Simple Summary
Epstein–Barr Virus (EBV) is a common virus that is readily controlled by a healthy immune system and rarely causes serious problems in infected people. However, patients with certain genetic defects of their immune system might have difficulties controlling EBV and often develop severe and life-threatening conditions, such as severe inflammation and malignancies. In this review, we provide a summary of inherited immune diseases that lead to a high susceptibility to EBV infection and discuss how this infection is associated with cancer development.
Abstract
Epstein–Barr Virus (EBV) is a ubiquitous virus affecting more than 90% of the world’s population. Upon infection, it establishes latency in B cells. It is a rather benign virus for immune-competent individuals, in whom infections usually go unnoticed. Nevertheless, EBV has been extensively associated with tumorigenesis. Patients suffering from certain inborn errors of immunity are at high risk of developing malignancies, while infection in the majority of immune-competent individuals does not seem to lead to immune dysregulation. Herein, we discuss how inborn mutations in TNFRSF9, CD27, CD70, CORO1A, CTPS1, ITK, MAGT1, RASGRP1, STK4, CARMIL2, SH2D1A, and XIAP affect the development, differentiation, and function of key factors involved in the immunity against EBV, leading to increased susceptibility to lymphoproliferative disease and lymphoma.
1. Introduction
Epstein–Barr Virus (EBV) is a gammaherpesvirus with a prevalence of over 90% in the adult population. In immune-competent patients, EBV establishes a life-long latent infection [1,2,3]. Most individuals are infected during childhood with few or no overt symptoms. Adolescents and young adults usually develop infectious mononucleosis (IM), a self-limiting illness with fever, sore throat, lymphadenopathy, hepatosplenomegaly, and fatigue, caused by acute inflammation and hyperactivation of CD8+ T cells [4,5].
Primary EBV infection occurs mainly through the oropharyngeal epithelium transmitted by saliva [6]. The lytic infection of the epithelium is followed by a high tropism of the virus towards B cells in which it switches to its latent program. Naïve B cells are driven by EBV into full latency (stage III, during which all latency genes are expressed Epstein–Barr nuclear antigen (EBNA)-1, 2, 3A, 3B, 3C, and LP, Latent membrane protein (LMP)-1, 2A and 2B, EBV-encoded small RNAs (EBERs), and Bam-HI A rightward transcripts (BARTs)). During further progression, EBV gradually reduces the number of encoded genes. Naïve B cells migrate to the germinal center and undergo further expansion. At the germinal center stage, B cells show a restricted gene expression profile (EBNA-1, LMP-1, 2A and 2B, EBERs, and BARTs), known as latency II, mediating survival and differentiation of EBV-infected B cells into memory cells. Finally, EBV-infected memory B cells, the site of virus persistence, further restrict their expression program to EBERs and BARTs only (latency 0), or additionally EBNA-1 (latency I) during homeostatic proliferation.
Occasionally, EBV turns to its lytic program in plasma cells, leading to the production of new virions, repeated epithelial infection, and shedding of viral particles into the saliva [7,8,9,10]. Viral antigens expressed by EBV during its lytic and latent stages are highly immunogenic and induce a strong response against infected cells. Hence, the downregulation of such molecules is essential to escape immune surveillance and provide virus persistence [11,12,13].
Natural killer (NK) and T cells play a major role in controlling EBV. Viral infection decreases MHC class I expression, but natural killer (NK) cells can recognize this state and destroy the cells. EBV lytic infection causes suppression of MHC class I expression and induction of expression of CD112 and UL16 binding protein 1, NK cell activation receptor ligands [14]. Thus, lytic infected cells are eliminated by NK cells, but most EBV infections evade NK cell attack by shifting to latent infection [14,15,16]. In humanized mice, which were challenged with EBV, depletion of NK cells caused exacerbated IM symptoms, with higher viral loads, larger spleens, increased weight loss, and more tumor burden [15].
Cytotoxic CD8+ T cell responses play an even bigger part in the immune response to EBV, addressing both lytic and latent stages of infection [17]. During IM, EBV-specific CD8+ T cells targeting mainly lytic proteins can expand up to 50% of the circulating CD8+ T cell pool. [18,19]. CD4+ T cells recognize a variety of EBV epitopes; however, their expansion is much less [20,21]. Interestingly, some CD4+ T cells develop a cytotoxic phenotype, with expression of granzyme and perforin, and are able to lyse lymphoblastoid cell lines (LCLs) and EBV loaded peripheral blood mononuclear cells (PBMCs) [22,23,24]. Recently, the impact of γδ and natural killer T (NKT) cells on immunity against EBV could be partially delineated. A comprehensive review of the T cell response to EBV, including unconventional populations, was conducted by Long et al. [25].
The importance of T cells to control EBV can be observed in several conditions in which effector cells are compromised, such as aging, human immunodeficiency virus (HIV) infection, transplantation, or as reviewed here, inborn errors. In those individuals, persistent reactivation and proliferation of EBV-infected cells are associated with severe pathologies that can have lethal outcomes [26,27,28,29]. In this review, we will discuss genetic diseases, which lead to uncontrolled EBV-associated immune dysregulation.
2. Inborn Errors of Immunity (IEI)
IEI (also known as primary immunodeficiencies) are a heterogeneous group of diseases, in which patients manifest with increased susceptibility to infections or other immunological disturbances such as autoimmunity, autoinflammation, or immune dysregulation [30]. These conditions result from germline mutations affecting the development, differentiation, and/or function of the immune system. More than 430 genes have been associated with specific diseases; due to next-generation sequencing technologies, this number is constantly growing [30]. IEI are expected to affect 1/1000 to 1/5000 births [31].
Interestingly, while many IEI show a broad susceptibility to several pathogens including EBV, few have a restricted vulnerability to EBV only [17]. Mutations in genes involving non-redundant mechanisms of immunity against EBV lead to this EBV predisposition syndrome (Figure 1).
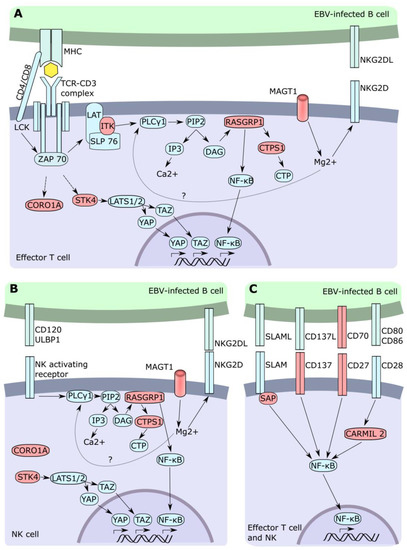
Figure 1.
T cell and NK cell signaling following EBV-infected cell recognition. Cascade associated with TCR (A), NK activating receptor (B), and co-stimulatory (C) stimulation. Red color describes a gene with mutations associated with EBV.
Although EBV has been associated with various malignancies [32,33,34], a healthy immune system is usually capable of controlling the infection. Most individuals remain asymptomatic, and EBV-associated cancer in immunocompetent individuals is relatively rare [35]. Disturbances of host immunity can tilt this balance to favor the virus, allowing its full oncogenic potential. Besides the persistent inflammatory environment caused by EBV viremia and the expression of oncogenic EBV proteins and nucleic acids, there is an inability to kill transformed cells due to defects in cytotoxicity in certain IEI [36]. In the IEI discussed below, the mechanisms involved in the immunity against EBV are dysfunctional leading to immune dysregulation and malignancies.
3. CD27-CD70 Deficiency
CD27 is a co-stimulatory receptor expressed constitutively in a variety of lymphocytes, such as NK and T cells [37]. It binds to CD70 resulting in nuclear factor kappa-light chain-enhancer of activated B cells (NF-kB) pathway activation [38]. CD70, on the other hand, is only transiently inducible upon stimulation on T, B, NK, and dendritic cells [39,40,41,42]. However, upon infection with EBV, CD70 is upregulated on B cells [43]. High levels of CD70 are also observed in B and T cell lymphoma and many solid tumors [44]. Murine models have shed light on the CD27–CD70 interaction. CD27 co-stimulation induces T cell development [45], increases CD8+ T cell activation [46,47,48] and cell survival, and further contributes to the differentiation of CD8+ T cells into memory cells [37,47,49].
Hypogammaglobulinemia clinical features of CD27- and CD70-deficient patients mainly result from EBV-associated immune dysregulation. They present with severe IM, lymphoproliferative disease (LPD), lymphoma, and hemophagocytic lymphohistiocytosis (HLH). In a retrospective study, nearly half (11/21) of CD70-deficient and 36% (12/33) of CD27-deficient patients developed lymphomas, with Hodgkin’s lymphoma (HL) being the most common malignancy (Table 1) [50,51]. The exact mechanisms involved in the defective immune response are still unknown. However, T cells from those patients show an altered phenotype, decreased EBV-specific expansion, and reduced cytotoxicity towards EBV-transformed B cells [43,50,52]. Additionally, CD27 and CD70 might play an important role in immune control of malignancies, even irrespective of EBV infection. In fact, the axis has the potential to induce expansion of effector T cells, break tolerance, and activate response in non-immunogenic tumors; several drugs—such as Varlilumab, SGN-CD70, SGN-7, and MDX-1203—targeting the CD27–CD70 axis are currently being tested in cancer therapy [44,53].

Table 1.
Inborn errors of immunity with high EBV susceptibility and disease.
4. CD137 (TNFRSF9, 4-1BB) Deficiency
CD137 (also known as 4-1BB and TNFRSF9) shows many similarities with CD27. Both receptors are part of the TNFR superfamily and act as co-stimulatory receptors, increasing T cell proliferation, survival, cytokine production, and cytotoxicity. Unlike CD27, which is constitutively expressed by resting T cells, CD137 is induced after cell activation [47,108]. The expression of those receptors at different stages could explain why CD27 engagement favors the formation of effector T cells, while CD137 induces a more robust long-term immunity and secondary response [46]. CD137 ligand is expressed by dendritic cells, macrophages, and activated T and B cells, including EBV-infected B cells [47,108].
EBV-specific T cells from CD137-deficient patients presented lower interferon (IFN)-γ and perforin expression and showed impaired expansion in response to EBV-infected B cells compared to healthy cells. Similar results were also observed following CD137 blockage in T cells from healthy donors, highlighting its non-redundant role in the immune response against EBV [62,63]. Susceptibility to EBV was a common clinical feature among the patients described. Chronic EBV viremia. EBV-associated HLH and lymphoma were present in the majority of patients (Table 1) [61,62,63]. Interestingly, Rodriguez et al. suggested an incomplete clinical penetrance in one of two siblings described. Though both were carrying the same mutation in TNFRSF9 and were EBV viremic, only one sibling developed symptoms. Importantly, specific CD8+ T cell responses towards LCL were impaired in both kindreds. The symptomatic sibling further showed digenic mutations in the PIK3CD gene (causative of activated PI3 kinase delta syndrome) that might have further contributed to the EBV-related clinical phenotype [61].
5. ITK Deficiency
Interleukin-2 inducible T cell kinase (ITK) is a member of the Tec family tyrosine kinases with a crucial role in mediating antigen receptor signaling in T cells. Following T cell receptor (TCR) engagement, the CD3 immunoreceptor tyrosine-based activation motifs (ITAMs) are phosphorylated by lymphocyte-specific protein tyrosine kinase (Lck). It allows zeta-chain-associated protein kinase 70 (Zap-70) to bind to phosphorylated ITAMs and subsequently to phosphorylate adapters of linker for activation of T cells (LAT) and the SH2 domain-containing leukocyte protein of 76kDa (SLP-76). ITK is recruited to the phosphorylated LAT/SLP-76 adapter complex, and together they activate phospholipase Cγ1 (PLCγ1). Activated PLCγ1 hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to produce the second messenger molecules inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 induces intracellular Ca2+ release, while DAG induces NF-κB and MAPK/ERK pathways [57,109].
ITK is not indispensable for TCR downstream signaling, it rather acts as an amplifier. Therefore, although some processes are barely affected, the development and differentiation of T cells might follow abnormal paths. ITK deficiency leads to a skewed Th1 response in detriment of the Th2 response, favors Treg differentiation over Th17, induces development of “innate like” CD8+ T cells, and abrogates development of NKT cells [54,110,111,112,113,114,115,116]. ITK-deficient CD8+ T cells show delayed effector function upon activation, decreased proliferation, and intrinsic defects in degranulation. Interestingly, those defects could be rescued by increasing costimulatory signals, such as prolonged IL-2 stimulation or the addition of IL-12 [117]. Intraperitoneal infection of ITK−/− mice with murine gammaherpesvirus-68 (MHV-68) leads to latent intestinal infection, which develops into lethal colitis [118].
T cells from ITK-deficient patients were also reported to have low or delayed Ca2+ flux upon TCR stimulation with anti-CD3 [119]. Clinical features include hypogammaglobulinemia, EBV viremia, EBV-induced LPD, and lymphoma. Most commonly, HL has been observed in the reported patients (38%, 8/21). To date, no asymptomatic and/or EBV-naïve patient has been identified; therefore, it is still not clear whether the increased risk of developing lymphomas is also present in the absence of EBV. Nevertheless, the high incidence of HL, together with the fact that all HL and HL-like patients were EBV seropositive and expressing latency II proteins, suggest that ITK is involved in the immune control of EBV-associated oncogenesis [120].
6. RASGRP1 Deficiency
Similar to ITK, the nucleotide exchange factor RAS guanyl-releasing protein 1 (RASGRP1) is a secondary TCR messenger. Following increased DAG production by PLCγ1, RASGRP1 is recruited to the membrane and activates the small G protein RAS that in turn activates the cascade of MAP kinase (also known as Raf-MEK-ERK kinases) [121]. RASGRP1 is expressed on lymphocytes and its deficiency in NK and CD8+ T cells leads to defective proliferation and cytotoxic function [104,105]. Salzer et al. showed that although those cells had an increased expression of perforin and granzyme B, the release of cytotoxic granules was impaired [105]. However, this was not observed in other studies [104]. CD27/CD70-induced proliferation was also disturbed in RASGRP1-deficient T cells [104]. Given the importance of this pathway to control EBV-infected and transformed cells (discussed above), the same mechanism could lead to EBV susceptibility in RASGRP1-deficient patients. T cells from these individuals show a reduced cytidine triphosphate (CTP) synthase 1 (CTPS1) expression, an enzyme with a key role in DNA replication (discussed below). Deficiency of CTPS1 expression has been attributed to defective T cell proliferation in those individuals [104].
RASGRP1-deficient patients commonly show recurrent infections, inverted CD4+: CD8+ T cell ratio, poor T cell proliferation, defective NK cell function, autoimmunity, and EBV-associated lymphoma. Six out of nine patients developed EBV-associate malignancies, among them two were diagnosed with diffuse large B cell lymphoma (DLBCL), two with HL, one with low-grade lymphoma, and one with polymorphic B cell lymphoma [101,102,103,104,105].
7. CTPS1 Deficiency
CTPS1 is a key enzyme for de novo synthesis of CTP, a limiting nucleotide in cells, and therefore, essential for DNA replication. Resting T cells express rather low levels of CTPS1, but it is readily upregulated after TCR stimulation in accordance with its requirement for DNA synthesis and proliferation [98,122,123]. As expected, CTPS1-deficient cells exhibit impaired proliferation in response to TCR engagement (IL-2-induced proliferation remains unaffected), but other T cell functions, such as cytokine production and cytotoxicity, are not affected [98]. In light of the massive T cell proliferation required in EBV control [18,19], it is not surprising that CTPS1-deficient patients manifest with EBV-associated immune dysregulation. Common clinical features include severe IM and EBV-induced LPD, three out of 28 patients also developed EBV-driven lymphomas. Recurrent viral infections with other viruses, such as varicella-zoster virus (VZV) and human herpesvirus 6 (HHV6), were also common [96,97,98,99,100].
8. MAGT1 Deficiency (XMEN Syndrome)
Mutations in the gene encoding the magnesium transporter protein 1 (MAGT1) are causative of XMEN (X-linked immunodeficiency with magnesium defect, EBV infection, and neoplasia) syndrome. This disease was initially described in 2011 in two males showing recurrent pulmonary infections, low CD4+ T cells, and EBV-induced LPD and lymphoma [124]. Following TCR activation, T cells show a rapid Mg2+ influx that was abrogated in MAGT1 deficiency. It was thought that Mg2+ was a second intracellular messenger of the TCR linked with PLCγ1 activation and subsequently Ca2+ influx upon TCR activation [124,125,126]. Recently, Ravell et al. revealed another function of MAGT1, which could not yet be clearly attributed to intracellular Mg2+ transport. It was shown that defects in MAGT1 cause glycosylation errors in specific subsets of glycoproteins, including NKG2D and CD70 expressed by immune cells [59]. NKG2D expressed in NK and CD8+ T cells plays a crucial role in killing EBV-infected and transformed cells, and its decreased expression makes it a perfect biomarker [127,128,129]. If poorly glycosylated, these receptors are prematurely degraded, leading to a low surface expression and subsequent disturbed effector function against EBV-infected targets [59,130]. Hence, besides recurrent infections, CD4+ T cell lymphopenia, hypogammaglobulinemia, and lymphadenopathy, the clinical phenotype of patients with MAGT1 deficiency includes high susceptibility to EBV-induced LPD and malignancies (14 of 37 reported patients). Again, HL was the most prevalent lymphoma (7 of 14 patients) (Table 1). Interestingly, EBV-naïve patients also frequently suffered from lymphadenopathy.
9. Coronin 1A Deficiency
Coronin 1A (coded by CORO1A) belongs to a family of coronins that is highly expressed in leukocytes. They are actin-binding proteins, which regulate cytoskeletal remodeling in response to extracellular signals. They modulate processes such as migration, phagocytosis, and cell polarization [131,132]. One of the most striking phenotypes of coronin 1A deficiency is the lack of naïve T cells. Interestingly, effector and memory T cell survival is barely affected and the intact thymus in these patients suggests normal T cell development [64,65,66,67,68]. It was initially believed that the accumulation of F-actin due to lack of coronin 1A activity and subsequent apoptosis was responsible for naïve T cell reduction [132]. Further studies did not confirm this but associated this finding with poor Ca2+ mobilization [133,134]. Finally, T cell lymphopenia was also thought to be a consequence of impaired thymic egress. Nevertheless, this hypothesis was based on defective egress observed in a murine model with a gain-of-function mutation (E26K) [64], while most patients have been harboring loss-of-function mutations in CORO1A.
Besides some individuals who manifest with a profound T cell reduction, i.e., complete SCID (severe combined immunodeficiency) phenotype, most coronin 1A-deficient patients reported to date suffered from recurrent (viral) infections, and an inability to control EBV, leading to EBV-associated LPD and lymphomas (Table 1). Although the mechanisms which make coronin 1A-deficient patients prone to EBV infection are still not clear, it is likely that a reduced EBV-specific CD8+ T cell expansion due to T cell lymphopenia plays a major role. Additionally, coronin 1A-deficient NK cells show diminished cytotoxicity and impaired degranulation caused by the accumulation of F-actin at the immunological synapse [135].
10. STK4 (MST1) Deficiency
Serine-threonine kinase 4 (STK4, also known as mammalian sterile 20-like 1, MST1), is a key kinase involved in the signaling of the canonical and non-canonical Hippo pathway. In the canonical path, STK4 phosphorylates large tumor suppressor kinases (LATS1/2), which further activate the yes-associated protein (YAP) and transcriptional co-activator PDZ-binding motif (TAZ). Activated YAP and TAZ act as transcription factors and induce the expression of various genes controlling cell growth, proliferation, and differentiation [136]. Through the non-canonical Hippo pathway, STK4 exerts a variety of other functions on immune cells, such as extravasation and vesicle trafficking of neutrophils [74,137], humoral immunity [77] and T cell migration, development, and function [76,138,139]. T cells from STK4-deficient patients show reduced proliferation upon stimulation [69,70,71,72,73,74,75,76,77,78,79,80]. Nehme et al. could link decreased T cell proliferation with elevated T cell apoptosis due to increased FAS expression on the T cell surface [78]. T cells from STK4-deficient patients also exhibit defective transwell migration in response to the chemokines CCL19, CCL20, and CXCL11, which is linked to lower expression of CCR7 and L-selectin in T cells [74,78].
STK4-deficient patients suffer from recurrent bacterial, fungal, and/or viral infections, including EBV-associated LPD, intermittent neutropenia, T and B cell lymphopenia, and increased risk of autoimmune diseases and lymphoma. Although around half of reported STK4-deficient patients have manifested with EBV-LPD and viremia, there is a further EBV independent risk of developing malignancies. Out of six lymphomas reported in five of the 28 patients, three were tested EBV-negative [79,80]. Several studies have associated STK4 with tumorigenesis in mice [138]. Kim et al. showed that chromosomal instability present in STK4 knockout mice accelerated lymphoma development following mutagen treatment or p53 deletion [140]. Additionally, analysis of publicly available datasets of B, T, and NK cell lymphoma showed a significant decrease in STK4 expression in those malignancies [80]. Therefore, the lack of the antitumor capacities of STK4 should be considered as an additional risk factor in lymphoma development.
11. CARMIL2 (RLTPR) Deficiency
Capping protein regulator and myosin 1 linker 2 (CARMIL2, also known as RLTPR) is a protein expressed in many cell types, including lymphoid tissue and the gastrointestinal tract. It controls actin polymerization; hence it regulates a variety of functions as cell polarization and migration [141]. Despite its functions associated with actin, CARMIL2 acts as a messenger downstream of CD28, bridging the co-stimulatory receptor to the NF-kB pathway. CARMIL2-defective T cells demonstrate reduced proliferation, differentiation, and effector function following TCR-dependent CD28 co-stimulation [87,89]. The clinical features of CARMIL2 deficiency include recurrent and/or chronic bacterial, viral, and fungal infections, inflammatory bowel disease, and cutaneous manifestations. Patients present with low-level EBV-viremia. Interestingly, EBV-LPD or lymphoma has never been observed, instead 20% of the patients (8/44) developed EBV-associated smooth muscle tumors (SMT) [82,83,84,85,86,87,88,89,90,91,92,93,94,95]. The mechanisms are still unknown.
12. SH2D1A (XLP1 Syndrome) and XIAP Deficiency (XLP2 Syndrome)
X-linked lymphoproliferative disease type 1 (XLP1) is caused by mutations in SH2D1A, which encodes the signaling lymphocyte activation molecular (SLAM)-associated protein (SAP) [142,143,144]. SAP binds to the cytoplasmic domain of SLAM family receptors and regulates downstream intracellular signaling pathways following activation of SLAM receptors to their cognate ligands [145,146]. Engagement of the SLAM receptors 2B4 and NTB-A on SAP-sufficient CD8+ T and NK cells increases their cytotoxic effect. However, in SAP-deficient cells, stimulation of those receptors showed an inhibitory effect [147,148,149]. Furthermore, SAP signaling was only indispensable in response to B cells, as SAP-deficient CD8+ T cells were still able to kill other cell types, i.e., fibroblasts, monocytes, or dendritic cells [147,150]. This might explain why individuals with XLP1 do not show any susceptibility to other common viruses, such as cytomegalovirus (CMV), varicella-zoster virus (VZV), and human papillomavirus (HPV). While EBV shows a high tropism towards B cells, other viruses infect different cell types that are unaffected by the loss of SAP.
SAP−/− mice infected with MHV-68 develop hypogammaglobulinemia and chronic inflammation with exacerbated proliferation of virus-specific CD8+ T cells and consequently increased tissue damage [151,152]. Similar symptoms were observed in XLP1 patients, who often manifest with severe EBV-induced IM and HLH, B cell lymphoma, and hypogammaglobulinemia. Surprisingly, although 25% of the cases of XLP1 develop B cell lymphoma, no significant difference between EBV-negative and EBV-positive individuals was observed. It is suggested that defects on NK and NKT cells, in addition to poor responsiveness of CD8+ T cells against B cells, play a pivotal role in the development of B cell lymphoma, rather than the ability of EBV to induce transformation [106].
In X-linked inhibitor of apoptosis protein (XIAP)-deficient patients, cytotoxicity of NK and CD8+ T cells are unaffected but CD8+ T cells lacking XIAP show increased apoptosis followed stimulation [153]. XIAP patients show high frequencies of EBV-related HLH; however, in contrast to SAP deficiency, inflammatory bowel disease manifestations are common, while B cell lymphomas are rare [154,155].
13. Conclusions
Since EBV was firstly identified 60 years ago, the immunological sequelae of EBV infection in immunocompetent and immunocompromised individuals has to a large extent been revealed [7,156]. Its association with malignancies, especially in numerous EBV-susceptible IEIs, is undisputed.
A shared characteristic observed in most IEIs susceptible to EBV is a CD8+ T cell dysfunction to various degrees (Figure 1). Although NK cells are also commonly affected, this feature was not observed in all genetic entities.
Furthermore, although the study of these IEIs contributed immensely to the knowledge of the interaction between the immune system and EBV, the exact mechanisms underlying lymphoma development are still not completely understood. Further studies will elucidate whether the high frequency of malignancies observed in some IEIs are linked to: (1) the uncontrolled EBV-infection, (2) the inability of the organism to control transformed cells independently of EBV, or most likely (3) a combination of both factors.
As the primary target of EBV is the B cell, it is not surprising that lymphoproliferative diseases in patients with IEIs are usually of B cell origin. However, our review did not include the rare but equally important as well as often fatal clinical manifestations of T/NK cell proliferative diseases. Fujiwara and Nakamura provide an in-depth review of the unique characteristics of chronic active EBV infection in IEIs with EBV-positive T/NK cell LPDs in a recent special issue of this journal [157].
The discovery of “new” IEIs have rapidly increased in the past years due to the increased application of next-generation sequences [158]. Novel discoveries should continue to rise as this technique becomes widely applied and new enhanced diagnoses are developed. As new cases arise, IEIs will remain a unique source of information to understand non-redundant pathways involved in the immunity against EBV and EBV-associated tumors. These studies will contribute to the development of better therapies, not only for individuals presenting those rare genetic diseases but also for more common diseases, such as severe IM, HLH, and cancer in immunocompetent people.
Author Contributions
Conceptualization, S.G.; writing—original draft, C.N.R.L.; writing—review and editing, S.G. and C.N.R.L. All authors have read and agreed to the published version of the manuscript.
Funding
C.N.R.L. and S.G. are generously supported by a grant of the Elterninitiative Kinderkrebsklinik e.V.
Acknowledgments
We thank all laboratory and clinical members of the Department of Pediatric Oncology, Hematology, and Clinical Immunology at the University Hospital Düsseldorf for their dedicated work with children and adolescents with inborn errors of immunity and malignancies. We thank all patients and their families for their endless willingness to support research in order to help other patients with these rare diseases.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pereira, M.S.; Blake, J.M.; Macrae, A.D. EB Virus Antibody at Different Ages. BMJ 1969, 4, 526–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, C.D.; Swerdlow, A.J.; Macsween, K.F.; Harrison, N.; Williams, H.; McAulay, K.; Thomas, R.; Reid, S.; Conacher, M.; Britton, K.; et al. A Study of Risk Factors for Acquisition of Epstein-Barr Virus and Its Subtypes. J. Infect. Dis. 2007, 195, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de-Thé, G.; Geser, A.; Day, N.E.; Tukei, P.M.; Williams, E.H.; Beri, D.P.; Smith, P.G.; Dean, A.G.; Bornkamm, G.W.; Feorino, P.; et al. Epidemiological Evidence for Causal Relationship between Epstein-Barr Virus and Burkitt’s Lymphoma from Ugandan Prospective Study. Nature 1978, 274, 756–761. [Google Scholar] [CrossRef]
- Dunmire, S.K.; Hogquist, K.A.; Balfour, H.H. Infectious Mononucleosis. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 390, pp. 211–240. [Google Scholar]
- Dunnet, W.N. Infectious Mononucleosis. BMJ 1963, 1, 1187–1191. [Google Scholar] [CrossRef] [PubMed]
- Temple, R.M.; Zhu, J.; Budgeon, L.; Christensen, N.D.; Meyers, C.; Sample, C.E. Efficient Replication of Epstein–Barr Virus in Stratified Epithelium In Vitro. Proc. Natl. Acad. Sci. USA 2014, 111, 16544–16549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein–Barr Virus: More than 50 Years Old and Still Providing Surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Rezk, S.A.; Zhao, X.; Weiss, L.M. Epstein-Barr Virus (EBV)–Associated Lymphoid Proliferations, a 2018 Update. Hum. Pathol. 2018, 79, 18–41. [Google Scholar] [CrossRef]
- Kang, M.-S.; Kieff, E. Epstein–Barr Virus Latent Genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [Green Version]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, D.A. The Expression Pattern of Epstein-Barr Virus Latent Genes In Vivo Is Dependent upon the Differentiation Stage of the Infected B Cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Murray, R.J.; Kurilla, M.G.; Brooks, J.M.; Thomas, W.A.; Rowe, M.; Kieff, E.; Rickinson, A.B. Identification of Target Antigens for the Human Cytotoxic T Cell Response to Epstein-Barr Virus (EBV): Implications for the Immune Control of EBV-Positive Malignancies. J. Exp. Med. 1992, 176, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Blake, N.; Haigh, T.; Shaka’a, G.; Croom-Carter, D.; Rickinson, A. The Importance of Exogenous Antigen in Priming the Human CD8+ T Cell Response: Lessons from the EBV Nuclear Antigen EBNA1. J. Immunol. 2000, 165, 7078–7087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, R.; Burrows, S.R.; Kurilla, M.G.; Jacob, C.A.; Misko, I.S.; Sculley, T.B.; Kieff, E.; Moss, D.J. Localization of Epstein-Barr Virus Cytotoxic T Cell Epitopes Using Recombinant Vaccinia: Implications for Vaccine Development. J. Exp. Med. 1992, 176, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Pappworth, I.Y.; Wang, E.C.; Rowe, M. The Switch from Latent to Productive Infection in Epstein-Barr Virus-Infected B Cells Is Associated with Sensitization to NK Cell Killing. J. Virol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Chijioke, O.; Müller, A.; Feederle, R.; Barros, M.H.M.; Krieg, C.; Emmel, V.; Marcenaro, E.; Leung, C.S.; Antsiferova, O.; Landtwing, V.; et al. Human Natural Killer Cells Prevent Infectious Mononucleosis Features by Targeting Lytic Epstein-Barr Virus Infection. Cell Rep. 2013, 5, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Azzi, T.; Lünemann, A.; Murer, A.; Ueda, S.; Béziat, V.; Malmberg, K.-J.; Staubli, G.; Gysin, C.; Berger, C.; Münz, C.; et al. Role for Early-Differentiated Natural Killer Cells in Infectious Mononucleosis. Blood 2014, 124, 2533–2543. [Google Scholar] [CrossRef] [Green Version]
- Latour, S.; Fischer, A. Signaling Pathways Involved in the T-cell-mediated Immunity against Epstein-Barr Virus: Lessons from Genetic Diseases. Immunol. Rev. 2019, 291, 174–189. [Google Scholar] [CrossRef]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The Immunology of Epstein-Barr Virus–Induced Disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Hislop, A.D. Tonsillar Homing of Epstein-Barr Virus-Specific CD8+ T Cells and the Virus-Host Balance. J. Clin. Investig. 2005, 115, 2546–2555. [Google Scholar] [CrossRef] [Green Version]
- Long, H.M.; Chagoury, O.L.; Leese, A.M.; Ryan, G.B.; James, E.; Morton, L.T.; Abbott, R.J.M.; Sabbah, S.; Kwok, W.; Rickinson, A.B. MHC II Tetramers Visualize Human CD4+ T Cell Responses to Epstein–Barr Virus Infection and Demonstrate Atypical Kinetics of the Nuclear Antigen EBNA1 Response. J. Exp. Med. 2013, 210, 933–949. [Google Scholar] [CrossRef]
- Long, H.M.; Haigh, T.A.; Gudgeon, N.H.; Leen, A.M.; Tsang, C.-W.; Brooks, J.; Landais, E.; Houssaint, E.; Lee, S.P.; Rickinson, A.B.; et al. CD4+ T-Cell Responses to Epstein-Barr Virus (EBV) Latent-Cycle Antigens and the Recognition of EBV-Transformed Lymphoblastoid Cell Lines. J. Virol. 2005, 79, 4896–4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.K.P.; Hui, K.F.; Ning, R.J.; Xu, X.Q.; Chan, K.H.; Chiang, A.K.S. Emergence of CD4+ and CD8+ Polyfunctional T Cell Responses against Immunodominant Lytic and Latent EBV Antigens in Children with Primary EBV Infection. Front. Microbiol. 2018, 9, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meckiff, B.J.; Ladell, K.; McLaren, J.E.; Ryan, G.B.; Leese, A.M.; James, E.A.; Price, D.A.; Long, H.M. Primary EBV Infection Induces an Acute Wave of Activated Antigen-Specific Cytotoxic CD4+ T Cells. J. Immunol. 2019, 203, 1276–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, H.M.; Leese, A.M.; Chagoury, O.L.; Connerty, S.R.; Quarcoopome, J.; Quinn, L.L.; Shannon-Lowe, C.; Rickinson, A.B. Cytotoxic CD4+ T Cell Responses to EBV Contrast with CD8 Responses in Breadth of Lytic Cycle Antigen Choice and in Lytic Cycle Recognition. J. Immunol. 2011, 187, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Long, H.M.; Meckiff, B.J.; Taylor, G.S. The T-Cell Response to Epstein-Barr Virus–New Tricks from an Old Dog. Front. Immunol. 2019, 10, 2193. [Google Scholar] [CrossRef]
- Ru, Y.; Chen, J.; Wu, D. Epstein-Barr Virus Post-Transplant Lymphoproliferative Disease (PTLD) after Hematopoietic Stem Cell Transplantation. Eur. J. Haematol. 2018, 101, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Shindiapina, P.; Ahmed, E.H.; Mozhenkova, A.; Abebe, T.; Baiocchi, R.A. Immunology of EBV-Related Lymphoproliferative Disease in HIV-Positive Individuals. Front. Oncol. 2020, 10, 1723. [Google Scholar] [CrossRef]
- Castillo, J.J.; Beltran, B.E.; Miranda, R.N.; Young, K.H.; Chavez, J.C.; Sotomayor, E.M. EBV-Positive Diffuse Large B-Cell Lymphoma of the Elderly: 2016 Update on Diagnosis, Risk-Stratification, and Management. Am. J. Hematol. 2016, 91, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangye, S.G. Genetic Susceptibility to EBV Infection: Insights from Inborn Errors of Immunity. Hum. Genet. 2020, 139, 885–901. [Google Scholar] [CrossRef]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020, 40, 24–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Frange, P.; Blanche, S.; Casanova, J.L. Pathogenesis of Infections in HIV-Infected Individuals: Insights from Primary Immunodeficiencies. Curr. Opin. Immunol. 2017, 48, 122–133. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, Y.; Wang, C.; Gan, R. Signaling Pathways of EBV-Induced Oncogenesis. Cancer Cell Int. 2021, 21, 93. [Google Scholar] [CrossRef]
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular Mechanisms of EBV-Driven Cell Cycle Progression and Oncogenesis. Med. Microbiol. Immunol. 2019, 208, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Leong, M.M.L.; Lung, M.L. The Impact of Epstein-Barr Virus Infection on Epigenetic Regulation of Host Cell Gene Expression in Epithelial and Lymphocytic Malignancies. Front. Oncol. 2021, 11, 201. [Google Scholar] [CrossRef]
- Cohen, J.I.; Kimura, H.; Nakamura, S.; Ko, Y.-H.; Jaffe, E.S. Epstein–Barr Virus-Associated Lymphoproliferative Disease in Non-Immunocompromised Hosts: A Status Report and Summary of an International Meeting, 8–9 September 2008. Ann. Oncol. 2009, 20, 1472–1482. [Google Scholar] [CrossRef]
- Kebudi, R.; Kiykim, A.; Sahin, M.K. Primary Immunodeficiency and Cancer in Children; A Review of the Literature. Curr. Pediatr. Rev. 2019, 15, 245–250. [Google Scholar] [CrossRef]
- Grant, E.J.; Nüssing, S.; Sant, S.; Clemens, E.B.; Kedzierska, K. The Role of CD27 in Anti-Viral T-Cell Immunity. Curr. Opin. Virol. 2017, 22, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Akiba, H.; Nakano, H.; Nishinaka, S.; Shindo, M.; Kobata, T.; Atsuta, M.; Morimoto, C.; Ware, C.F.; Malinin, N.L.; Wallach, D.; et al. CD27, a Member of the Tumor Necrosis Factor Receptor Superfamily, Activates NF-ΚB and Stress-Activated Protein Kinase/c-Jun N-Terminal Kinase via TRAF2, TRAF5, and NF-ΚB-Inducing Kinase. J. Biol. Chem. 1998, 273, 13353–13358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hintzen, R.Q.; Lens, S.M.; Beckmann, M.P.; Goodwin, R.G.; Lynch, D.; van Lier, R.A. Characterization of the Human CD27 Ligand, a Novel Member of the TNF Gene Family. J. Immunol. 1994, 152, 1762–1773. [Google Scholar]
- Orengo, A.M.; Cantoni, C.; Neglia, F.; Biassoni, R.; Ferrini, S. Reciprocal Expression of CD70 and of Its Receptor, CD27, in Human Long Term-Activated T and Natural Killer (NK) Cells: Inverse Regulation by Cytokines and Role in Induction of Cytotoxicity. Clin. Exp. Immunol. 1997, 107, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Tesselaar, K.; Xiao, Y.; Arens, R.; van Schijndel, G.M.W.; Schuurhuis, D.H.; Mebius, R.E.; Borst, J.; van Lier, R.A.W. Expression of the Murine CD27 Ligand CD70 In Vitro and In Vivo. J. Immunol. 2003, 170, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Nolte, M.A.; van Olffen, R.W.; van Gisbergen, K.P.J.M.; van Lier, R.A.W. Timing and Tuning of CD27-CD70 Interactions: The Impact of Signal Strength in Setting the Balance between Adaptive Responses and Immunopathology. Immunol. Rev. 2009, 229, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Izawa, K.; Martin, E.; Soudais, C.; Bruneau, J.; Boutboul, D.; Rodriguez, R.; Lenoir, C.; Hislop, A.D.; Besson, C.; Touzot, F.; et al. Inherited CD70 Deficiency in Humans Reveals a Critical Role for the CD70-CD27 Pathway in Immunity to Epstein-Barr Virus Infection. J. Exp. Med. 2017, 214, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Deschoolmeester, V.; Zwaenepoel, K.; Rolfo, C.; Silence, K.; Rottey, S.; Lardon, F.; Smits, E.; Pauwels, P. CD70: An Emerging Target in Cancer Immunotherapy. Pharmacol. Ther. 2015, 155, 1–10. [Google Scholar] [CrossRef]
- Gravestein, L.A.; van Ewijk, W.; Ossendorp, F.; Borst, J. CD27 Cooperates with the Pre-T Cell Receptor in the Regulation of Murine T Cell Development. J. Exp. Med. 1996, 184, 675–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, J.E.; Kerr, J.P.; Rogel, A.; Taraban, V.Y.; Buchan, S.L.; Johnson, P.W.M.; Al-Shamkhani, A. Differential Impact of CD27 and 4-1BB Costimulation on Effector and Memory CD8 T Cell Generation Following Peptide Immunization. J. Immunol. 2014, 193, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, M. The Role of TNF Superfamily Members in T-Cell Function and Diseases. Nat. Rev. Immunol. 2009, 9, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Rowley, T.F.; Al-Shamkhani, A. Stimulation by Soluble CD70 Promotes Strong Primary and Secondary CD8+ Cytotoxic T Cell Responses In Vivo. J. Immunol. 2004, 172, 6039–6046. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, J.; Gravestein, L.A.; Tesselaar, K.; van Lier, R.A.W.; Schumacher, T.N.M.; Borst, J. CD27 Is Required for Generation and Long-Term Maintenance of T Cell Immunity. Nat. Immunol. 2000, 1, 433–440. [Google Scholar] [CrossRef]
- Ghosh, S.; Köstel Bal, S.; Edwards, E.S.J.; Pillay, B.; Jiménez Heredia, R.; Erol Cipe, F.; Rao, G.; Salzer, E.; Zoghi, S.; Abolhassani, H.; et al. Extended Clinical and Immunological Phenotype and Transplant Outcome in CD27 and CD70 Deficiency. Blood 2020, 136, 2638–2655. [Google Scholar] [CrossRef]
- Khodzhaev, K.; Bay, S.B.; Kebudi, R.; Altindirek, D.; Kaya, A.; Erbilgin, Y.; Ng, O.H.; Kiykim, A.; Erol, F.C.; Zengin, F.S.; et al. Lymphoma Predisposing Gene in an Extended Family: CD70 Signaling Defect. J. Clin. Immunol. 2020, 40, 883–892. [Google Scholar] [CrossRef]
- Abolhassani, H.; Edwards, E.S.J.; Ikinciogullari, A.; Jing, H.; Borte, S.; Buggert, M.; Du, L.; Matsuda-Lennikov, M.; Romano, R.; Caridha, R.; et al. Combined Immunodeficiency and Epstein-Barr Virus–Induced B Cell Malignancy in Humans with Inherited CD70 Deficiency. J. Exp. Med. 2017, 214, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. Therapeutic Targeting of CD70 and CD27. Expert Opin. Ther. Targets 2016, 20, 959–973. [Google Scholar] [CrossRef]
- Eken, A.; Cansever, M.; Somekh, I.; Mizoguchi, Y.; Zietara, N.; Okus, F.Z.; Erdem, S.; Canatan, H.; Akyol, S.; Ozcan, A.; et al. Genetic Deficiency and Biochemical Inhibition of ITK Affect Human Th17, Treg, and Innate Lymphoid Cells. J. Clin. Immunol. 2019, 39, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Howe, M.K.; Dowdell, K.; Roy, A.; Niemela, J.E.; Wilson, W.; McElwee, J.J.; Hughes, J.D.; Cohen, J.I. Magnesium Restores Activity to Peripheral Blood Cells in a Patient with Functionally Impaired Interleukin-2-Inducible T Cell Kinase. Front. Immunol. 2019, 10, 2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssefian, L.; Vahidnezhad, H.; Yousefi, M.; Saeidian, A.H.; Azizpour, A.; Touati, A.; Nikbakht, N.; Hesari, K.K.; Adib-Sereshki, M.M.; Zeinali, S.; et al. Inherited Interleukin 2–Inducible T-Cell (ITK) Kinase Deficiency in Siblings with Epidermodysplasia Verruciformis and Hodgkin Lymphoma. Clin. Infect. Dis. 2019, 68, 1938–1941. [Google Scholar] [CrossRef]
- Ghosh, S.; Drexler, I.; Bhatia, S.; Gennery, A.R.; Borkhardt, A. Interleukin-2-Inducible T-Cell Kinase Deficiency-New Patients, New Insight? Front. Immunol. 2018, 9, 979. [Google Scholar] [CrossRef]
- Fang, M.; Abolhassani, H.; Pan-Hammarström, Q.; Sandholm, E.; Liu, X.; Hammarström, L. Compound Heterozygous Mutations of IL2-Inducible T Cell Kinase in a Swedish Patient: The Importance of Early Genetic Diagnosis. J. Clin. Immunol. 2019, 39, 131–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravell, J.C.; Matsuda-Lennikov, M.; Chauvin, S.D.; Zou, J.; Biancalana, M.; Deeb, S.J.; Price, S.; Su, H.C.; Notarangelo, G.; Jiang, P.; et al. Defective Glycosylation and Multisystem Abnormalities Characterize the Primary Immunodeficiency XMEN Disease. J. Clin. Investig. 2020, 130, 507–522. [Google Scholar] [CrossRef] [Green Version]
- Ravell, J.C.; Chauvin, S.D.; He, T.; Lenardo, M. An Update on XMEN Disease. J. Clin. Immunol. 2020, 40, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Fournier, B.; Cordeiro, D.J.; Winter, S.; Izawa, K.; Martin, E.; Boutboul, D.; Lenoir, C.; Fraitag, S.; Kracker, S.; et al. Concomitant PIK3CD and TNFRSF9 Deficiencies Cause Chronic Active Epstein-Barr Virus Infection of T Cells. J. Exp. Med. 2019, 216, 2800–2818. [Google Scholar] [CrossRef]
- Somekh, I.; Thian, M.; Medgyesi, D.; Gülez, N.; Magg, T.; Duque, A.G.; Stauber, T.; Lev, A.; Genel, F.; Unal, E.; et al. CD137 Deficiency Causes Immune Dysregulation with Predisposition to Lymphomagenesis. Blood 2019, 134, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Alosaimi, M.F.; Hoenig, M.; Jaber, F.; Platt, C.D.; Jones, J.; Wallace, J.; Debatin, K.; Schulz, A.; Jacobsen, E.; Möller, P.; et al. Immunodeficiency and EBV-Induced Lymphoproliferation Caused by 4-1BB Deficiency. J. Allergy Clin. Immunol. 2019, 144, 574–583.e5. [Google Scholar] [CrossRef] [Green Version]
- Shiow, L.R.; Roadcap, D.W.; Paris, K.; Watson, S.R.; Grigorova, I.L.; Lebet, T.; An, J.; Xu, Y.; Jenne, C.N.; Föger, N.; et al. The Actin Regulator Coronin 1A Is Mutant in a Thymic Egress–Deficient Mouse Strain and in a Patient with Severe Combined Immunodeficiency. Nat. Immunol. 2008, 9, 1307–1315. [Google Scholar] [CrossRef]
- Stray-Pedersen, A.; Jouanguy, E.; Crequer, A.; Bertuch, A.A.; Brown, B.S.; Jhangiani, S.N.; Muzny, D.M.; Gambin, T.; Sorte, H.; Sasa, G.; et al. Compound Heterozygous CORO1A Mutations in Siblings with a Mucocutaneous-Immunodeficiency Syndrome of Epidermodysplasia Verruciformis-HPV, Molluscum Contagiosum and Granulomatous Tuberculoid Leprosy. J. Clin. Immunol. 2014, 34, 871–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, C.S.; Massaad, M.J.; Bainter, W.; Ohsumi, T.K.; Föger, N.; Chan, A.C.; Akarsu, N.A.; Aytekin, C.; Ayvaz, D.Ç.; Tezcan, I.; et al. Recurrent Viral Infections Associated with a Homozygous CORO1A Mutation That Disrupts Oligomerization and Cytoskeletal Association. J. Allergy Clin. Immunol. 2016, 137, 879–888.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punwani, D.; Pelz, B.; Yu, J.; Arva, N.C.; Schafernak, K.; Kondratowicz, K.; Makhija, M.; Puck, J.M. Coronin-1A: Immune Deficiency in Humans and Mice. J. Clin. Immunol. 2015, 35, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moshous, D.; Martin, E.; Carpentier, W.; Lim, A.; Callebaut, I.; Canioni, D.; Hauck, F.; Majewski, J.; Schwartzentruber, J.; Nitschke, P.; et al. Whole-Exome Sequencing Identifies Coronin-1A Deficiency in 3 Siblings with Immunodeficiency and EBV-Associated B-Cell Lymphoproliferation. J. Allergy Clin. Immunol. 2013, 131, 1594–1603.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignesh, P.; Rawat, A.; Kumrah, R.; Singh, A.; Gummadi, A.; Sharma, M.; Kaur, A.; Nameirakpam, J.; Jindal, A.; Suri, D.; et al. Clinical, Immunological, and Molecular Features of Severe Combined Immune Deficiency: A Multi-Institutional Experience from India. Front. Immunol. 2021, 11, 3747. [Google Scholar] [CrossRef] [PubMed]
- Abdollahpour, H.; Appaswamy, G.; Kotlarz, D.; Diestelhorst, J.; Beier, R.; Schäffer, A.A.; Gertz, E.M.; Schambach, A.; Kreipe, H.H.; Pfeifer, D.; et al. The Phenotype of Human STK4 Deficiency. Blood 2012, 119, 3450–3457. [Google Scholar] [CrossRef] [PubMed]
- Al-Saud, B.; Alajlan, H.; Sabar, H.; Anwar, S.; Alruwaili, H.; Al-Hussain, T.; Alamri, N.; Alazami, A.M. STK4 Deficiency in a Patient with Immune Complex Glomerulonephritis, Salt-Losing Tubulopathy, and Castleman’s-Like Disease. J. Clin. Immunol. 2019, 39, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, F.; Klein, C.; Poorpooneh, M.; Sherkat, R.; Khoshnevisan, R. A Case Report of Sinusoidal Diffuse Large B-Cell Lymphoma in a STK4 Deficient Patient. Medicine 2020, 99, e18601. [Google Scholar] [CrossRef]
- Crequer, A.; Picard, C.; Patin, E.; D’Amico, A.; Abhyankar, A.; Munzer, M.; Debré, M.; Zhang, S.-Y.; de Saint-Basile, G.; Fischer, A.; et al. Inherited MST1 Deficiency Underlies Susceptibility to EV-HPV Infections. PLoS ONE 2012, 7, e44010. [Google Scholar] [CrossRef]
- Dang, T.S.; Willet, J.D.; Griffin, H.R.; Morgan, N.V.; O’Boyle, G.; Arkwright, P.D.; Hughes, S.M.; Abinun, M.; Tee, L.J.; Barge, D.; et al. Defective Leukocyte Adhesion and Chemotaxis Contributes to Combined Immunodeficiency in Humans with Autosomal Recessive MST1 Deficiency. J. Clin. Immunol. 2016, 36, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halacli, S.O.; Ayvaz, D.C.; Sun-Tan, C.; Erman, B.; Uz, E.; Yilmaz, D.Y.; Ozgul, K.; Tezcan, İ.; Sanal, O. STK4 (MST1) Deficiency in Two Siblings with Autoimmune Cytopenias: A Novel Mutation. Clin. Immunol. 2015, 161, 316–323. [Google Scholar] [CrossRef]
- Jørgensen, S.E.; Al-Mousawi, A.; Assing, K.; Hartling, U.; Grosen, D.; Fisker, N.; Nielsen, C.; Jakobsen, M.A.; Mogensen, T.H. STK4 Deficiency Impairs Innate Immunity and Interferon Production through Negative Regulation of TBK1-IRF3 Signaling. J. Clin. Immunol. 2021, 41, 109–124. [Google Scholar] [CrossRef]
- Moran, I.; Avery, D.T.; Payne, K.; Lenthall, H.; Davies, E.G.; Burns, S.; Ip, W.; Oleastro, M.M.; Reisli, I.; Guner, S.; et al. B Cell–Intrinsic Requirement for STK4 in Humoral Immunity in Mice and Human Subjects. J. Allergy Clin. Immunol. 2019, 143, 2302–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nehme, N.T.; Schmid, J.P.; Debeurme, F.; André-Schmutz, I.; Lim, A.; Nitschke, P.; Rieux-Laucat, F.; Lutz, P.; Picard, C.; Mahlaoui, N.; et al. MST1 Mutations in Autosomal Recessive Primary Immunodeficiency Characterized by Defective Naive T-Cell Survival. Blood 2012, 119, 3458–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radwan, N.; El-Owaidy, R.; El-Sayed, Z.A.; Abdel-Baky, A.; El-Haddad, A.; Rashad, H.; Khorshed, E.N.; Platt, C.D.; Wallace, J.G.; Chou, J.; et al. A Case of STK4 Deficiency with Complications Evoking Mycobacterial Infection. J. Clin. Immunol. 2020, 40, 665–669. [Google Scholar] [CrossRef]
- Schipp, C.; Schlütermann, D.; Hönscheid, A.; Nabhani, S.; Höll, J.; Oommen, P.T.; Ginzel, S.; Fleckenstein, B.; Stork, B.; Borkhardt, A.; et al. EBV Negative Lymphoma and Autoimmune Lymphoproliferative Syndrome Like Phenotype Extend the Clinical Spectrum of Primary Immunodeficiency Caused by STK4 Deficiency. Front. Immunol. 2018, 9, 2400. [Google Scholar] [CrossRef] [Green Version]
- Sherkat, R.; Sabri, M.R.; Dehghan, B.; Bigdelian, H.; Reisi, N.; Afsharmoghadam, N.; Rahimi, H.; Rahmanian, N.; Klein, C. EBV Lymphoproliferative-Associated Disease and Primary Cardiac T-Cell Lymphoma in a STK4 Deficient Patient. Medicine 2017, 96, e8852. [Google Scholar] [CrossRef] [PubMed]
- Marangi, G.; Garcovich, S.; Sante, G.; Orteschi, D.; Frangella, S.; Scaldaferri, F.; Genuardi, M.; Peris, K.; Gurrieri, F.; Zollino, M. Complex Muco-Cutaneous Manifestations of CARMIL2-Associated Combined Immunodeficiency: A Novel Presentation of Dysfunctional Epithelial Barriers. Acta Derm. Venereol. 2020, 100, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosa, L.; Batura, V.; Colavito, D.; Fiedler, K.; Gaio, P.; Guo, C.; Li, Q.; Marzollo, A.; Mescoli, C.; Nambu, R.; et al. Novel CARMIL2 Loss-of-Function Variants Are Associated with Pediatric Inflammatory Bowel Disease. Sci. Rep. 2021, 11, 5945. [Google Scholar] [CrossRef] [PubMed]
- Maccari, M.E.; Speckmann, C.; Heeg, M.; Reimer, A.; Casetti, F.; Has, C.; Ehl, S.; Castro, C.N. Profound Immunodeficiency with Severe Skin Disease Explained by Concomitant Novel CARMIL2 and PLEC1 Loss-of-Function Mutations. Clin. Immunol. 2019, 208, 108228. [Google Scholar] [CrossRef]
- Yonkof, J.R.; Gupta, A.; Rueda, C.M.; Mangray, S.; Prince, B.T.; Rangarajan, H.G.; Alshahrani, M.; Varga, E.; Cripe, T.P.; Abraham, R.S. A Novel Pathogenic Variant in CARMIL2 (RLTPR) Causing CARMIL2 Deficiency and EBV-Associated Smooth Muscle Tumors. Front. Immunol. 2020, 11, 884. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Uner, A.; Saglam, A.; Chadburn, A.; Crane, G.M. Peripheral Eosinophilia in Primary Immunodeficiencies of Actin Dysregulation: A Case Series of Wiskott-Aldrich Syndrome, CARMIL2 and DOCK8 Deficiency and Review of the Literature. Ann. Diagn. Pathol. 2019, 43, 151413. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, C.S.; Ling, Y.; Bousfiha, A.; Camcioglu, Y.; Jacquot, S.; Payne, K.; Crestani, E.; Roncagalli, R.; Belkadi, A.; et al. Dual T Cell- and B Cell-Intrinsic Deficiency in Humans with Biallelic RLTPR Mutations. J. Exp. Med. 2016, 213, 2413–2435. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Al-Helale, M.; Alhissi, S.; Al-Saud, B.; Alajlan, H.; Monies, D.; Shah, Z.; Abouelhoda, M.; Arnaout, R.; Al-Dhekri, H.; et al. Novel CARMIL2 Mutations in Patients with Variable Clinical Dermatitis, Infections, and Combined Immunodeficiency. Front. Immunol. 2018, 9, 203. [Google Scholar] [CrossRef] [Green Version]
- Schober, T.; Magg, T.; Laschinger, M.; Rohlfs, M.; Linhares, N.D.; Puchalka, J.; Weisser, T.; Fehlner, K.; Mautner, J.; Walz, C.; et al. A Human Immunodeficiency Syndrome Caused by Mutations in CARMIL2. Nat. Commun. 2017, 8, 14209. [Google Scholar] [CrossRef] [Green Version]
- Sorte, H.S.; Osnes, L.T.; Fevang, B.; Aukrust, P.; Erichsen, H.C.; Backe, P.H.; Abrahamsen, T.G.; Kittang, O.B.; Øverland, T.; Jhangiani, S.N.; et al. A Potential Founder Variant in CARMIL2/RLTPR in Three Norwegian Families with Warts, Molluscum Contagiosum, and T-Cell Dysfunction. Mol. Genet. Genom. Med. 2016, 4, 604–616. [Google Scholar] [CrossRef]
- Atschekzei, F.; Jacobs, R.; Wetzke, M.; Sogkas, G.; Schröder, C.; Ahrenstorf, G.; Dhingra, A.; Ott, H.; Baumann, U.; Schmidt, R.E. A Novel CARMIL2 Mutation Resulting in Combined Immunodeficiency Manifesting with Dermatitis, Fungal, and Viral Skin Infections as Well as Selective Antibody Deficiency. J. Clin. Immunol. 2019, 39, 274–276. [Google Scholar] [CrossRef]
- Shamriz, O.; Simon, A.J.; Lev, A.; Megged, O.; Ledder, O.; Picard, E.; Joseph, L.; Molho-Pessach, V.; Tal, Y.; Millman, P.; et al. Exogenous Interleukin-2 Can Rescue In-Vitro T Cell Activation and Proliferation in Patients with a Novel Capping Protein Regulator and Myosin 1 Linker 2 Mutation. Clin. Exp. Immunol. 2020, 200, 215–227. [Google Scholar] [CrossRef]
- Magg, T.; Shcherbina, A.; Arslan, D.; Desai, M.M.; Wall, S.; Mitsialis, V.; Conca, R.; Unal, E.; Karacabey, N.; Mukhina, A.; et al. CARMIL2 Deficiency Presenting as Very Early Onset Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 1788–1795. [Google Scholar] [CrossRef]
- Kurolap, A.; Eshach Adiv, O.; Konnikova, L.; Werner, L.; Gonzaga-Jauregui, C.; Steinberg, M.; Mitsialis, V.; Mory, A.; Nunberg, M.Y.; Wall, S.; et al. A Unique Presentation of Infantile-Onset Colitis and Eosinophilic Disease without Recurrent Infections Resulting from a Novel Homozygous CARMIL2 Variant. J. Clin. Immunol. 2019, 39, 430–439. [Google Scholar] [CrossRef]
- Shayegan, L.H.; Garzon, M.C.; Morel, K.D.; Borlack, R.; Vuguin, P.M.; Margolis, K.G.; Demirdag, Y.Y.; Pereira, E.M.; Lauren, C.T. CARMIL2-related Immunodeficiency Manifesting with Photosensitivity. Pediatr. Dermatol. 2020, 37, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Trück, J.; Kelly, D.F.; Taylor, J.M.; Kienzler, A.K.; Lester, T.; Seller, A.; Pollard, A.J.; Patel, S.Y. Variable Phenotype and Discrete Alterations of Immune Phenotypes in CTP Synthase 1 Deficiency: Report of 2 Siblings. J. Allergy Clin. Immunol. 2016, 138, 1722–1725.e6. [Google Scholar] [CrossRef] [Green Version]
- Kucuk, Z.Y.; Zhang, K.; Filipovich, L.; Bleesing, J.J.H. CTP Synthase 1 Deficiency in Successfully Transplanted Siblings with Combined Immune Deficiency and Chronic Active EBV Infection. J. Clin. Immunol. 2016, 36, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Palmic, N.; Sanquer, S.; Lenoir, C.; Hauck, F.; Mongellaz, C.; Fabrega, S.; Nitschké, P.; Esposti, M.D.; Schwartzentruber, J.; et al. CTP Synthase 1 Deficiency in Humans Reveals Its Central Role in Lymphocyte Proliferation. Nature 2014, 510, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Minet, N.; Boschat, A.-C.; Sanquer, S.; Sobrino, S.; Lenoir, C.; de Villartay, J.P.; Leites-de-Moraes, M.; Picard, C.; Soudais, C.; et al. Impaired Lymphocyte Function and Differentiation in CTPS1-Deficient Patients Result from a Hypomorphic Homozygous Mutation. JCI Insight 2020, 5, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Nademi, Z.; Wynn, R.F.; Slatter, M.; Hughes, S.M.; Bonney, D.; Qasim, W.; Latour, S.; Trück, J.; Patel, S.; Abinun, M.; et al. Hematopoietic Stem Cell Transplantation for Cytidine Triphosphate Synthase 1 (CTPS1) Deficiency. Bone Marrow Transplant. 2019, 54, 130–133. [Google Scholar] [CrossRef]
- Somekh, I.; Marquardt, B.; Liu, Y.; Rohlfs, M.; Hollizeck, S.; Karakukcu, M.; Unal, E.; Yilmaz, E.; Patiroglu, T.; Cansever, M.; et al. Novel Mutations in RASGRP1 Are Associated with Immunodeficiency, Immune Dysregulation, and EBV-Induced Lymphoma. J. Clin. Immunol. 2018, 38, 699–710. [Google Scholar] [CrossRef]
- Mao, H.; Yang, W.; Latour, S.; Yang, J.; Winter, S.; Zheng, J.; Ni, K.; Lv, M.; Liu, C.; Huang, H.; et al. RASGRP1 Mutation in Autoimmune Lymphoproliferative Syndrome-like Disease. J. Allergy Clin. Immunol. 2018, 142, 595–604.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, C.D.; Fried, A.J.; Hoyos-Bachiloglu, R.; Usmani, G.N.; Schmidt, B.; Whangbo, J.; Chiarle, R.; Chou, J.; Geha, R.S. Combined Immunodeficiency with EBV Positive B Cell Lymphoma and Epidermodysplasia Verruciformis Due to a Novel Homozygous Mutation in RASGRP1. Clin. Immunol. 2017, 183, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.; Martin, E.; Boutboul, D.; Lenoir, C.; Boudjemaa, S.; Petit, A.; Picard, C.; Fischer, A.; Leverger, G.; Latour, S. Loss of RASGRP 1 in Humans Impairs T-cell Expansion Leading to Epstein-Barr Virus Susceptibility. EMBO Mol. Med. 2018, 10, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Salzer, E.; Cagdas, D.; Hons, M.; MacE, E.M.; Garncarz, W.; Petronczki, Ö.Y.; Platzer, R.; Pfajfer, L.; Bilic, I.; Ban, S.A.; et al. RASGRP1 Deficiency Causes Immunodeficiency with Impaired Cytoskeletal Dynamics. Nat. Immunol. 2016, 17, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G. XLP: Clinical Features and Molecular Etiology Due to Mutations in SH2D1A Encoding SAP. J. Clin. Immunol. 2014, 34, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Latour, S.; Winter, S. Inherited Immunodeficiencies with High Predisposition to Epstein–Barr Virus-Driven Lymphoproliferative Diseases. Front. Immunol. 2018, 9, 1103. [Google Scholar] [CrossRef]
- Wortzman, M.E.; Clouthier, D.L.; McPherson, A.J.; Lin, G.H.Y.; Watts, T.H. The Contextual Role of TNFR Family Members in CD8+ T-Cell Control of Viral Infections. Immunol. Rev. 2013, 255, 125–148. [Google Scholar] [CrossRef]
- Andreotti, A.H.; Schwartzberg, P.L.; Joseph, R.E.; Berg, L.J. T-Cell Signaling Regulated by the Tec Family Kinase, Itk. Cold Spring Harb. Perspect. Biol. 2010, 2, a002287. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, A.H.; Joseph, R.E.; Conley, J.M.; Iwasa, J.; Berg, L.J.; Carver, R.J. Annual Review of Immunology Multidomain Control over TEC Kinase Activation State Tunes the T Cell Response. Ann. Rev. Immunol. 2018, 36, 549–578. [Google Scholar] [CrossRef]
- Mamontov, P.; Eberwine, R.A.; Perrigoue, J.; Das, A.; Friedman, J.R.; Mora, J.R. A Negative Role for the Interleukin-2-Inducible T-Cell Kinase (ITK) in Human Foxp3+ T REG Differentiation. PLoS ONE 2019, 14, e0215963. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Jeong, A.-R.; Kannan, A.K.; Huang, L.; August, A. IL-2–Inducible T Cell Kinase Tunes T Regulatory Cell Development and Is Required for Suppressive Function. J. Immunol. 2014, 193, 2267–2272. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Rodriguez, J.; Wohlfert, E.A.; Handon, R.; Meylan, F.; Wu, J.Z.; Anderson, S.M.; Kirby, M.R.; Belkaid, Y.; Schwartzberg, P.L. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J. Exp. Med. 2014, 211, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.-M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broussard, C.; Fleischecker, C.; Horai, R.; Chetana, M.; Venegas, A.M.; Sharp, L.L.; Hedrick, S.M.; Fowlkes, B.J.; Schwartzberg, P.L. Altered Development of CD8+ T Cell Lineages in Mice Deficient for the Tec Kinases Itk and Rlk. Immunity 2006, 25, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Huck, K.; Feyen, O.; Niehues, T.; Rüschendorf, F.; Hübner, N.; Laws, H.J.; Telieps, T.; Knapp, S.; Wacker, H.H.; Meindl, A.; et al. Girls Homozygous for an IL-2–Inducible T Cell Kinase Mutation That Leads to Protein Deficiency Develop Fatal EBV-Associated Lymphoproliferation. J. Clin. Investig. 2009, 119, 1350–1358. [Google Scholar] [CrossRef]
- Kapnick, S.M.; Stinchcombe, J.C.; Griffiths, G.M.; Schwartzberg, P.L. Inducible T Cell Kinase Regulates the Acquisition of Cytolytic Capacity and Degranulation in CD8+ CTLs. J. Immunol. 2017, 198, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-S.; Ha, S.; Shin, H.M.; Reboldi, A.; Hall, J.A.; Huh, J.R.; Usherwood, E.J.; Berg, L.J. CD8+ T Cells Require ITK-Mediated TCR Signaling for Migration to the Intestine. Immunohorizons 2020, 4, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Linka, R.M.; Risse, S.L.; Bienemann, K.; Werner, M.; Linka, Y.; Krux, F.; Synaeve, C.; Deenen, R.; Ginzel, S.; Dvorsky, R.; et al. Loss-of-Function Mutations within the IL-2 Inducible Kinase ITK in Patients with EBV-Associated Lymphoproliferative Diseases. Leukemia 2012, 26, 963–971. [Google Scholar] [CrossRef]
- Bienemann, K.; Borkhardt, A.; Klapper, W.; Oschlies, I. High Incidence of Epstein-Barr Virus (EBV)-Positive Hodgkin Lymphoma and Hodgkin Lymphoma-like B-Cell Lymphoproliferations with EBV Latency Profile 2 in Children with Interleukin-2-Inducible T-Cell Kinase Deficiency. Histopathology 2015, 67, 607–616. [Google Scholar] [CrossRef]
- Huang, W.; Lei, X.; Ridder, G.; John Strauss, Y.Z. MTOR and Other Effector Kinase Signals That Impact T Cell Function and Activity Darienne. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Traut, T.W. Physiological Concentrations of Purines and Pyrimidines. Mol. Cell. Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, L.D.; Bofill, M.; Ruckemann, K.; Simmonds, H.A. Importance of Ribonucleotide Availability to Proliferating T-Lymphocytes from Healthy Humans. J. Biol. Chem. 1995, 270, 29682–29689. [Google Scholar] [CrossRef] [Green Version]
- Li, F.-Y.; Lenardo, M.J.; Chaigne-Delalande, B. Loss of MAGT1 Abrogates the Mg2+ Flux Required for T Cell Signaling and Leads to a Novel Human Primary Immunodeficiency. Magnes. Res. 2011, 24, 109–114. [Google Scholar] [CrossRef]
- Li, F.-Y.; Chaigne-Delalande, B.; Kanellopoulou, C.; Davis, J.C.; Matthews, H.F.; Douek, D.C.; Cohen, J.I.; Uzel, G.; Su, H.C.; Lenardo, M.J. Second Messenger Role for Mg2+ Revealed by Human T-Cell Immunodeficiency. Nature 2011, 475, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Y.; Chaigne-Delalande, B.; Su, H.; Uzel, G.; Matthews, H.; Lenardo, M.J. XMEN Disease: A New Primary Immunodeficiency Affecting Mg2+ Regulation of Immunity against Epstein-Barr Virus. Blood 2014, 123, 2148–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, M.; Cao, T.M.; Baker, J.A.; Verneris, M.R.; Soares, L.; Negrin, R.S. Silencing Human NKG2D, DAP10, and DAP12 Reduces Cytotoxicity of Activated CD8+ T Cells and NK Cells. J. Immunol. 2005, 175, 7819–7828. [Google Scholar] [CrossRef] [Green Version]
- Chaigne-Delalande, B.; Li, F.Y.; O’Connor, G.M.; Lukacs, M.J.; Jiang, P.; Zheng, L.; Shatzer, A.; Biancalana, M.; Pittaluga, S.; Matthews, H.F.; et al. Mg2+ Regulates Cytotoxic Functions of NK and CD8 T Cells in Chronic EBV Infection through NKG2D. Science 2013, 341, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Qi, Y.; Im, W. Effects of N-Glycosylation on Protein Conformation and Dynamics: Protein Data Bank Analysis and Molecular Dynamics Simulation Study. Sci. Rep. 2015, 5, 8926. [Google Scholar] [CrossRef]
- Pieters, J.; Müller, P.; Jayachandran, R. On Guard: Coronin Proteins in Innate and Adaptive Immunity. Nat. Rev. Immunol. 2013, 13, 510–518. [Google Scholar] [CrossRef]
- Foger, N. Requirement for Coronin 1 in T Lymphocyte Trafficking and Cellular Homeostasis. Science 2006, 313, 839–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, P.; Massner, J.; Jayachandran, R.; Combaluzier, B.; Albrecht, I.; Gatfield, J.; Blum, C.; Ceredig, R.; Rodewald, H.-R.; Rolink, A.G.; et al. Regulation of T Cell Survival through Coronin-1–Mediated Generation of Inositol-1,4,5-Trisphosphate and Calcium Mobilization after T Cell Receptor Triggering. Nat. Immunol. 2008, 9, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Mueller, P.; Liu, X.; Pieters, J. Migration and Homeostasis of Naive T Cells Depends on Coronin 1-Mediated Prosurvival Signals and Not on Coronin 1-Dependent Filamentous Actin Modulation. J. Immunol. 2011, 186, 4039–4050. [Google Scholar] [CrossRef] [Green Version]
- Mace, E.M.; Orange, J.S. Lytic Immune Synapse Function Requires Filamentous Actin Deconstruction by Coronin 1A. Proc. Natl. Acad. Sci. USA 2014, 111, 6708–6713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.-X.; Zhao, B.; Guan, K.-L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, A.R.M.; Pruenster, M.; Rohwedder, I.; Ramadass, M.; Schäfer, K.; Harrison, U.; Gouveia, G.; Nussbaum, C.; Immler, R.; Wiessner, J.R.; et al. MST1-Dependent Vesicle Trafficking Regulates Neutrophil Transmigration through the Vascular Basement Membrane. J. Clin. Investig. 2016, 126, 4125–4139. [Google Scholar] [CrossRef]
- Kurz, A.R.M.; Catz, S.D.; Sperandio, M. Noncanonical Hippo Signalling in the Regulation of Leukocyte Function. Trends Immunol. 2018, 39, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Du, X.; Shi, H.; Deng, K.; Chi, H.; Tao, W. Mammalian Sterile 20-like Kinase 1 (Mst1) Enhances the Stability of Forkhead Box P3 (Foxp3) and the Function of Regulatory T Cells by Modulating Foxp3 Acetylation. J. Biol. Chem. 2015, 290, 30762–30770. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-S.; Lee, D.-H.; Kim, S.K.; Shin, S.Y.; Seo, E.-J.; Lim, D.-S. Mammalian Sterile 20–like Kinase 1 Suppresses Lymphoma Development by Promoting Faithful Chromosome Segregation. Cancer Res. 2012, 72, 5386–5395. [Google Scholar] [CrossRef] [Green Version]
- Stark, B.C.; Lanier, M.H.; Cooper, J.A. CARMIL Family Proteins as Multidomain Regulators of Actin-Based Motility. Mol. Biol. Cell 2017, 28, 1713–1723. [Google Scholar] [CrossRef]
- Sayos, J.; Wu, C.; Morra, M.; Wang, N.; Zhang, X.; Allen, D.; van Schaik, S.; Notarangelo, L.; Geha, R.; Roncarolo, M.G.; et al. The X-Linked Lymphoproliferative-Disease Gene Product SAP Regulates Signals Induced through the Co-Receptor SLAM. Nature 1998, 395, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Coffey, A.J.; Brooksbank, R.A.; Brandau, O.; Oohashi, T.; Howell, G.R.; Bye, J.M.; Cahn, A.P.; Durham, J.; Heath, P.; Wray, P.; et al. Host Response to EBV Infection in X-Linked Lymphoproliferative Disease Results from Mutations in an SH2-Domain Encoding Gene. Nat. Genet. 1998, 20, 129–135. [Google Scholar] [CrossRef]
- Nichols, K.E.; Harkin, D.P.; Levitz, S.; Krainer, M.; Kolquist, K.A.; Genovese, C.; Bernard, A.; Ferguson, M.; Zuo, L.; Snyder, E.; et al. Inactivating Mutations in an SH2 Domain-Encoding Gene in X-Linked Lymphoproliferative Syndrome. Proc. Natl. Acad. Sci. USA 1998, 95, 13765–13770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.S.; Nichols, K.E.; Tangye, S.G. Regulation of Cellular and Humoral Immune Responses by the SLAM and SAP Families of Molecules. Annu. Rev. Immunol. 2007, 25, 337–379. [Google Scholar] [CrossRef] [PubMed]
- Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L. SLAM Family Receptors and SAP Adaptors in Immunity. Annu. Rev. Immunol. 2011, 29, 665–705. [Google Scholar] [CrossRef] [PubMed]
- Palendira, U.; Low, C.; Chan, A.; Hislop, A.D.; Ho, E.; Phan, T.G.; Deenick, E.; Cook, M.C.; Riminton, D.S.; Choo, S.; et al. Molecular Pathogenesis of EBV Susceptibility in XLP as Revealed by Analysis of Female Carriers with Heterozygous Expression of SAP. PLoS Biol. 2011, 9, e1001187. [Google Scholar] [CrossRef] [PubMed]
- Parolini, S.; Bottino, C.; Falco, M.; Augugliaro, R.; Giliani, S.; Franceschini, R.; Ochs, H.D.; Wolf, H.; Bonnefoy, J.-Y.; Biassoni, R.; et al. X-Linked Lymphoproliferative Disease. J. Exp. Med. 2000, 192, 337–346. [Google Scholar] [CrossRef]
- Dupré, L.; Andolfi, G.; Tangye, S.G.; Clementi, R.; Locatelli, F.; Aricò, M.; Aiuti, A.; Roncarolo, M.-G. SAP Controls the Cytolytic Activity of CD8+ T Cells against EBV-Infected Cells. Blood 2005, 105, 4383–4389. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Palendira, U.; Leese, A.M.; Arkwright, P.D.; Rohrlich, P.S.; Tangye, S.G.; Gaspar, H.B.; Lankester, A.C.; Moretta, A.; Rickinson, A.B. Impaired Epstein-Barr Virus-Specific CD8+ T-Cell Function in X-Linked Lymphoproliferative Disease Is Restricted to SLAM Family-Positive B-Cell Targets. Blood 2010, 116, 3249–3257. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Al-Alem, U.; Liang, J.; Tong, W.-M.; Li, C.; Badiali, M.; Médard, J.J.; Sumegi, J.; Wang, Z.-Q.; Romeo, G. Mice Deficient in the X-Linked Lymphoproliferative Disease Gene Sap Exhibit Increased Susceptibility to Murine Gammaherpesvirus-68 and Hypo-Gammaglobulinemia. J. Med. Virol. 2003, 71, 446–455. [Google Scholar] [CrossRef]
- Chen, G.; Tai, A.K.; Lin, M.; Chang, F.; Terhorst, C.; Huber, B.T. Signaling Lymphocyte Activation Molecule-Associated Protein Is a Negative Regulator of the CD8 T Cell Response in Mice. J. Immunol. 2005, 175, 2212–2218. [Google Scholar] [CrossRef] [Green Version]
- Rigaud, S.; Fondanèche, M.C.; Lambert, N.; Pasquier, B.; Mateo, V.; Soulas, P.; Galicier, L.; le Deist, F.; Rieux-Laucat, F.; Revy, P.; et al. XIAP Deficiency in Humans Causes an X-Linked Lymphoproliferative Syndrome. Nature 2006, 444, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.P.; Canioni, D.; Moshous, D.; Touzot, F.; Mahlaoui, N.; Hauck, F.; Kanegane, H.; Lopez-Granados, E.; Mejstrikova, E.; Pellier, I.; et al. Clinical Similarities and Differences of Patients with X-Linked Lymphoproliferative Syndrome Type 1 (XLP-1/SAP Deficiency) versus Type 2 (XLP-2/XIAP Deficiency). Blood 2011, 117, 1522–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speckmann, C.; Lehmberg, K.; Albert, M.H.; Damgaard, R.B.; Fritsch, M.; Gyrd-Hansen, M.; Rensing-Ehl, A.; Vraetz, T.; Grimbacher, B.; Salzer, U.; et al. X-Linked Inhibitor of Apoptosis (XIAP) Deficiency: The Spectrum of Presenting Manifestations beyond Hemophagocytic Lymphohistiocytosis. Clin. Immunol. 2013, 149, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 283, 702–703. [Google Scholar] [CrossRef]
- Fujiwara, S.; Nakamura, H. Chronic Active Epstein–Barr Virus Infection: Is It Immunodeficiency, Malignancy, or Both? Cancers 2020, 12, 3202. [Google Scholar] [CrossRef]
- Meyts, I.; Bosch, B.; Bolze, A.; Boisson, B.; Itan, Y.; Belkadi, A.; Pedergnana, V.; Moens, L.; Picard, C.; Cobat, A.; et al. Exome and Genome Sequencing for Inborn Errors of Immunity. J. Allergy Clin. Immunol. 2016, 138, 957–969. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).