
Precision Medicine for Colorectal Cancer with Liquid Biopsy and Immunotherapy




Abstract
:Simple Summary
Abstract
1. Introduction
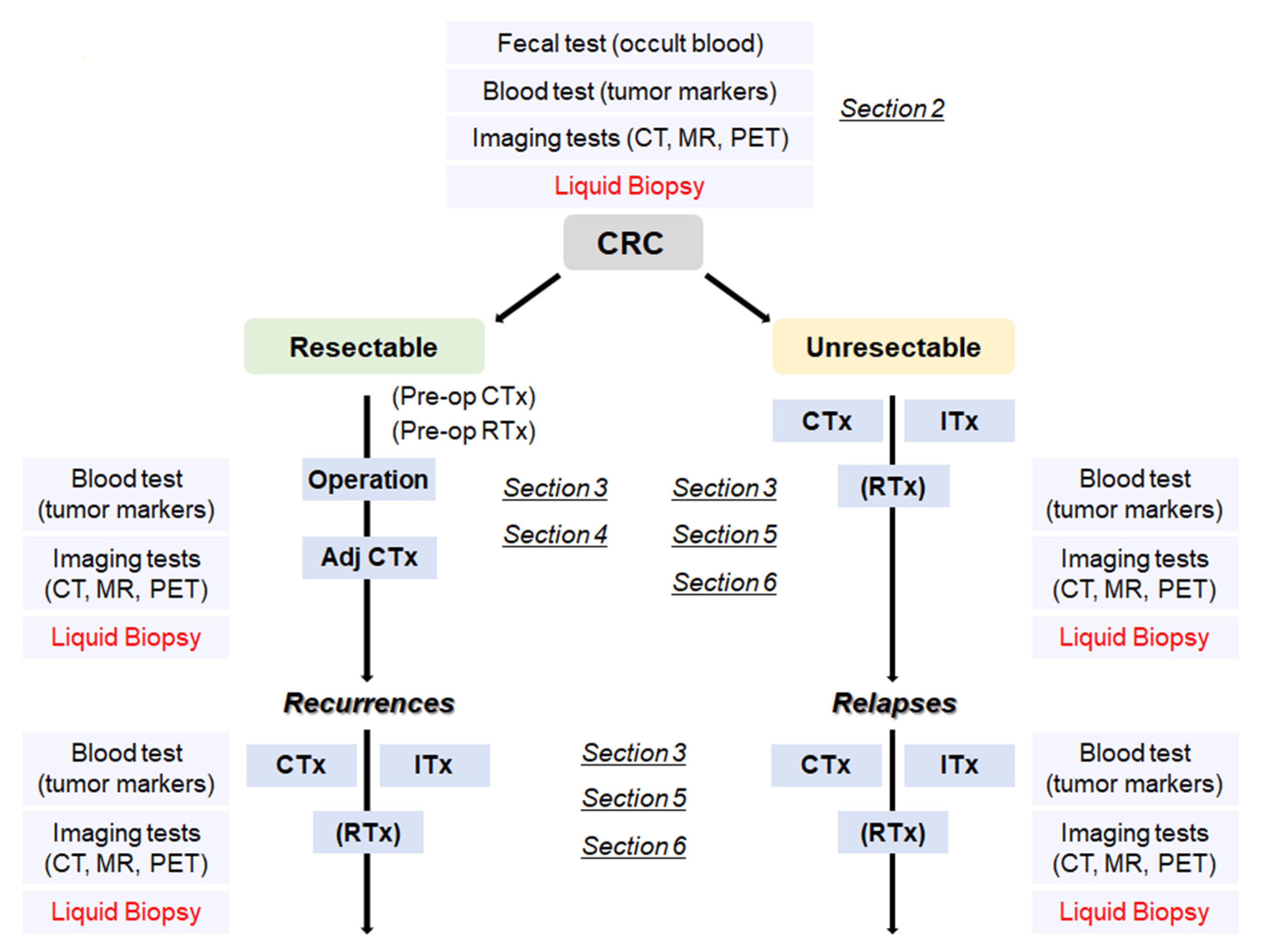
2. Cancer Screening Using Liquid Biopsy
3. Genomic Analysis for Selection of Molecular-Targeted Drugs
4. Detection of Minimal Residual Disease (MRD) by Liquid Biopsy after the Curative Resection
5. Estimation of Treatment Responsiveness, Detection of Acquired Mutations, and Early Detection of Disease Progression with Liquid Biopsy
6. Personalized Immunotherapy Based on Genome Information of Tumors
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vega, P.; Valentin, F.; Cubiella, J. Colorectal cancer diagnosis: Pitfalls and opportunities. World J. Gastrointest. Oncol. 2015, 7, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Brenner, H.; Stock, C.; Hoffmeister, M. Effect of screening sigmoidoscopy and screening colonoscopy on colorectal cancer incidence and mortality: Systematic review and meta-analysis of randomised controlled trials and observational studies. BMJ 2014, 348, g2467. [Google Scholar] [CrossRef] [Green Version]
- Elmunzer, B.J.; Singal, A.G.; Sussman, J.B.; Deshpande, A.R.; Sussman, D.A.; Conte, M.L.; Dwamena, B.A.; Rogers, M.A.; Schoenfeld, P.S.; Inadomi, J.M.; et al. Comparing the effectiveness of competing tests for reducing colorectal cancer mortality: A network meta-analysis. Gastrointest. Endosc. 2015, 81, 700–709.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandel, J.S.; Bond, J.H.; Church, T.R.; Snover, D.C.; Bradley, G.M.; Schuman, L.M.; Ederer, F. Reducing mortality from colorectal cancer by screening for fecal occult blood. Minnesota Colon Cancer Control Study. N. Engl. J. Med. 1993, 328, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardcastle, J.D.; Chamberlain, J.O.; Robinson, M.H.; Moss, S.M.; Amar, S.S.; Balfour, T.W.; James, P.D.; Mangham, C.M. Randomised controlled trial of faecal-occult-blood screening for colorectal cancer. Lancet 1996, 348, 1472–1477. [Google Scholar] [CrossRef]
- Kronborg, O.; Fenger, C.; Olsen, J.; Jorgensen, O.D.; Sondergaard, O. Randomised study of screening for colorectal cancer with faecal-occult-blood test. Lancet 1996, 348, 1467–1471. [Google Scholar] [CrossRef]
- Allison, J.E.; Tekawa, I.S.; Ransom, L.J.; Adrain, A.L. A comparison of fecal occult-blood tests for colorectal-cancer screening. N. Engl. J. Med. 1996, 334, 155–159. [Google Scholar] [CrossRef]
- Brenner, H.; Tao, S. Superior diagnostic performance of faecal immunochemical tests for haemoglobin in a head-to-head comparison with guaiac based faecal occult blood test among 2235 participants of screening colonoscopy. Eur. J. Cancer 2013, 49, 3049–3054. [Google Scholar] [CrossRef]
- Imperiale, T.F.; Gruber, R.N.; Stump, T.E.; Emmett, T.W.; Monahan, P.O. Performance Characteristics of Fecal Immunochemical Tests for Colorectal Cancer and Advanced Adenomatous Polyps: A Systematic Review and Meta-analysis. Ann. Intern. Med. 2019, 170, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Niedermaier, T.; Balavarca, Y.; Brenner, H. Stage-Specific Sensitivity of Fecal Immunochemical Tests for Detecting Colorectal Cancer: Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2020, 115, 56–69. [Google Scholar] [CrossRef] [Green Version]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef] [Green Version]
- Liles, E.G.; Coronado, G.D.; Perrin, N.; Harte, A.H.; Nungesser, R.; Quigley, N.; Potter, N.T.; Weiss, G.; Koenig, T.; deVos, T. Uptake of a colorectal cancer screening blood test is higher than of a fecal test offered in clinic: A randomized trial. Cancer Treat. Res. Commun. 2017, 10, 27–31. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Consortium, C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.O.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.; Geiger, S.; Keil, A.; Bias, H.; Schatz, P.; deVos, T.; Dhein, J.; Zimmermann, M.; Tauber, R.; Wiedenmann, B. Improving compliance to colorectal cancer screening using blood and stool based tests in patients refusing screening colonoscopy in Germany. BMC Gastroenterol. 2014, 14, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, N.T.; Hurban, P.; White, M.N.; Whitlock, K.D.; Lofton-Day, C.E.; Tetzner, R.; Koenig, T.; Quigley, N.B.; Weiss, G. Validation of a real-time PCR-based qualitative assay for the detection of methylated SEPT9 DNA in human plasma. Clin. Chem. 2014, 60, 1183–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.A.; Barclay, R.L.; Mergener, K.; Weiss, G.; Konig, T.; Beck, J.; Potter, N.T. Plasma Septin9 versus fecal immunochemical testing for colorectal cancer screening: A prospective multicenter study. PLoS ONE 2014, 9, e98238. [Google Scholar] [CrossRef]
- Lennon, A.M.; Buchanan, A.H.; Kinde, I.; Warren, A.; Honushefsky, A.; Cohain, A.T.; Ledbetter, D.H.; Sanfilippo, F.; Sheridan, K.; Rosica, D.; et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020, 369, 6499. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Klein, E.A.; Seiden, M.; Hubbell, E.; Venn, O.; Jamshidi, A.; Zhang, N.; Beausang, J.F.; Gross, S.; Kurtzman, K.N.; et al. Simultaneous multi-cancer detection and tissue of origin (TOO) localization using targeted bisulfite sequencing of plasma cell-free DNA (cfDNA). J. Glob. Oncol. 2019, 5, 44. [Google Scholar] [CrossRef]
- Liu, M.C.; Klein, E.; Hubbell, E.; Maddala, T.; Aravanis, A.M.; Beausang, J.F.; Filippova, D.; Gross, S.; Jamshidi, A.; Kurtzman, K.; et al. Plasma cell-free DNA (cfDNA) assays for early multi-cancer detection: The circulating cell-free genome atlas (CCGA) study. Ann. Oncol. 2018, 29, viii14. [Google Scholar] [CrossRef]
- Lieu, C.H.; Corcoran, R.B.; Overman, M.J. Integrating Biomarkers and Targeted Therapy Into Colorectal Cancer Management. Am. Soc. Clin. Oncol. Educ. Book. 2019, 39, 207–215. [Google Scholar] [CrossRef]
- Neumann, J.; Zeindl-Eberhart, E.; Kirchner, T.; Jung, A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol. Res. Pract. 2009, 205, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, E.; Martino, C.; Sartore-Bianchi, A.; Penaullt-Llorca, F.; Viale, G.; Risio, M.; Rugge, M.; Grigioni, W.; Bencardino, K.; Lonardi, S.; et al. Assessment of a HER2 scoring system for colorectal cancer: Results from a validation study. Mod. Pathol. 2015, 28, 1481–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siena, S.; Sartore-Bianchi, A.; Marsoni, S.; Hurwitz, H.I.; McCall, S.J.; Penault-Llorca, F.; Srock, S.; Bardelli, A.; Trusolino, L. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann. Oncol. 2018, 29, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.; Tran, B.; Ensor, J.; Gibbs, P.; Wong, H.L.; Wong, S.F.; Vilar, E.; Tie, J.; Broaddus, R.; Kopetz, S.; et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann. Oncol. 2014, 25, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Koopman, M.; Kortman, G.A.; Mekenkamp, L.; Ligtenberg, M.J.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.; van Krieken, J.H. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Bokemeyer, C.; Bondarenko, I.; Makhson, A.; Hartmann, J.T.; Aparicio, J.; de Braud, F.; Donea, S.; Ludwig, H.; Schuch, G.; Stroh, C.; et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmuller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Kohne, C.H.; Lang, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. 2011, 29, 2011–2019. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Lenz, H.J.; Kohne, C.H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Bokemeyer, C.; Kohne, C.H.; Ciardiello, F.; Lenz, H.J.; Heinemann, V.; Klinkhardt, U.; Beier, F.; Duecker, K.; van Krieken, J.H.; Tejpar, S. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 2015, 51, 1243–1252. [Google Scholar] [CrossRef]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Ballard, J.; Blake, J.F.; Bouhana, K.; Brandhuber, B.J.; Briere, D.M.; Burgess, L.E.; Burkard, M.R.; et al. Discovery of Tetrahydropyridopyrimidines as Irreversible Covalent Inhibitors of KRAS-G12C with In Vivo Activity. ACS Med. Chem. Lett. 2018, 9, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Solomon, M.; Li, L.S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Zuberi, M.; Khan, I.; O’Bryan, J.P. Inhibition of RAS: Proven and potential vulnerabilities. Biochem. Soc. Trans. 2020, 48, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRASG12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR Blockade Reverts Resistance to KRAS(G12C) Inhibition in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef]
- Paraiso, K.H.; Fedorenko, I.V.; Cantini, L.P.; Munko, A.C.; Hall, M.; Sondak, V.K.; Messina, J.L.; Flaherty, K.T.; Smalley, K.S. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 2010, 102, 1724–1730. [Google Scholar] [CrossRef]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Ahronian, L.G.; Sennott, E.M.; Van Allen, E.M.; Wagle, N.; Kwak, E.L.; Faris, J.E.; Godfrey, J.T.; Nishimura, K.; Lynch, K.D.; Mermel, C.H.; et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov. 2015, 5, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B.; Andre, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahuja, K.B.; Nguyen, T.T.; Jaiswal, B.S.; Prabhash, K.; Thaker, T.M.; Senger, K.; Chaudhuri, S.; Kljavin, N.M.; Antony, A.; Phalke, S.; et al. Actionable Activating Oncogenic ERBB2/HER2 Transmembrane and Juxtamembrane Domain Mutations. Cancer Cell 2018, 34, 792–806.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Lonardi, S.; Martino, C.; Fenocchio, E.; Tosi, F.; Ghezzi, S.; Leone, F.; Bergamo, F.; Zagonel, V.; Ciardiello, F.; et al. Pertuzumab and trastuzumab emtansine in patients with HER2-amplified metastatic colorectal cancer: The phase II HERACLES-B trial. ESMO Open 2020, 5, e000911. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- Siena, S.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Fakih, M.; Elez, E.; et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2021, 22, 779–789. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Dudley, J.C.; Lin, M.T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jager, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Andre, T.; Amonkar, M.; Norquist, J.M.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.; Garcia-Carbonero, R.; et al. Health-related quality of life in patients with microsatellite instability-high or mismatch repair deficient metastatic colorectal cancer treated with first-line pembrolizumab versus chemotherapy (KEYNOTE-177): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 665–677. [Google Scholar] [CrossRef]
- Trullas, A.; Delgado, J.; Genazzani, A.; Mueller-Berghaus, J.; Migali, C.; Muller-Egert, S.; Zander, H.; Enzmann, H.; Pignatti, F. The EMA assessment of pembrolizumab as monotherapy for the first-line treatment of adult patients with metastatic microsatellite instability-high or mismatch repair deficient colorectal cancer. ESMO Open 2021, 6, 100145. [Google Scholar] [CrossRef]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.S.; Zhang, W.; Wang, X.; Jiang, P.; Traugh, N.; Li, Z.; Meyer, C.; Stewig, B.; Xie, Y.; Bu, X.; et al. Therapeutically Increasing MHC-I Expression Potentiates Immune Checkpoint Blockade. Cancer Discov. 2021, 11, 1524–1541. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Dahlman, K.H.; Knol, J.; Gilbert, J.; Puzanov, I.; Means-Powell, J.; Balko, J.M.; Lovly, C.M.; Murphy, B.A.; Goff, L.W.; et al. Enabling a genetically informed approach to cancer medicine: A retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next-generation sequencing panel. Oncologist 2014, 19, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimberidou, A.M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Luthra, R.; et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockley, T.L.; Oza, A.M.; Berman, H.K.; Leighl, N.B.; Knox, J.J.; Shepherd, F.A.; Chen, E.X.; Krzyzanowska, M.K.; Dhani, N.; Joshua, A.M.; et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: The Princess Margaret IMPACT/COMPACT trial. Genome Med. 2016, 8, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meric-Bernstam, F.; Brusco, L.; Shaw, K.; Horombe, C.; Kopetz, S.; Davies, M.A.; Routbort, M.; Piha-Paul, S.A.; Janku, F.; Ueno, N.; et al. Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. J. Clin. Oncol. 2015, 33, 2753–2762. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Stephenson, J.J., Jr.; Rosen, P.; Loesch, D.M.; Borad, M.J.; Anthony, S.; Jameson, G.; Brown, S.; Cantafio, N.; Richards, D.A.; et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef] [PubMed]
- Laskin, J.; Jones, S.; Aparicio, S.; Chia, S.; Ch’ng, C.; Deyell, R.; Eirew, P.; Fok, A.; Gelmon, K.; Ho, C.; et al. Lessons learned from the application of whole-genome analysis to the treatment of patients with advanced cancers. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000570. [Google Scholar] [CrossRef] [Green Version]
- Sholl, L.M.; Do, K.; Shivdasani, P.; Cerami, E.; Dubuc, A.M.; Kuo, F.C.; Garcia, E.P.; Jia, Y.; Davineni, P.; Abo, R.P.; et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 2016, 1, e87062. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Ohhara, Y.; Takada, K.; Tanabe, H.; Hatanaka, K.; Amano, T.; Hatanaka, K.C.; Hatanaka, Y.; Mitamura, T.; Kato, M.; et al. Clinical significance of comprehensive genomic profiling tests covered by public insurance in patients with advanced solid cancers in Hokkaido, Japan. Jpn. J. Clin. Oncol. 2021, 51, 753–761. [Google Scholar] [CrossRef]
- Takeda, M.; Takahama, T.; Sakai, K.; Shimizu, S.; Watanabe, S.; Kawakami, H.; Tanaka, K.; Sato, C.; Hayashi, H.; Nonagase, Y.; et al. Clinical Application of the FoundationOne CDx Assay to Therapeutic Decision-Making for Patients with Advanced Solid Tumors. Oncologist 2021, 26, e588–e596. [Google Scholar] [CrossRef]
- Kondo, T.; Matsubara, J.; Quy, P.N.; Fukuyama, K.; Nomura, M.; Funakoshi, T.; Doi, K.; Sakamori, Y.; Yoshioka, M.; Yokoyama, A.; et al. Comprehensive genomic profiling for patients with chemotherapy-naive advanced cancer. Cancer Sci. 2021, 112, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Daniels, G.A.; Piccioni, D.E.; Fanta, P.T.; Schwab, R.B.; Shimabukuro, K.A.; Parker, B.A.; Kurzrock, R. On the Road to Precision Cancer Medicine: Analysis of Genomic Biomarker Actionability in 439 Patients. Mol. Cancer Ther. 2015, 14, 1488–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirshfield, K.M.; Tolkunov, D.; Zhong, H.; Ali, S.M.; Stein, M.N.; Murphy, S.; Vig, H.; Vazquez, A.; Glod, J.; Moss, R.A.; et al. Clinical Actionability of Comprehensive Genomic Profiling for Management of Rare or Refractory Cancers. Oncologist 2016, 21, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Daniels, G.A.; Piccioni, D.E.; Kesari, S.; Helsten, T.L.; Bazhenova, L.A.; Romero, J.; Fanta, P.T.; et al. Precision Oncology: The UC San Diego Moores Cancer Center PREDICT Experience. Mol. Cancer Ther. 2016, 15, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Lazar, V.; Leyland-Jones, B.; Schilsky, R.L.; Mendelsohn, J.; Kurzrock, R. Association of Biomarker-Based Treatment Strategies With Response Rates and Progression-Free Survival in Refractory Malignant Neoplasms: A Meta-analysis. JAMA Oncol. 2016, 2, 1452–1459. [Google Scholar] [CrossRef] [Green Version]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Vacante, M.; Ciuni, R.; Basile, F.; Biondi, A. The Liquid Biopsy in the Management of Colorectal Cancer: An Overview. Biomedicines 2020, 8, 308. [Google Scholar] [CrossRef]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [Green Version]
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.B.; Siu, L.L.; et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 2019, 30, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra392. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.M.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating Tumor DNA Measurements As Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N.; et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med. 2020, 26, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): A multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef]
- Vidula, N.; Niemierko, A.; Malvarosa, G.; Yuen, M.; Lennerz, J.; Iafrate, A.J.; Wander, S.A.; Spring, L.; Juric, D.; Isakoff, S.; et al. Tumor Tissue- versus Plasma-based Genotyping for Selection of Matched Therapy and Impact on Clinical Outcomes in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2021, 27, 3404–3413. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantel, K.; Alix-Panabieres, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.R.; Mojtahed, A.; Schneider, J.L.; Kanter, K.; Van Seventer, E.E.; Fetter, I.J.; Thabet, A.; Fish, M.G.; Teshome, B.; Fosbenner, K.; et al. Serial ctDNA Monitoring to Predict Response to Systemic Therapy in Metastatic Gastrointestinal Cancers. Clin. Cancer Res. 2020, 26, 1877–1885. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, N.; Gimeno-Valiente, F.; Gambardella, V.; Zuniga, S.; Rentero-Garrido, P.; Huerta, M.; Rosello, S.; Martinez-Ciarpaglini, C.; Carbonell-Asins, J.A.; Carrasco, F.; et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann. Oncol. 2019, 30, 1804–1812. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Suzuki, T.; Yoshimura, Y.; Yahata, M.; Yew, P.Y.; Nakamura, T.; Nakamura, Y.; Park, J.H.; Matsuo, R. Detection of circulating tumor DNA in patients of operative colorectal and gastric cancers. Oncotarget 2020, 11, 3198–3207. [Google Scholar] [CrossRef]
- Couraud, S.; Vaca-Paniagua, F.; Villar, S.; Oliver, J.; Schuster, T.; Blanche, H.; Girard, N.; Tredaniel, J.; Guilleminault, L.; Gervais, R.; et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: A proof-of-concept study from BioCAST/IFCT-1002. Clin. Cancer Res. 2014, 20, 4613–4624. [Google Scholar] [CrossRef] [Green Version]
- Fontanges, Q.; De Mendonca, R.; Salmon, I.; Le Mercier, M.; D’Haene, N. Clinical Application of Targeted Next Generation Sequencing for Colorectal Cancers. Int. J. Mol. Sci. 2016, 17, 2117. [Google Scholar] [CrossRef]
- Zviran, A.; Schulman, R.C.; Shah, M.; Hill, S.T.K.; Deochand, S.; Khamnei, C.C.; Maloney, D.; Patel, K.; Liao, W.; Widman, A.J.; et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med. 2020, 26, 1114–1124. [Google Scholar] [CrossRef]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, P.V.; Berchuck, J.E.; Korthauer, K.; Spisak, S.; Nassar, A.H.; Abou Alaiwi, S.; Chakravarthy, A.; Shen, S.Y.; Bakouny, Z.; Boccardo, F.; et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 2020, 26, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, F.; Chakravarthy, A.; Feng, S.; Shen, S.Y.; Nejad, R.; Zuccato, J.A.; Voisin, M.R.; Patil, V.; Horbinski, C.; Aldape, K.; et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat. Med. 2020, 26, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhu, D.; Shao, F.; Chen, S.; Guo, Y.; Li, K.; Wang, Y.; Ding, R.; Gao, L.; Ma, W.; et al. Efficient detection and post-surgical monitoring of colon cancer with a multi-marker DNA methylation liquid biopsy. Proc. Natl. Acad. Sci. USA 2021, 118, e2017421118. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Mahoney, D.W.; Foote, P.H.; Burger, K.N.; Doering, K.A.; Taylor, W.R.; Then, S.S.; Cao, X.; McGlinch, M.; Berger, C.K.; et al. Novel Methylated DNA Markers in the Surveillance of Colorectal Cancer Recurrence. Clin. Cancer Res. 2021, 27, 141–149. [Google Scholar] [CrossRef]
- Pantel, K.; Hayes, D.F. Disseminated breast tumour cells: Biological and clinical meaning. Nat. Rev. Clin. Oncol. 2018, 15, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Ulz, P.; Thallinger, G.G.; Auer, M.; Graf, R.; Kashofer, K.; Jahn, S.W.; Abete, L.; Pristauz, G.; Petru, E.; Geigl, J.B.; et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 2016, 48, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Krimmel, J.D.; Schmitt, M.W.; Harrell, M.I.; Agnew, K.J.; Kennedy, S.R.; Emond, M.J.; Loeb, L.A.; Swisher, E.M.; Risques, R.A. Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proc. Natl. Acad. Sci. USA 2016, 113, 6005–6010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Lippman, S.M.; Flaherty, K.T.; Kurzrock, R. The Conundrum of Genetic “Drivers” in Benign Conditions. J. Natl. Cancer Inst. 2016, 108, djw036. [Google Scholar] [CrossRef]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef]
- Chan, H.T.; Nagayama, S.; Chin, Y.M.; Otaki, M.; Hayashi, R.; Kiyotani, K.; Fukunaga, Y.; Ueno, M.; Nakamura, Y.; Low, S.K. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol. Oncol. 2020, 14, 1719–1730. [Google Scholar] [CrossRef]
- Bardelli, A.; Pantel, K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.Y.; Lin, M.; Zhang, H.B. Diagnostic value of carcinoembryonic antigen and carcinoma antigen 19-9 for colorectal carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 9404–9409. [Google Scholar]
- Bagaria, B.; Sood, S.; Sharma, R.; Lalwani, S. Comparative study of CEA and CA19-9 in esophageal, gastric and colon cancers individually and in combination (ROC curve analysis). Cancer Biol. Med. 2013, 10, 148–157. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Li, L.; Christie, M.; Simons, K.; Elsaleh, H.; Kosmider, S.; Wong, R.; Yip, D.; et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: A prospective biomarker study. Gut 2019, 68, 663–671. [Google Scholar] [CrossRef]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Grasselli, J.; Elez, E.; Caratu, G.; Matito, J.; Santos, C.; Macarulla, T.; Vidal, J.; Garcia, M.; Vieitez, J.M.; Paez, D.; et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2017, 28, 1294–1301. [Google Scholar] [CrossRef]
- Schmiegel, W.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normanno, N.; Tejpar, S.; Morgillo, F.; De Luca, A.; Van Cutsem, E.; Ciardiello, F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [Google Scholar] [CrossRef]
- Sorich, M.J.; Wiese, M.D.; Rowland, A.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann. Oncol. 2015, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.H.; Cunningham, D.; Werner, B.; Vlachogiannis, G.; Spiteri, I.; Heide, T.; Mateos, J.F.; Vatsiou, A.; Lampis, A.; Damavandi, M.D.; et al. Longitudinal Liquid Biopsy and Mathematical Modeling of Clonal Evolution Forecast Time to Treatment Failure in the PROSPECT-C Phase II Colorectal Cancer Clinical Trial. Cancer Discov. 2018, 8, 1270–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Siravegna, G.; Lazzari, L.; Crisafulli, G.; Sartore-Bianchi, A.; Mussolin, B.; Cassingena, A.; Martino, C.; Lanman, R.B.; Nagy, R.J.; Fairclough, S.; et al. Radiologic and Genomic Evolution of Individual Metastases during HER2 Blockade in Colorectal Cancer. Cancer Cell 2018, 34, 148–162.e7. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, H.; Kuziora, M.; Quinn, K.J.; Helman, E.; Ye, J.; Liu, F.; Scheuring, U.; Peters, S.; Rizvi, N.A.; Brohawn, P.Z.; et al. A Blood-based Assay for Assessment of Tumor Mutational Burden in First-line Metastatic NSCLC Treatment: Results from the MYSTIC Study. Clin. Cancer Res. 2021, 27, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Bando, H.; Tsukada, Y.; Inamori, K.; Yuki, S.; Komatsu, Y.; Homma, S.; Uemura, M.; Kato, T.; Kotani, D.; et al. Voltage: Investigator-initiated clinical trial of nivolumab monotherapy and subsequent radical surgery following preoperative chemoradiotherapy in patients with microsatellite stable locally advanced rectal cancer. J. Clin. Oncol. 2019, 37, 3606. [Google Scholar] [CrossRef]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Forget, M.A.; Haymaker, C.; Hess, K.R.; Meng, Y.J.; Creasy, C.; Karpinets, T.; Fulbright, O.J.; Roszik, J.; Woodman, S.E.; Kim, Y.U.; et al. Prospective Analysis of Adoptive TIL Therapy in Patients with Metastatic Melanoma: Response, Impact of Anti-CTLA4, and Biomarkers to Predict Clinical Outcome. Clin. Cancer Res. 2018, 24, 4416–4428. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcotte, S.; Gros, A.; Hogan, K.; Tran, E.; Hinrichs, C.S.; Wunderlich, J.R.; Dudley, M.E.; Rosenberg, S.A. Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: Implications for adoptive cell transfer therapy. J. Immunol. 2013, 191, 2217–2225. [Google Scholar] [CrossRef] [Green Version]
- Marits, P.; Karlsson, M.; Dahl, K.; Larsson, P.; Wanders, A.; Thorn, M.; Winqvist, O. Sentinel node lymphocytes: Tumour reactive lymphocytes identified intraoperatively for the use in immunotherapy of colon cancer. Br. J. Cancer 2006, 94, 1478–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, M.; Marits, P.; Dahl, K.; Dagoo, T.; Enerback, S.; Thorn, M.; Winqvist, O. Pilot study of sentinel-node-based adoptive immunotherapy in advanced colorectal cancer. Ann. Surg. Oncol. 2010, 17, 1747–1757. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Y.H.; Liu, X.H.; Yang, Y.; Li, B.; Tang, J.L.; Zeng, Q.X.; Hu, J.; Zeng, X.N.; Zhang, L.; Wang, Z.J.; et al. Phase I/II study of adjuvant immunotherapy with sentinel lymph node T lymphocytes in patients with colorectal cancer. Cancer Immunol. Immunother. 2015, 64, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, T.; Miyauchi, E.; Hsu, Y.W.; Nagayama, S.; Kiyotani, K.; Zewde, M.; Park, J.H.; Kato, T.; Harada, M.; Matsui, S.; et al. TCR sequencing analysis of cancer tissues and tumor draining lymph nodes in colorectal cancer patients. Oncoimmunology 2019, 8, e1588085. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.; Malekzadeh, P.; et al. Immunologic Recognition of a Shared p53 Mutated Neoantigen in a Patient with Metastatic Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef]
- Sim, M.J.W.; Lu, J.; Spencer, M.; Hopkins, F.; Tran, E.; Rosenberg, S.A.; Long, E.O.; Sun, P.D. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl. Acad. Sci. USA 2020, 117, 12826–12835. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Screening Method | Type of Sample | Description | Overall Performance |
|---|---|---|---|
| High sensitivity Guaiac-based fecal occult blood test (gFOBT) -FDA approved | Stool |
| In randomized controlled trials, screening with FOBT reduced CRC mortality rates by 15% to 33% [4,5,6] Specificity: 86.7% to 97.7%; Sensitivity: 33.3% to 79.4% [7,8] |
| Fecal Immunochemical test (FIT) -FDA approved | Stool |
| Randomized control: On-going Specificity: 81% to 96%; Sensitivity: 65% to 95% [9,10] |
| Multitarget stool DNA test (Cologuard) -FDA approved | Stool |
| Specificity: 86.6%; Sensitivity: 92.3% [11] |
| Epi proColon -FDA approved | Blood |
| Specificity: 80 to 82%; Sensitivity: 68 to 72% [12] |
| CancerSEEK | Blood |
| All 8 cancer types including CRC Specificity > 99% Sensitivity: 33% to 70% [13] |
| GRAIL (CCGA and STRIVE) | Blood |
| All 12 cancer types including CRC Specificity: 98.3% to 99.8% Stage I–III sensitivity: 60.7% to 73.3% [14] |
| DELFI | Blood |
| Specificity: 95%; Sensitivity: 81% [15] |
| Gene | Biomarkers | Frequencies (%) | Anticancer Agents |
|---|---|---|---|
| KRAS | Wild type | 60 [23] | Cetuximab Panitumumab |
| KRAS | G12C | 8 [23] | Sotorasib (AMG510) Adagrasib (MRTX849) |
| BRAF | V600E | 10 [24] | Vemurafenib Dabrafenib Ecorafenib |
| HER2 | Amplification | 2–3 [25,26] | Pertuzumab Trastuzumab Lapatinib |
| MLH1 MSH2 MSH6 PMS2 | MSI-H | 10–15 [27,28] | Nivolumab Pembrolizumab Ipilimumab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagayama, S.; Low, S.-K.; Kiyotani, K.; Nakamura, Y. Precision Medicine for Colorectal Cancer with Liquid Biopsy and Immunotherapy. Cancers 2021, 13, 4803. https://doi.org/10.3390/cancers13194803
Nagayama S, Low S-K, Kiyotani K, Nakamura Y. Precision Medicine for Colorectal Cancer with Liquid Biopsy and Immunotherapy. Cancers. 2021; 13(19):4803. https://doi.org/10.3390/cancers13194803
Chicago/Turabian StyleNagayama, Satoshi, Siew-Kee Low, Kazuma Kiyotani, and Yusuke Nakamura. 2021. "Precision Medicine for Colorectal Cancer with Liquid Biopsy and Immunotherapy" Cancers 13, no. 19: 4803. https://doi.org/10.3390/cancers13194803
APA StyleNagayama, S., Low, S.-K., Kiyotani, K., & Nakamura, Y. (2021). Precision Medicine for Colorectal Cancer with Liquid Biopsy and Immunotherapy. Cancers, 13(19), 4803. https://doi.org/10.3390/cancers13194803