [116] and ∆133p53 [117,118] have opened new perspective to the field. The tissue-specific expression of p53 isoforms [15] could indeed partially account for the diversity of p53 aggregation phenotypes observed.
3.2. p53 Aggregation: In Vitro Dynamics
Despite the fact that only 36 proteins have been shown to form pathogenic amyloids in vivo, it is important to note that in vitro amyloid formation is observed for many more protein sequences. It is actually a property shared by many, if not all, natural polypeptide chains, once placed in the appropriate conditions [119,120,121,122,123,124,125,126], and it seems that p53 is no exception to the rule. Indeed, a large body of literature reports that, when subjected to high pressure, high temperature, zinc absence, low pH or RNA molecules, full-length p53, N-terminal, core or C-terminal fragments of p53, can be led to convert into either amorphous aggregates or ThT positive amyloid fibers in vitro [106,113,127,128,129,130,131,132,133,134,135].
The nucleation-dependent mechanism has three characteristics: the existence of a lag time, critical concentration and seeding. The lag time corresponds to the time required for nucleus formation during which the protein appears to be soluble. In contrast with nucleation-dependent polymerization, the growth of a linear polymer does not require nucleation and is characterized by the sequential buildup of intermediates. No lag time is observed, and supersaturated solutions rapidly aggregate. This process can be difficult to distinguish from a nucleation-dependent process with a very short lag time or from seeded growth [136]. The in vitro kinetics of p53 aggregation indeed differs from that of the classical nucleation-growth formation of amyloid fibrils, as the initiation of p53 aggregation happens to be relatively rapid [106,129,137] and as such is reminiscent of that of a linear polymer. The kinetics of p53 aggregation involves the formation of small aggregates that rapidly form amyloid structures that bind ThT and subsequently grow into larger amorphous aggregates. The progress curves fit to a two-step sequential pattern [138,139,140] in which a destabilized mutant p53 may co-aggregate with wild-type p53 and its paralogs p63 and p73. In this model, a mutant preferentially adopts an unfolded structure and would primarily react with another fast-unfolding mutant protein while only occasionally trapping a slow-unfolding wild-type protein. The mutant population rapidly self-aggregates before much of the wild-type p53 protein is depleted. However, as wild-type p53 is incorporated in hetero-tetramers by mutants, the continual production of mutant p53 in a cancer cell would gradually trap more and more wild-type p53 [141], its paralogs p63 and p73 [14,139] and MDM2 [142]. The trapping dynamics could also involve molecular chaperone HSP70 as it is proposed to stabilize mutant p.R175H and increase its aggregation [143]. This may account for the observations of co-aggregates in cell lines and in tumor tissue samples, and possibly causing a dominant-negative effect by directly impairing DNA binding activity [141,144,145].
3.3. p53 Aggregation: The Seeding Attempts
As nucleation is rate-limiting at low saturation levels, adding a seed (preformed nucleus) greatly accelerates the polymerization of molecules from solution [120,136,146,147,148,149]. However, in vitro p53 preformed aggregates did not significantly seed the aggregation of bulk proteins and stoichiometric amounts of aggregation-prone mutants induced only small amounts of wild-type p53 to co-aggregate [138,139,140]. p53 seeding experiments are indeed usually based on large amounts of aggregated proteins (10% of the dilution of aggregated proteins) [106] whereas very small amounts of prion or prionoid aggregates (1% to 0.05%) seed aggregation of bulk protein [147,150,151]. In 2013, based on prior demonstration of amyloid aggregation of p53 in vitro, Forget et al. tackled the question of whether or not an in vitro aggregated full-length p53 could be able to propagate its conformation to endogenous wild-type p53 in cells. These p53 aggregates were shown to penetrate cells via a nonspecific macropinocytosis pathway and to induce the co-aggregation of endogenous wild-type p53 in equally amorphous aggregates [152]. However, this report shows a single cell exhibiting p53 aggregates which hardly demonstrates that p53 can induce the aggregation of endogenous p53 proteins. Co-culture experiments were also used by Gosh et al. to demonstrate the cell-to-cell transmission of amyloid aggregates of p53 that were previously induced by the P8 peptide (PILTIITL, corresponding to the residues 250–257 of p53) [113]. The in vitro mechanism of p53 aggregation corresponds more to trapping by cross-reaction and co-aggregation rather than classical seeding and growth (see below; [139]).
3.4. p53 Aggregation: The Trapping Evidence
A consequence of prion self-propagation is that PrPC protein overexpression leads to an increased propagation of PrPSc prion due to the increased availability of its substrate [153,154]. This is also a feature of prionoids. However, the dominant-negative inhibition of mutants and isoforms of p53 has been shown to be dose-dependent in Soas-2 cells [155] as well as in S. cerevisiæ [14,156], two models lacking endogenous p53. Conversely, the expression of wild-type p53 has been shown to suppress the growth of tumor cell lines bearing dominant negative p53 mutants [157,158] and to overwhelm the inhibition exerted by mutant p53 on its transcription activity in baker’s yeast [14].
Then, in order to challenge the main ability of prions that is self-propagation, wild-type p53 has been co-expressed in yeast with a mutant p53 (p.R175H or p.R248Q) which expression has been placed under the control of a galactose-inducible promoter in a typical prion-propagation assay [159,160]. When co-expressed with mutant p53, wild-type p53 transcriptional functions were inhibited by the dominant-negative mutant p53. However, as soon as mutant p53 expression was shut off, wild-type p53 fully recovered its transcriptional abilities [14]. These results thus show that mutant p53 did not print its [p53MUT] conformation on wild-type p53 in a prion-like manner in S. cerevisiae, although recent data also suggest that when fused to EYFP, p53 overexpression leads, in some rare yeast cells, to the formation of aggregates that are transmitted across generations [161]. Altogether, these data show that the dominant-negative effect of mutant p53 is dose-dependent and can be reduced or even neutralized by increasing the wild-type/mutant p53 ratio.
The fact that increasing wild-type p53 expression level can overwhelm mutant p53 dominant-negative effect also led to p53-based gene therapy trials relying on adenoviruses. They aimed at restoring p53 tumor suppressive function in cancer cells by thwarting mutant p53 with a therapeutic wild-type p53 gene. Advexin and Gendicine [162] appeared as the main therapeutic candidates, having been used in numerous trials (including phase III). Chinese FDA approved the use of Gendicine in 2003 for head and neck tumors and in 2005 for naso-pharyngeal cancers [163]. To date, more than 30,000 patients have received Gendicine in association with chemo- or radiotherapy with promising results and relatively few side effects [164].
Other therapeutic strategies aim at reactivating mutant p53 tumor suppressor functions. Quinuclidines PRIMA-1 and its analog APR-246 (phase I and phase II clinical trials) have demonstrated their ability to inhibit cell proliferation and increase apoptosis in cancer cell lines and other models, although it has been reported that they could display non-p53 related activity [165]. The ReACp53 peptide targets the aggregation ability of mutant p53 and restores a partial function of the mutant protein, thereby inducing tumor shrinking both in vitro and in vivo [166,167]. These strategies, among others [168], seem to lead to mutant p53 refolding, allowing for a functional recovery of the protein.
Both the successful therapeutic approaches set up to target mutant p53 and the unsuccessful attempts of propagating [p53MUT] conformational phenotype strongly indicate that the p53 protein, be it in a wild-type or mutant state, does not behave as a prion nor as a prionoid.
4. Conclusions
Almost 40 years in the making, p53 remains an intense research item, as theories and evidence on its behavior and interactions keep blossoming. From an oncogene, to a tumor suppressor, an amyloid, a prion, p53 has often been studied through the lens of scientific trends [169,170] and once in the air, the idea that p53 behaves in a prion-like manner consistently seeded into the literature.
Although the presented data clearly state that p53 does not behave like a prion or a prionoid, we do not challenge its ability to, potentially, form amyloid fibers in vivo, should concrete evidence be provided. Indeed, human diseases such as Amyloidosis A exhibit amyloid aggregates but no prion or prionoid features [171]. In the case of p53, the formation of amyloid fibers could result from the natural tetramerization between mutant and wild-type p53 serving as a nucleation starting point. Hetero-tetramerization between wild-type p53 and p63/p73 remains unlikely due to the divergence in their tetramerization sequences [20,172]. However, aggregation-prone regions buried in the core domain of p53, which are exposed in several p53 mutants or isoforms, could allow the mutant protein to interact with p63, p73 and MDM2 [14,111,142,173,174,175,176]. These interactions happen preferentially when mutant p53 is able to tetramerize, suggesting that tetramers of mutant p53 may present structural properties allowing them to interact with their paralogs (Figure 2b) [14]. Interaction between p53 and its paralogs is a credible explanation for gain of function properties of p53 mutants evidenced in engineered mice models of Li-Fraumeni Syndrome p53R270H/+ and p53R172H/+ which harbor mutations equivalent to human R273H and R175H, respectively. On the one hand, these mice developed allele-specific tumor spectra distinct from p53+/− mice and on the other hand developed novel tumors when compared to p53−/− mice [177,178].
Given the prevalence of p53 mutations and the frequent accumulation of mutant p53 in cancers, it could be asked whether and how the (co)-aggregation of p53 family members would provide a decisive advantage to malignant cells. Before undergoing loss of heterozygosity, cancerous cells can see a decreased p53 function mainly due to the dominant-negative effect of mutant p53 if the mutant is stabilized. The mechanisms underlying mutant p53 stabilization still need to be elucidated. MDM2 seems indeed able to degrade mutant p53 proteins in vitro and the stabilization of p53 could be related to other factors specifically related to cancerous cell types, the mutation involved and/or loss of heterozygosity itself [105,179]. At later stages, the wild-type allele being often lost, mutant p53 may remain deleterious by trapping p63 and p73 isoforms, thereby maintaining the formation of aggregates. Although induced p53 oligomers have been described as cytotoxic for cells in culture [112] as others types of protein aggregates [85,180], p53 aggregation does not appear to be toxic for cells in vitro nor in vivo, suggesting, once again that there is more to p53 aggregation that meets the eye. Hence, conformational changes, especially amyloid-like, are not necessarily associated with a pathological condition, whereas in the case of prions and prionoids, they definitely are.
In the absence of amyloid characteristics of p53 aggregates, its polymer-like aggregation kinetics, the inability of self-perpetuation of the mutant conformation, and the lack of proven seeding capacities, the qualification of “prion p53” appears largely overstated. The prion p53 theory seems to rather result from an all-too-tempting intellectual jump from the observation of protein aggregates to infectious prion.
Funding
This research was funded by Institut National du Cancer [INCa N°2017-69] (PR, OB), Institut de France—Fondation NRJ (CV), Inserm (CV and GF), UBO (CV and GF), Association Défi Organisation (CV).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
Philippe Fort and Véronique Gire for insightful proofreading and suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Chène, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Dobbelstein, M.; Levine, A.J. Mdm2: Open questions. Cancer Sci. 2020, 111, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; A Liebermann, D.; Hoffman, B.; Reed, J.C. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 259–265. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Sato, Y.; Tsurumi, T. Genome guardian p53 and viral infections. Rev. Med Virol. 2012, 23, 213–220. [Google Scholar] [CrossRef]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in the cross fire: p53 in inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. P53 and The Immune Response: 40 Years of Exploration—A Plan for the Future. Int. J. Mol. Sci. 2020, 21, 541. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C. p53 isoforms change p53 paradigm. Mol. Cell. Oncol. 2014, 1, e969136. [Google Scholar] [CrossRef] [PubMed]
- Billant, O.; Léon, A.; Le Guellec, S.; Friocourt, G.; Blondel, M.; Voisset, C. The dominant-negative interplay between p53, p63 and p73: A family affair. Oncotarget 2016, 7, 69549–69564. [Google Scholar] [CrossRef]
- Anbarasan, T.; Bourdon, J.-C. The emerging landscape of p53 isoforms in physiology, cancer and degenerative diseases. Int. J. Mol. Sci. 2019, 20, 6257. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Kaghad, M.; Bonnet, H.; Ferrara, P.; McKeon, F.; Caput, D.; Yang, A.; Creancier, L.; Biscan, J.-C.; Valent, A.; Minty, A.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Senoo, M.; Seki, N.; Ohira, M.; Sugano, S.; Watanabe, M.; Tachibana, M.; Tanaka, T.; Shinkai, Y.; Kato, H. A second p53-related protein, p73l, with high homology to p73. Biochem. Biophys. Res. Commun. 1998, 250, 536. [Google Scholar] [CrossRef]
- Khoury, M.P.; Bourdon, J.-C. p53 isoforms: An intracellular microprocessor? Genes Cancer 2011, 2, 453–465. [Google Scholar] [CrossRef]
- Belyi, V.A.; Ak, P.; Markert, E.; Wang, H.; Hu, W.; Puzio-Kuter, A.; Levine, A.J. The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol. 2009, 2, a001198. [Google Scholar] [CrossRef]
- Dotsch, V.; Bernassola, F.; Coutandin, D.; Candi, E.; Melino, G. p63 and p73, the Ancestors of p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a004887. [Google Scholar] [CrossRef] [PubMed]
- Deyoung, M.P.; Ellisen, L.W. p63 and p73 in human cancer: Defining the network. Oncogene 2007, 26, 5169–5183. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.A.S.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, C.; Xu, Y.L.C.C.Z.; Scuoppo, C.; Rillahan, C.D.; Gao, J.; Spitzer, B.; Bosbach, B.; Kastenhuber, E.R.; Baslan, T.; et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nat. Cell Biol. 2016, 531, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and Degradation of Mutant p53. Mol. Cell. Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef]
- Finlay, C.A.; Hinds, P.W. Activating mutations for transformation by p53 produce a gene product that forms an hsc7o-p53 complex with an altered half-life. Mol. Cell. Biol. 1988, 8, 9. [Google Scholar] [CrossRef]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties onTP53mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2009, 2, a001008. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Ding, Y.; Viox, D.J.; Jiang, M.; Zheng, Y.; Liao, W.; Chen, X.; Xiang, W.; Yi, Y. Evaluation of public cancer datasets and signatures identifies TP53 mutant signatures with robust prognostic and predictive value. BMC Cancer 2015, 15, 179. [Google Scholar] [CrossRef]
- Bargonetti, J.; Prives, C. Gain-of-function mutant p53: History and speculation. J. Mol. Cell Biol. 2019, 11, 605–609. [Google Scholar] [CrossRef]
- Srivastava, S.; Wang, S.; A Tong, Y.; Hao, Z.M.; Chang, E.H. Dominant negative effect of a germ-line mutant p53: A step fostering tumorigenesis. Cancer Res. 1993, 53, 4452–4455. [Google Scholar] [PubMed]
- E Hegi, M.; A Klein, M.; Rüedi, D.; Chène, P.; Hamou, M.F.; Aguzzi, A. p53 transdominance but no gain of function in mouse brain tumor model. Cancer Res. 2000, 60, 3019–3024. [Google Scholar]
- De Vries, A.; Flores, E.R.; Miranda, B.; Hsieh, H.-M.; Van Oostrom, C.T.M.; Sage, J.; Jacks, T. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc. Natl. Acad. Sci. USA 2002, 99, 2948–2953. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Teoh, W.W.; Phang, B.H.; Tong, W.-M.; Wang, Z.Q.; Sabapathy, K. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell 2012, 22, 751–764. [Google Scholar] [CrossRef]
- Kyle, R.A. Amyloidosis: A convoluted story. Br. J. Haematol. 2001, 114, 529–538. [Google Scholar] [CrossRef]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.; Merlini, G.; Saraiva, M.J.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Otzen, D.; Riek, R. Functional amyloids. Cold Spring Harb. Perspect. Biol. 2019, 11, a033860. [Google Scholar] [CrossRef]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Ness, F.; Tuite, M. Analysis of the generation and segregation of propagons: Entities that propagate the [PSI+] prion in yeast. Genetics 2003, 165, 23–33. [Google Scholar] [PubMed]
- Aguzzi, A.; Lakkaraju, A.K. Cell Biology of Prions and Prionoids: A Status Report. Trends Cell Biol. 2016, 26, 40–51. [Google Scholar] [CrossRef]
- Scialò, C.; De Cecco, E.; Manganotti, P.; Legname, G. Prion and prion-like protein strains: Deciphering the molecular basis of heterogeneity in neurodegeneration. Viruses 2019, 11, 261. [Google Scholar] [CrossRef]
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef] [PubMed]
- MacLea, K.S. What makes a prion. Int. Rev. Cell. Mol. Biol. 2017, 329, 227–276. [Google Scholar] [CrossRef] [PubMed]
- Koutsoumanis, K.; Benestad, S.L.; Comoy, E.; Allende, A.; Nonno, R.; Felicio, T.D.S.; Ortiz-Pelaez, A.; Simmons, M.M.; Alvarez-Ordoňez, A.; Bolton, D.; et al. Update on chronic wasting disease (CWD) III. EFSA J. 2019, 17, e05863. [Google Scholar] [CrossRef]
- Will, R.G.; Ironside, J.W. Sporadic and infectious human prion diseases. Cold Spring Harb. Perspect. Med. 2016, 7, a024364. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A. Beyond the prion principle. Nat. Cell Biol. 2009, 459, 924–925. [Google Scholar] [CrossRef]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef]
- Mukherjee, A.; Soto, C. Prion-like protein aggregates and type 2 diabetes. Cold Spring Harb. Perspect. Med. 2017, 7, a024315. [Google Scholar] [CrossRef] [PubMed]
- Westermark, G.T.; Fändrich, M.; Westermark, P. AA amyloidosis: Pathogenesis and targeted therapy. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 321–344. [Google Scholar] [CrossRef]
- Ashe, K.H.; Aguzzi, A. Prions, prionoids and pathogenic proteins in Alzheimer disease. Prion 2013, 7, 55–59. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M.-Y. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef]
- Eraña, H.; Venegas, V.; Moreno, J.; Castilla, J. Prion-like disorders and Transmissible Spongiform Encephalopathies: An overview of the mechanistic features that are shared by the various disease-related misfolded proteins. Biochem. Biophys. Res. Commun. 2017, 483, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nat. Cell Biol. 2005, 435, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morales-Scheihing, D.; Salvadores, N.; Moreno-Gonzalez, I.; Gonzalez, C.; Taylor-Presse, K.; Mendez, N.; Shahnawaz, M.; Gaber, A.O.; Sabek, O.M.; et al. Induction of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. J. Exp. Med. 2017, 214, 2591–2610. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Vigouret, J.-M.; Paganetti, P.; Walsh, D.M.; Mathews, P.M.; Ghiso, J.; Staufenbiel, M.; Walker, L.C.; Jucker, M.; et al. Exogenous induction of cerebral -amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.S.; Wangsanut, T.; Tayler, K.K.; Wiltgen, B.J.; Hatami, A.; Rönicke, R.; et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nat. Cell Biol. 2012, 485, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kaufman, S.K.; Miller, T.M.; Grinberg, L.T.; Seeley, W.W.; Diamond, M.I.; Devos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef]
- Watts, J.C.; Condello, C.; Stöhr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of A prions from Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.F.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.M.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nat. Cell Biol. 2015, 525, 247–250. [Google Scholar] [CrossRef]
- Frontzek, K.; I Lutz, M.; Aguzzi, A.; Kovacs, G.G.; Budka, H. Amyloid-β pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt-Jakob disease after dural grafting. Swiss Med. Wkly. 2016, 146, w14287. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.L.; Adlard, P.; Peden, A.H.; Lowrie, S.; Le Grice, M.; Burns, K.; Jackson, R.J.; Yull, H.; Keogh, M.J.; Wei, W.; et al. Amyloid-β accumulation in the CNS in human growth hormone recipients in the UK. Acta Neuropathol. 2017, 134, 221–240. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; Trojanowski, J.Q. Pathological a-synuclein transmission initiates parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 6. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Sazdovitch, V.; Ando, K.; Seilhean, D.; Privat, N.; Yilmaz, Z.; Peckeu, L.; Amar, E.; Comoy, E.; Maceski, A.; et al. Neuropathology of iatrogenic Creutzfeldt–Jakob disease and immunoassay of French cadaver-sourced growth hormone batches suggest possible transmission of tauopathy and long incubation periods for the transmission of Abeta pathology. Acta Neuropathol. 2017, 135, 201–212. [Google Scholar] [CrossRef]
- Lundmark, K.; Westermark, G.T.; Nyström, S.; Murphy, C.L.; Solomon, A.; Westermark, P. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 6979–6984. [Google Scholar] [CrossRef]
- Solomon, A.; Richey, T.; Murphy, C.L.; Weiss, D.T.; Wall, J.S.; Westermark, G.T.; Westermark, P. Amyloidogenic potential of foie gras. Proc. Natl. Acad. Sci. USA 2007, 104, 10998–11001. [Google Scholar] [CrossRef]
- Zhang, B.; Une, Y.; Fu, X.; Yan, J.; Ge, F.; Yao, J.; Sawashita, J.; Mori, M.; Tomozawa, H.; Kametani, F.; et al. Fecal transmission of AA amyloidosis in the cheetah contributes to high incidence of disease. Proc. Natl. Acad. Sci. USA 2008, 105, 7263–7268. [Google Scholar] [CrossRef]
- Murakami, T.; Ishiguro, N.; Higuchi, K. Transmission of systemic AA amyloidosis in animals. Veter. Pathol. 2013, 51, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Perrin, M.H.; Eisenberg, D.; Rivier, J.; Sawchenko, P.; Vale, W.; Riek, R.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, O.; Bouma, B.; Kroon-Batenburg, L.M.; Reijerkerk, A.; Wu, Y.-P.; Voest, E.E.; Gebbink, M.F. Tissue-type plasminogen activator is a multiligand cross-β structure receptor. Curr. Biol. 2002, 12, 1833–1839. [Google Scholar] [CrossRef]
- Watt, B.; Van Niel, G.; Raposo, G.; Marks, M.S. PMEL: A pigment cell-specific model for functional amyloid formation. Pigment. Cell Melanoma Res. 2013, 26, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Vogler, C.; Spalek, K.; Aerni, A.; Demougin, P.; Müller, A.; Huynh, K.-D.; Papassotiropoulos, A.; De Quervain, D. CPEB3 is associated with human episodic memory. Front. Behav. Neurosci. 2009, 3, 4. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Chan, F.K.-M.; Wu, H.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Molina-García, L.; Gasset-Rosa, F.; Álamo, M.M.-D.; Fernández-Tresguerres, M.E.; De La Espina, S.M.D.; Lurz, R.; Giraldo, R. Functional amyloids as inhibitors of plasmid DNA replication. Sci. Rep. 2016, 6, 25425. [Google Scholar] [CrossRef]
- Si, K.; Choi, Y.-B.; White-Grindley, E.; Majumdar, A.; Kandel, E.R. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 2010, 140, 421–435. [Google Scholar] [CrossRef]
- Caudron, F.; Barral, Y. A super-assembly of whi3 encodes memory of deceptive encounters by single cells during yeast courtship. Cell 2013, 155, 1244–1257. [Google Scholar] [CrossRef]
- Schlissel, G.; Krzyzanowski, M.K.; Caudron, F.; Barral, Y.; Rine, J. Aggregation of the Whi3 protein, not loss of heterochromatin, causes sterility in old yeast cells. Science 2017, 355, 1184–1187. [Google Scholar] [CrossRef]
- Boke, E.; Ruer, M.; Wühr, M.; Coughlin, M.; Lemaitre, R.; Gygi, S.P.; Alberti, S.; Drechsel, D.; Hyman, A.A.; Mitchison, T.J. Amyloid-like self-assembly of a cellular compartment. Cell 2016, 166, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Boke, E.; Mitchison, T.J. The balbiani body and the concept of physiological amyloids. Cell Cycle 2016, 16, 153–154. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kleino, A.; Ramia, N.F.; Li, J.; Silverman, N.; Bozkurt, G.; Shen, Y.; Nailwal, H.; Huang, J.; Napetschnig, J.; Gangloff, M.; et al. Peptidoglycan-Sensing Receptors Trigger the Formation of Functional Amyloids of the Adaptor Protein Imd to Initiate Drosophila NF-κB Signaling. Immunity 2017, 47, 635–647.e6. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Saupe, S.J. The HET-S/s prion motif in the control of programmed cell death. Cold Spring Harb. Perspect. Biol. 2016, 8, a023515. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, M.; Li, C. The unexposed secrets of prion protein oligomers. J. Mol. Neurosci. 2015, 56, 932–937. [Google Scholar] [CrossRef]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-synuclein oligomers and fibrils: A spectrum of species, a spectrum of toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef]
- Silveira, J.R.; Raymond, G.J.; Hughson, A.G.; Race, R.E.; Sim, V.L.; Hayes, S.F.; Caughey, B. The most infectious prion protein particles. Nat. Cell Biol. 2005, 437, 257–261. [Google Scholar] [CrossRef]
- Milner, J. Flexibility: The key to p53 function? Trends Biochem. Sci. 1995, 20, 49–51. [Google Scholar] [CrossRef]
- Milner, J.; Medcalf, E. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 1991, 65, 765–774. [Google Scholar] [CrossRef]
- Ory, K.; Legros, Y.; Auguin, C.; Soussi, T. Analysis of the most representative tumour-derived p53 mutants reveals that changes in protein conformation are not correlated with loss of transactivation or inhibition of cell proliferation. EMBO J. 1994, 13, 3496–3504. [Google Scholar] [CrossRef]
- Gamble, J.; Milner, J. Evidence that immunological variants of p53 represent alternative protein conformation. Virology 1988, 162, 452–458. [Google Scholar] [CrossRef]
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Aloni-Grinstein, R.; Rotter, V. Mutant p53 oncogenicity: Dominant-negative or gain-of-function. Carcinogenesis 2020. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Milner, J. Evidence for allosteric variants of wild-type p53, a tumour suppressor protein. Br. J. Cancer 1990, 61, 548–552. [Google Scholar] [CrossRef]
- Hainaut, P.; Milner, J. Interaction of heat-shock protein 70 with p53 translated in vitro: Evidence for interaction with dimeric p53 and for a role in the regulation of p53 conformation. EMBO J. 1992, 11, 3513–3520. [Google Scholar] [CrossRef]
- Berg, F.M.V.D.; Tigges, A.J.; Schipper, M.E.I.; Hartog-Jager, F.C.A.D.; Kroes, W.G.M.; Walboomers, J.M.M. Expression of the nuclear oncogene p53 in colon tumours. J. Pathol. 1989, 157, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Iggo, R.; Gannon, J.; Lane, D.P. Genetic and immunochemical analysis of mutant p53 in human breast cancer cell lines. Oncogene 1990, 5, 893–899. [Google Scholar]
- Iggo, R.; Bartek, J.; Lane, D.P.; Gatter, K.; Harris, A.; Harris, A.L. Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet 1990, 335, 675–679. [Google Scholar] [CrossRef]
- Moll, U.M.; Riou, G.; Levine, A.J. Two distinct mechanisms alter p53 in breast cancer: Mutation and nuclear exclusion. Proc. Natl. Acad. Sci. USA 1992, 89, 7262–7266. [Google Scholar] [CrossRef]
- Moll, U.M.; Laquaglia, M.; Benard, J.; Riou, G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc. Natl. Acad. Sci. USA 1995, 92, 4407–4411. [Google Scholar] [CrossRef] [PubMed]
- Porter, P.L.; Gown, A.M.; Kramp, S.G.; Coltrera, M.D. Widespread p53 overexpression in human malignant tumors. An immunohistochemical study using methacarn-fixed, embedded tissue. Am. J. Pathol. 1992, 140, 145–153. [Google Scholar]
- Alsner, J.; Jensen, V.; Kyndi, M.; Offersen, B.V.; Vu, P.; Børresen-Dale, A.-L.; Overgaard, J. A comparison between p53 accumulation determined by immunohistochemistry and TP53 mutations as prognostic variables in tumours from breast cancer patients. Acta Oncol. 2008, 47, 600–607. [Google Scholar] [CrossRef]
- Iggo, R.D.; Rudewicz, J.; Monceau, E.; Sévenet, N.; Bergh, J.; Sjöblom, T.; Bonnefoi, H. Validation of a yeast functional assay for p53 mutations using clonal sequencing. J. Pathol. 2013, 231, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in cancer: Accumulation, gain-of-function, and therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef]
- Bom, A.P.D.A.; Rangel, L.P.; Gallo, C.V.D.M.; Cordeiro, Y.; Silva, J.L.; Costa, D.C.F.; De Oliveira, G.A.P.; Sanches, D.; Braga, C.A.; Gava, L.M.; et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrilsa. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C.G. Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem. Sci. 2004, 29, 542–547. [Google Scholar] [CrossRef]
- Liu, P.; Paulson, J.B.; Forster, C.L.; Shapiro, S.L.; Ashe, K.H.; Zahs, K.R. Characterization of a novel mouse model of alzheimer’s disease—amyloid pathology and unique β-amyloid oligomer profile. PLoS ONE 2015, 10, e0126317. [Google Scholar] [CrossRef]
- Levy, C.B.; Stumbo, A.C.; Bom, A.P.A.; Portari, E.A.; Carneiro, Y.; Silva, J.L.; De Moura-Gallo, C.V. Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 2011, 43, 60–64. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.-C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Clos, A.L.; Castillo-Carranza, D.; Sengupta, U.; Guerrero-Muñoz, M.; Kelly, B.; Wagner, R.; Kayed, R. Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity. Biochem. Biophys. Res. Commun. 2013, 430, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. p53 amyloid formation leading to its loss of function: Implications in cancer pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef]
- Navalkar, A.; Ghosh, S.; Pandey, S.; Paul, A.; Datta, D.; Maji, S.K. Prion-like p53 amyloids in cancer. Biochemistry 2020, 59, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.-I.; Merlini, G.; Saraiva, M.J.M.; Westermark, P. Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification International Society of Amyloidosis 2016 nomenclature guidelines. Amyloid 2016, 23, 209–213. [Google Scholar] [CrossRef]
- Dos Santos, N.M.; De Oliveira, G.A.P.; Rocha, M.R.; Pedrote, M.M.; Ferretti, G.D.D.S.; Rangel, L.P.; Morgado-Diaz, J.A.; Silva, J.L.; Gimba, E.R.P. Loss of the p53 transactivation domain results in high amyloid aggregation of the Δ40p53 isoform in endometrial carcinoma cells. J. Biol. Chem. 2019, 294, 9430–9439. [Google Scholar] [CrossRef]
- Vieler, M.; Sanyal, S. p53 isoforms and their implications in cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef]
- Lei, J.; Qi, R.; Tang, Y.; Wang, W.; Wei, G.; Nussinov, R.; Ma, B. Conformational stability and dynamics of the cancer-associated isoform Δ133p53β are modulated by p53 peptides and p53-specific DNA. FASEB J. 2018, 33, 4225–4235. [Google Scholar] [CrossRef]
- Horwich, A.L. Protein aggregation in disease: A role for folding intermediates forming specific multimeric interactions. J. Clin. Investig. 2002, 110, 1221–1232. [Google Scholar] [CrossRef]
- Chiti, F.; Webster, P.; Taddei, N.; Clark, A.; Stefani, M.; Ramponi, G.; Dobson, C.M. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA 1999, 96, 3590–3594. [Google Scholar] [CrossRef]
- Fändrich, M.; Fletcher, M.A.; Dobson, C.M. Amyloid fibrils from muscle myoglobin. Nat. Cell Biol. 2001, 410, 165–166. [Google Scholar] [CrossRef]
- Krebs, M.R.; Wilkins, D.K.; Chung, E.W.; Pitkeathly, M.C.; Chamberlain, A.K.; Zurdoa, J.; Robinson, C.V.; Dobson, C.M. Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the β-domain. J. Mol. Biol. 2000, 300, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, J.I.; Sunde, M.; Jones, J.A.; Campbell, I.D.; Dobson, C.M. Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. USA 1998, 95, 4224–4228. [Google Scholar] [CrossRef] [PubMed]
- Litvinovich, S.V.; A Brew, S.; Aota, S.; Akiyama, S.K.; Haudenschild, C.; Ingham, K.C. Formation of amyloid-like fibrils by self-association of a partially unfolded fibronectin type III module. J. Mol. Biol. 1998, 280, 245–258. [Google Scholar] [CrossRef]
- Clark, A.; Judge, F.; Richards, J.; Stubbs, J.; Suggett, A. Electron microscopy of network structures in thermally-induced globular protein gels. Int. J. Pept. Protein Res. 2009, 17, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Canet, D.; Sunde, M.; Last, A.M.; Miranker, A.; Spencer, A.; Robinson, C.V.; Dobson, C.M. Mechanistic Studies of the Folding of Human Lysozyme and the Origin of Amyloidogenic Behavior in Its Disease-Related Variants. Biochemistry 1999, 38, 6419–6427. [Google Scholar] [CrossRef]
- DiGiammarino, E.L.; Lee, A.S.; Cadwell, C.; Zhang, W.; Bothner, B.; Ribeiro, R.C.; Zambetti, G.; Kriwacki, R.W. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat. Genet. 2001, 9, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.S.; Loh, S.N. Structure, Function, and Aggregation of the Zinc-Free Form of the p53 DNA Binding Domain&dagger. Biochemistry 2003, 42, 2396–2403. [Google Scholar] [CrossRef]
- Ishimaru, D.; Andrade, L.R.; Foguel, D.; Silva, J.L.; Teixeira, L.S.P.; Quesado, P.A.; Maiolino, L.M.; Lopez, P.M.; Cordeiro, Y.; Costa, L.T.; et al. Fibrillar aggregates of the tumor suppressor p53 core domain&dagger. Biochemistry 2003, 42, 9022–9027. [Google Scholar] [CrossRef]
- Lee, A.S.; Galea, C.A.; DiGiammarino, E.L.; Jun, B.; Murti, G.; Ribeiro, R.C.; Zambetti, G.; Schultz, C.P.; Kriwacki, R.W. Reversible amyloid formation by the p53 tetramerization domain and a cancer-associated mutant. J. Mol. Biol. 2003, 327, 699–709. [Google Scholar] [CrossRef]
- Rigacci, S.; Bucciantini, M.; Relini, A.; Pesce, A.; Gliozzi, A.; Berti, A.; Stefani, M. The (1–63) region of the p53 transactivation domain aggregates in vitro into cytotoxic amyloid assemblies. Biophys. J. 2008, 94, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Vieira, T.C.R.G.; Gomes, M.P.B.; Bom, A.P.A.; Lima, L.M.T.R.; Freitas, M.S.; Ishimaru, D.; Cordeiro, Y.; Foguel, D. Ligand binding and hydration in protein misfolding: Insights from studies of prion and p53 tumor suppressor proteins&dagger. Accounts Chem. Res. 2010, 43, 271–279. [Google Scholar] [CrossRef]
- Kovachev, P.S.; Banerjee, D.; Rangel, L.P.; Eriksson, J.; Pedrote, M.M.; Martins-Dinis, M.M.D.C.; Edwards, K.; Cordeiro, Y.; Silva, J.L.; Sanyal, S. Distinct modulatory role of RNA in the aggregation of the tumor suppressor protein p53 core domain. J. Biol. Chem. 2017, 292, 9345–9357. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fersht, A. Multisite aggregation of p53 and implications for drug rescue. Proc. Natl. Acad. Sci. USA 2017, 114, E2634–E2643. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, G.A.P.; Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Vieira, T.C.R.G.; Cino, E.A.; Silva, J.L. The status of p53 oligomeric and aggregation states in cancer. Biomolecules 2020, 10, 548. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Wang, G.; Fersht, A.R. Mechanism of initiation of aggregation of p53 revealed by Φ-value analysis. Proc. Natl. Acad. Sci. USA 2015, 112, 2437–2442. [Google Scholar] [CrossRef]
- Wang, G.; Fersht, A.R. First-order rate-determining aggregation mechanism of p53 and its implications. Proc. Natl. Acad. Sci. USA 2012, 109, 13590–13595. [Google Scholar] [CrossRef]
- Wang, G.; Fersht, A.R. Propagation of aggregated p53: Cross-reaction and coaggregation vs. seeding. Proc. Natl. Acad. Sci. USA 2015, 112, 2443–2448. [Google Scholar] [CrossRef]
- Wilcken, R.; Wang, G.; Boeckler, F.M.; Fersht, A.R. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl. Acad. Sci. USA 2012, 109, 13584–13589. [Google Scholar] [CrossRef]
- Nicholls, C.D.; McLure, K.G.; Shields, M.A.; Lee, P.W.K. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers implications on the dominant negative effect. J. Biol. Chem. 2002, 277, 12937–12945. [Google Scholar] [CrossRef] [PubMed]
- Stindt, M.H.; Muller, P.A.J.; Ludwig, R.L.; Kehrloesser, S.; Dotsch, V.; Vousden, K.H. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 2014, 34, 4300–4310. [Google Scholar] [CrossRef] [PubMed]
- Wiech, M.; Olszewski, M.B.; Tracz-Gaszewska, Z.; Wawrzynow, B.; Zylicz, M.; Zylicz, A. Molecular Mechanism of Mutant p53 Stabilization: The Role of HSP70 and MDM2. PLoS ONE 2012, 7, e51426. [Google Scholar] [CrossRef] [PubMed]
- Chène, P. In vitro analysis of the dominant negative effect of p53 mutants. J. Mol. Biol. 1998, 281, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef]
- Soto, C.; Estrada, L.; Castilla, J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem. Sci. 2006, 31, 150–155. [Google Scholar] [CrossRef]
- Colby, D.W.; Zhang, Q.; Wang, S.; Groth, D.; Legname, G.; Riesner, D.; Prusiner, S.B. Prion detection by an amyloid seeding assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20914–20919. [Google Scholar] [CrossRef]
- Rochet, J.-C.; Lansbury, P.T. Amyloid fibrillogenesis: Themes and variations. Curr. Opin. Struct. Biol. 2000, 10, 60–68. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nat. Cell Biol. 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Nielsen, L.; Khurana, R.; Coats, A.; Frokjaer, S.; Brange, J.; Vyas, S.; Uversky, V.N.; Fink, A.L. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 2001, 40, 6036–6046. [Google Scholar] [CrossRef]
- Woods, L.A.; Platt, G.W.; Hellewell, A.L.; Hewitt, E.W.; Homans, S.W.; Ashcroft, A.E.; Radford, S.E. Ligand binding to distinct states diverts aggregation of an amyloid-forming protein. Nat. Chem. Biol. 2011, 7, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Forget, K.J.; Tremblay, G.; Roucou, X. p53 aggregates penetrate cells and induce the co-aggregation of intracellular p53. PLoS ONE 2013, 8, e69242. [Google Scholar] [CrossRef] [PubMed]
- Vilotte, J.-L.; Soulier, S.; Essalmani, R.; Stinnakre, M.-G.; Vaiman, D.; Lepourry, L.; Da Silva, J.C.; Besnard, N.; Dawson, M.; Buschmann, A.; et al. Markedly Increased Susceptibility to Natural Sheep Scrapie of Transgenic Mice Expressing Ovine PrP. J. Virol. 2001, 75, 5977–5984. [Google Scholar] [CrossRef]
- Glatzel, M.; Aguzzi, A. PrPC expression in the peripheral nervous system is a determinant of prion neuroinvasion. J. Gen. Virol. 2000, 81, 2813–2821. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Dong, Z.; Nakamura, K.; Colburn, N.H. Dosage-dependent dominance over wild-type p53 of a mutant p53 isolated from nasopharyngeal carcinoma 1. FASEB J. 1993, 7, 944–950. [Google Scholar] [CrossRef]
- Billant, O.; Blondel, M.; Voisset, C. p53, p63 and p73 in the wonderland of S. cerevisiae. Oncotarget 2017, 8, 57855–57869. [Google Scholar] [CrossRef] [PubMed]
- Finlay, C.A.; Levine, J. The P53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 11. [Google Scholar] [CrossRef]
- Isaacs, W.B.; Carter, B.S.; Ewing, C.M. Wild-type p53 suppresses growth of human prostate cancer cells containing mutant p53 alleles. Cancer Res. 1991, 51, 6. [Google Scholar]
- Bach, S.; Tribouillard, D.; Talarek, N.; Desban, N.; Gug, F.; Galons, H.; Blondel, M. A yeast-based assay to isolate drugs active against mammalian prions. Methods 2006, 39, 72–77. [Google Scholar] [CrossRef]
- Voisset, C.; Saupe, S.J.; Galons, H.; Blondel, M. Procedure for identification and characterization of drugs efficient against mammalian prion: From a yeast-based antiprion drug screening assay to in vivo mouse models. Infect. Disord. Drug Targets 2009, 9, 31–39. [Google Scholar] [CrossRef]
- Park, S.-K.; Park, S.; Pentek, C.; Liebman, S.W. Tumor suppressor protein p53 expressed in yeast can remain diffuse, form a prion or form unstable liquid-like droplets. Science 2020. [Google Scholar] [CrossRef]
- Devaraja, K. Current prospects of molecular therapeutics in head and neck squamous cell carcinoma. Pharm. Med. 2019, 33, 269–289. [Google Scholar] [CrossRef] [PubMed]
- Wold, W.S.M.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Zhang, W.-W.; Li, L.; Hu, A.; Xu, W.; Lam, D.M.-K.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; et al. The first approved gene therapy product for cancer ad-p53(gendicine): 12 years in the clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Pellegrini, M.; Memarzadeh, S.; Eisenberg, D.; Johnson, L.M.; Lindgren, A.G.; Nguyen, A.T.-Q.; Tiourin, E.; Soriaga, A.B.; et al. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef]
- Loh, S.N. Follow the mutations: Toward class-specific, small-molecule reactivation of P53. Biomolecules 2020, 10, 303. [Google Scholar] [CrossRef]
- Soussi, T. The history of p53. EMBO Rep. 2010, 11, 822–826. [Google Scholar] [CrossRef]
- Magzoub, M.; Miranker, A.D. Protein aggregation: P53 succumbs to peer pressure. Nat. Chem. Biol. 2011, 7, 248–249. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Wilcken, R.; Andreeva, A. Tracing the evolution of the p53 tetramerization domain. Structure 2014, 22, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef]
- Li, Y.; Prives, C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene 2007, 26, 2220–2225. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Di Agostino, S.; Coppari, E.; Bizzarri, A.R.; Blandino, G.; Cannistraro, S. Interaction of mutant p53 with p73: A surface plasmon resonance and atomic force spectroscopy study. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 1958–1964. [Google Scholar] [CrossRef]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of li-fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; El-Naggar, A.K.; Lozano, G.; Suh, Y.-A.; Liu, G.; Rao, V.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; et al. Gain of function of a p53 hot spot mutation in a mouse model of li-fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; A Mirza, S.; Xu, S.; Schulz-Heddergott, R.; Marchenko, N.D.; Moll, U.M. p53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis. 2017, 8, e2661. [Google Scholar] [CrossRef]
- Wells, C.; Brennan, S.E.; Keon, M.; Saksena, N.K. Prionoid proteins in the pathogenesis of neurodegenerative diseases. Front. Mol. Neurosci. 2019, 12, 271. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).