
Adoptive NK Cell Therapy: A Promising Treatment Prospect for Metastatic Melanoma

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
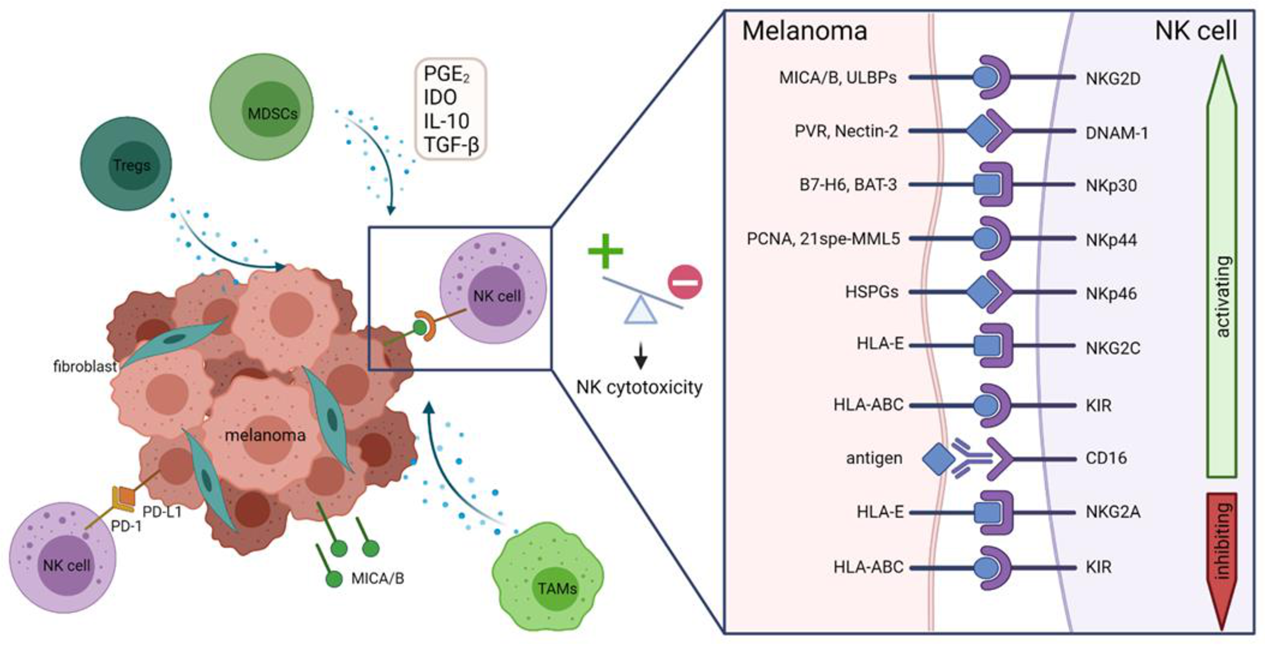
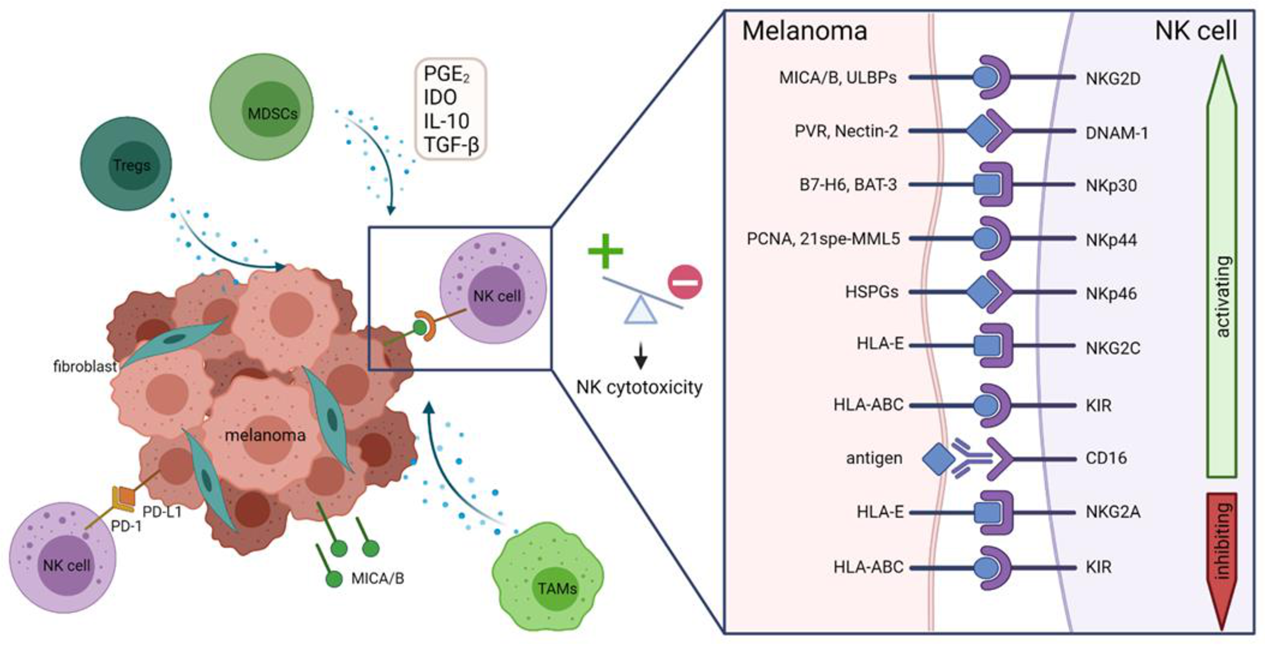
2. Dysregulation of the Immune System by Melanoma
3. Developments in ACT for Melanoma
4. Natural Killer Cells and Receptors Involved in Tumor Surveillance
5. The Possible Role of NK Cells in Immune Control of Melanoma
NK Cells as a Prognostic Factor
6. NK Cell Deficiency and Immunosuppression in Melanoma
6.1. Downregulation of Activating Receptors on NK Cells
6.2. Shedding or Downregulation of Activating Ligands on Melanoma Cells
6.3. The Suppressive Tumor Microenvironment
7. Clinical Exploration of Adoptive NK Cell Therapy for Melanoma
7.1. Autologous NK Cell Therapy
7.2. Allogeneic NK Cell Therapy
7.3. NK Cell Line Therapy
7.4. Ongoing Clinical Trials
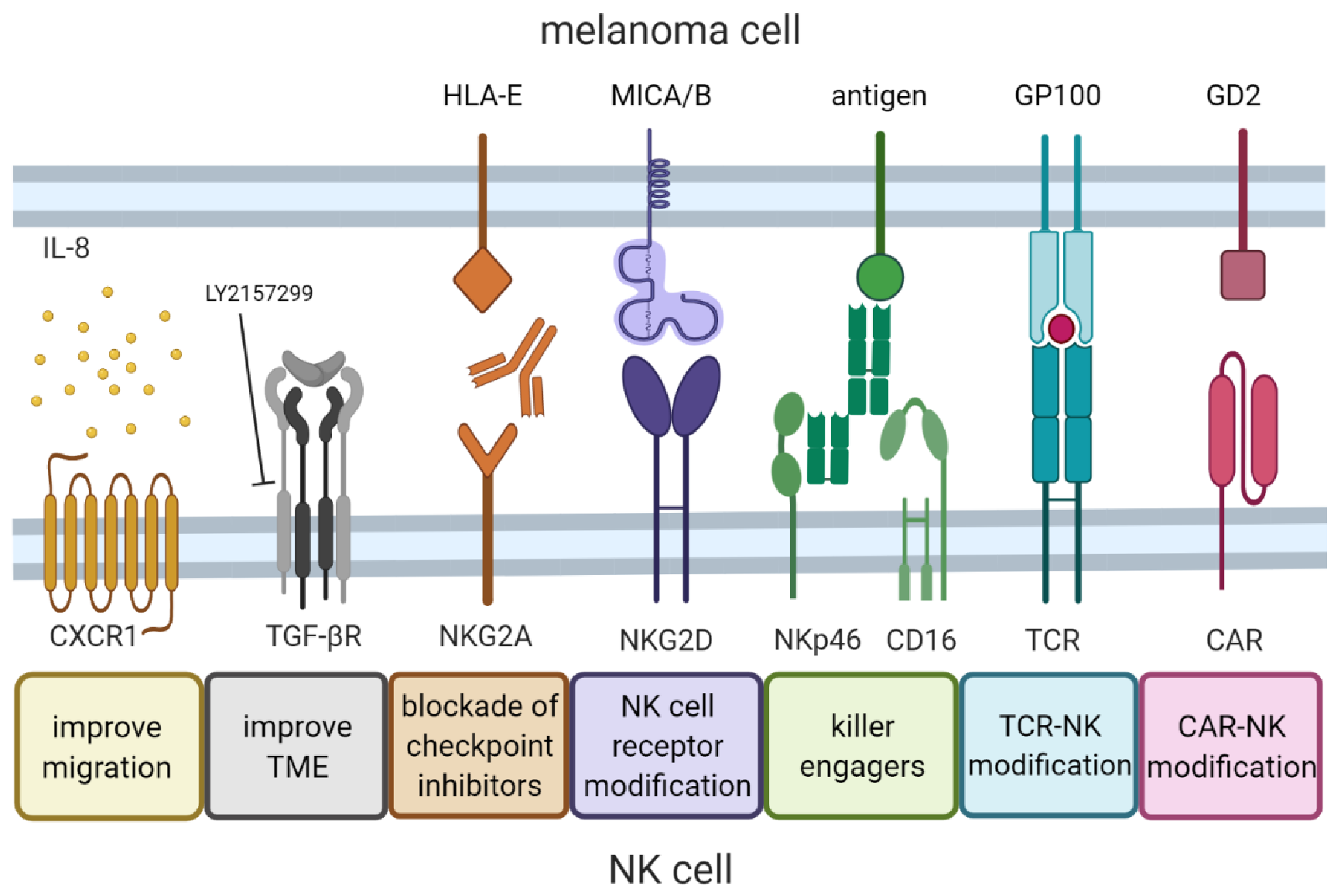
8. Future Developments in NK Cell Therapy for Melanoma
8.1. Migration to the Tumor Site
8.2. Inhibition of Immunosuppression in the Tumor Microenvironment
8.3. Killer Engagers
8.4. NK Cell Receptor Modification
8.5. CAR-NK Cells
8.6. TCR-NK Cells
8.7. Immune Checkpoint Inhibitors
8.8. Use of Nanoparticles
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Erdmann, F.; Lortet-Tieulent, J.; Schuz, J.; Zeeb, H.; Greinert, R.; Breitbart, E.W.; Bray, F. International trends in the incidence of malignant melanoma 1953-2008--are recent generations at higher or lower risk? Int. J. Cancer 2013, 132, 385–400. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Agarwala, S.S. Cutaneous melanoma: Available therapy for metastatic disease. Dermatol. Ther. 2006, 19, 19–25. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The First Effective Immunotherapy for Human Cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Azijli, K.; Stelloo, E.; Peters, G.J.; Eertwegh, A.J.M.V.D. New developments in the treatment of metastatic melanoma: Immune checkpoint inhibitors and targeted therapies. Anticancer. Res. 2014, 34, 1493–1505. [Google Scholar]
- Hazarika, M.; Chuk, M.K.; Theoret, M.R.; Mushti, S.; He, K.; Weis, S.L.; Putman, A.H.; Helms, W.S.; Cao, X.; Li, H.; et al. U.S. FDA Approval Summary: Nivolumab for Treatment of Unresectable or Metastatic Melanoma Following Progression on Ipilimumab. Clin. Cancer Res. 2017, 23, 3484–3488. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; McKee, A.E.; Ning, Y.-M.; Hazarika, M.; Theoret, M.; Johnson, J.R.; Xu, Q.C.; Tang, S.; Sridhara, R.; Jiang, X.; et al. FDA Approval Summary: Vemurafenib for Treatment of Unresectable or Metastatic Melanoma with the BRAFV600E Mutation. Clin. Cancer Res. 2014, 20, 4994–5000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuk, M.K.; Chang, J.T.; Theoret, M.R.; Sampene, E.; He, K.; Weis, S.L.; Helms, W.S.; Jin, R.; Li, H.; Yu, J.; et al. FDA Approval Summary: Accelerated Approval of Pembrolizumab for Second-Line Treatment of Metastatic Melanoma. Clin. Cancer Res. 2017, 23, 5666–5670. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Hershkovitz, L.; Levy, D.; Kubi, A.; Hovav, E.; Chermoshniuk, N.; et al. Clinical Responses in a Phase II Study Using Adoptive Transfer of Short-term Cultured Tumor Infiltration Lymphocytes in Metastatic Melanoma Patients. Clin. Cancer Res. 2010, 16, 2646–2655. [Google Scholar] [CrossRef] [Green Version]
- Golchin, A.; Farahany, T.Z. Biological Products: Cellular Therapy and FDA Approved Products. Stem Cell Rev. Rep. 2019, 15, 166–175. [Google Scholar] [CrossRef]
- Chhabra, N.; Kennedy, J. A Review of Cancer Immunotherapy Toxicity II: Adoptive Cellular Therapies, Kinase Inhibitors, Monoclonal Antibodies, and Oncolytic Viruses. J. Med. Toxicol. 2021, 1–13. [Google Scholar] [CrossRef]
- Méndez, R.; Aptsiauri, N.; Del Campo, A.; Maleno, I.; Cabrera, T.; Ruiz-Cabello, F.; Garrido, F.; Garcia-Lora, A. HLA and melanoma: Multiple alterations in HLA class I and II expression in human melanoma cell lines from ESTDAB cell bank. Cancer Immunol. Immunother. 2009, 58, 1507–1515. [Google Scholar] [CrossRef]
- Kärre, K. NK Cells, MHC Class I Molecules and the Missing Self. Scand. J. Immunol. 2002, 55, 221–228. [Google Scholar] [CrossRef]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Investig. 2009, 119, 1251–1263. [Google Scholar] [CrossRef] [Green Version]
- Pietra, G.; Manzini, C.; Vitale, M.; Balsamo, M.; Ognio, E.; Boitano, M.; Queirolo, P.; Moretta, L.; Mingari, M.C. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int. Immunol. 2009, 21, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Stehlin, J.S. Spontaneous regression of primary malignant melanomas with regional metastases. Cancer 1965, 18, 1399–1415. [Google Scholar] [CrossRef]
- Mackensen, A.; Ferradini, L.; Carcelain, G.; Triebel, F.; Faure, F.; Viel, S.; Hercend, T. Evidence for in situ amplification of cytotoxic T-lymphocytes with antitumor activity in a human regressive melanoma. Cancer Res. 1993, 53, 3569–3573. [Google Scholar]
- Halliday, G.M.; Patel, A.; Hunt, M.J.; Tefany, F.J.; Barnetson, R.S.C. Spontaneous regression of human melanoma/nonmelanoma skin cancer: Association with infiltrating CD4+ T cells. World J. Surg. 1995, 19, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Clemente, C.G.; Mihm, M.C., Jr.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Muul, L.M.; Spiess, P.J.; Director, E.P.; A Rosenberg, S. Identification of specific cytolytic immune responses against autologous tumor in humans bearing malignant melanoma. J. Immunol. 1987, 138, 989–995. [Google Scholar]
- Topalian, S.L.; Solomon, D.; A Rosenberg, S. Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J. Immunol. 1989, 142, 3714–3725. [Google Scholar]
- Kalialis, L.V.; Drzewiecki, K.T.; Klyver, H. Spontaneous regression of metastases from melanoma: Review of the literature. Melanoma Res. 2009, 19, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef] [PubMed]
- Cesana, G.C.; DeRaffele, G.; Cohen, S.; Moroziewicz, D.; Mitcham, J.; Stoutenburg, J.; Cheung, K.; Hesdorffer, C.; Kim-Schulze, S.; Kaufman, H.L. Characterization of CD4+CD25+ Regulatory T Cells in Patients Treated With High-Dose Interleukin-2 for Metastatic Melanoma or Renal Cell Carcinoma. J. Clin. Oncol. 2006, 24, 1169–1177. [Google Scholar] [CrossRef]
- Correll, A.; Tuettenberg, A.; Becker, C.; Jonuleit, H. Increased regulatory T-cell frequencies in patients with advanced melanoma correlate with a generally impaired T-cell responsiveness and are restored after dendritic cell-based vaccination. Exp. Dermatol. 2009, 19, e213–e221. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Inozume, T.; Yaguchi, T.; Furuta, J.; Harada, K.; Kawakami, Y.; Shimada, S. Melanoma Cells Control Antimelanoma CTL Responses via Interaction between TIGIT and CD155 in the Effector Phase. J. Investig. Dermatol. 2016, 136, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of Patients With Metastatic Melanoma With Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Dudley, M.E.; Wunderlich, J.; El-Gamil, M.; Li, Y.F.; Zhou, J.; Huang, J.; Powell, J., Jr.; Rosenberg, S.A. Cutting edge: Persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004, 173, 7125–7130. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of Tumor-Infiltrating Lymphocyte Cultures for Use in Adoptive Transfer Therapy for Melanoma Patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef]
- Kageshita, T.; Ishihara, T.; Campoli, M.; Ferrone, S. Selective monomorphic and polymorphic HLA class I antigenic determinant loss in surgically removed melanoma lesions. Tissue Antigens 2005, 65, 419–428. [Google Scholar] [CrossRef]
- Zhang, H.; Ye, Z.-L.; Yuan, Z.-G.; Luo, Z.-Q.; Jin, H.-J.; Zheng-Qiang, L. New Strategies for the Treatment of Solid Tumors with CAR-T Cells. Int. J. Biol. Sci. 2016, 12, 718–729. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef]
- Upshaw, J.L.; Arneson, L.N.; A Schoon, R.; Dick, C.J.; Billadeau, D.D.; Leibson, P.J. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 2006, 7, 524–532. [Google Scholar] [CrossRef]
- Krzewski, K.; Coligan, J. Human NK cell lytic granules and regulation of their exocytosis. Front. Immunol. 2012, 3, 335. [Google Scholar] [CrossRef] [Green Version]
- Billadeau, D.D.; Upshaw, J.L.; A Schoon, R.; Dick, C.J.; Leibson, P.J. NKG2D-DAP10 triggers human NK cell–mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Pillars Article: Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science. 1999. 285: 727-729. J. Immunol. 2018, 200, 2231–2233. [Google Scholar]
- Vetter, C.S.; Groh, V.; Straten, P.T.; Spies, T.; Bröcker, E.-B.; Becker, J.C. Expression of Stress-induced MHC Class I Related Chain Molecules on Human Melanoma. J. Investig. Dermatol. 2002, 118, 600–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groh, V.; Rhinehart, R.; Secrist, H.; Bauer, S.; Grabstein, K.H.; Spies, T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc. Natl. Acad. Sci. USA 1999, 96, 6879–6884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pende, D.; Rivera, P.; Marcenaro, S.; Chang, C.-C.; Biassoni, R.; Conte, R.; Kubin, M.; Cosman, D.; Ferrone, S.; Moretta, L.; et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: Analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002, 62, 6178–6186. [Google Scholar]
- Raulet, D.H.; Gasser, S.; Gowen, B.; Deng, W.; Jung, H. Regulation of Ligands for the NKG2D Activating Receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, A.; Campbell, D.; Hannum, C.; Yssel, H.; Franz-Bacon, K.; McClanahan, T.; Kitamura, T.; Nicholl, J.; Sutherland, G.R.; Lanier, L.L.; et al. DNAM-1, A Novel Adhesion Molecule Involved in the Cytolytic Function of T Lymphocytes. Immunity 1996, 4, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Tahara-Hanaoka, S.; Shibuya, K.; Onoda, Y.; Zhang, H.; Yamazaki, S.; Miyamoto, A.; Honda, S.; Lanier, L.L.; Shibuya, A. Functional characterization of DNAM-1 (CD226) interaction with its ligands PVR (CD155) and nectin-2 (PRR-2/CD112). Int. Immunol. 2004, 16, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Long, E.O. Complementary Phosphorylation Sites in the Adaptor Protein SLP-76 Promote Synergistic Activation of Natural Killer Cells. Sci. Signal. 2012, 5, ra49. [Google Scholar] [CrossRef] [Green Version]
- Moretta, A.; Biassoni, R.; Bottino, C.; Mingari, M.C.; Moretta, L. Natural cytotoxicity receptors that trigger human NK-cell-mediated cytolysis. Immunol. Today 2000, 21, 228–234. [Google Scholar] [CrossRef]
- Rosental, B.; Brusilovsky, M.; Hadad, U.; Oz, D.; Appel, M.; Afergan, F.; Yossef, R.; Rosenberg, L.A.; Aharoni, A.; Cerwenka, A.; et al. Proliferating Cell Nuclear Antigen Is a Novel Inhibitory Ligand for the Natural Cytotoxicity Receptor NKp44. J. Immunol. 2011, 187, 5693–5702. [Google Scholar] [CrossRef]
- Binici, J.; Hartmann, J.; Herrmann, J.; Schreiber, C.; Beyer, S.; Güler, G.; Vogel, V.; Tumulka, F.; Abele, R.; Mäntele, W.; et al. A Soluble Fragment of the Tumor Antigen BCL2-associated Athanogene 6 (BAG-6) Is Essential and Sufficient for Inhibition of NKp30 Receptor-dependent Cytotoxicity of Natural Killer Cells. J. Biol. Chem. 2013, 288, 34295–34303. [Google Scholar] [CrossRef] [Green Version]
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a Novel Triggering Surface Molecule Specifically Expressed by Activated Natural Killer Cells, Is Involved in Non–Major Histocompatibility Complex–restricted Tumor Cell Lysis. J. Exp. Med. 1998, 187, 2065–2072. [Google Scholar] [CrossRef]
- Sivori, S.; Vitale, M.; Morelli, L.; Sanseverino, L.; Augugliaro, R.; Bottino, C.; Moretta, L.; Moretta, A. p46, a Novel Natural Killer Cell–specific Surface Molecule That Mediates Cell Activation. J. Exp. Med. 1997, 186, 1129–1136. [Google Scholar] [CrossRef] [Green Version]
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and Molecular Characterization of Nkp30, a Novel Triggering Receptor Involved in Natural Cytotoxicity Mediated by Human Natural Killer Cells. J. Exp. Med. 1999, 190, 1505–1516. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, D.; Churov, A.; Fu, R. Research Progress on NK Cell Receptors and Their Signaling Pathways. Mediat. Inflamm. 2020, 2020, 1–14. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Q.; Mariuzza, R.A. Structure of the human activating natural cytotoxicity receptor NKp30 bound to its tumor cell ligand B7-H6. J. Exp. Med. 2011, 208, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Von Strandmann, E.P.; Simhadri, V.R.; Von Tresckow, B.; Sasse, S.; Reiners, K.S.; Hansen, H.P.; Rothe, A.; Borchmann, P.; McKinnon, P.J.; Hallek, M.; et al. Tumor Cell-Derived HLA-B-Associated Transkript 3 (BAT3) Is a Ligand for NKp30 and Activates NK Cells. Blood 2006, 108, 643. [Google Scholar] [CrossRef]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Strandmann, E.P.; Simhadri, V.R.; von Tresckow, B.; Sasse, S.; Reiners, K.S.; Hansen, H.P.; Rothe, A.; Böll, B.; Simhadri, V.L.; Borchmann, P.; et al. Human Leukocyte Antigen-B-Associated Transcript 3 Is Released from Tumor Cells and Engages the NKp30 Receptor on Natural Killer Cells. Immunity 2007, 27, 965–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baychelier, F.; Sennepin, A.; Ermonval, M.; Dorgham, K.; Debré, P.; Vieillard, V. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood 2013, 122, 2935–2942. [Google Scholar] [CrossRef] [Green Version]
- Gaggero, S.; Bruschi, M.; Petretto, A.; Parodi, M.; Del Zotto, G.; Lavarello, C.; Prato, C.; Santucci, L.; Barbuto, A.; Bottino, C.; et al. Nidogen-1 is a novel extracellular ligand for the NKp44 activating receptor. OncoImmunology 2018, 7, e1470730. [Google Scholar] [CrossRef]
- Niehrs, A.; Garcia-Beltran, W.F.; Norman, P.J.; Watson, G.M.; Hölzemer, A.; Chapel, A.; Richert, L.; Pommerening-Röser, A.; Körner, C.; Ozawa, M.; et al. A subset of HLA-DP molecules serve as ligands for the natural cytotoxicity receptor NKp44. Nat. Immunol. 2019, 20, 1129–1137. [Google Scholar] [CrossRef]
- Hecht, M.-L.; Rosental, B.; Horlacher, T.; Hershkovitz, O.; de Paz, J.L.; Noti, C.; Schauer, S.; Porgador, A.; Seeberger, P.H. Natural Cytotoxicity Receptors NKp30, NKp44 and NKp46 Bind to Different Heparan Sulfate/Heparin Sequences. J. Proteome Res. 2009, 8, 712–720. [Google Scholar] [CrossRef]
- Joncker, N.T.; Raulet, D.H. Regulation of NK cell responsiveness to achieve self-tolerance and maximal responses to diseased target cells. Immunol. Rev. 2008, 224, 85–97. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.J.; O’Callaghan, C.A.; Söderström, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Binstadt, B.; Billadeau, D.D.; Jevremovic, D.; Williams, B.L.; Fang, N.; Yi, T.; Koretzky, G.A.; Abraham, R.T.; Leibson, P.J. SLP-76 Is a Direct Substrate of SHP-1 Recruited to Killer Cell Inhibitory Receptors. J. Biol. Chem. 1998, 273, 27518–27523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebbins, C.C.; Watzl, C.; Billadeau, D.D.; Leibson, P.J.; Burshtyn, D.N.; Long, E.O. Vav1 Dephosphorylation by the Tyrosine Phosphatase SHP-1 as a Mechanism for Inhibition of Cellular Cytotoxicity. Mol. Cell. Biol. 2003, 23, 6291–6299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanier, L.L.; Corliss, B.; Wu, J.; Phillips, J.H. Association of DAP12 with Activating CD94/NKG2C NK Cell Receptors. Immunity 1998, 8, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Faure, M.; Long, E.O. KIR2DL4 (CD158d), an NK Cell-Activating Receptor with Inhibitory Potential. J. Immunol. 2002, 168, 6208–6214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watzl, C.; Long, E. Exposing tumor cells to killer cell attack. Nat. Med. 2000, 6, 867–868. [Google Scholar] [CrossRef]
- Lanier, L.L.; Yu, G.; Phillips, J.H. Analysis of Fc gamma RIII (CD16) membrane expression and association with CD3 zeta and Fc epsilon RI-gamma by site-directed mutation. J. Immunol. 1991, 146, 1571–1576. [Google Scholar]
- Bryceson, Y.; March, M.; Ljunggren, H.-G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Vitale, M.; Falco, M.; Castriconi, R.; Parolini, S.; Zambello, R.; Semenzato, G.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Identification of NKp80, a novel triggering molecule expressed by human NK cells. Eur. J. Immunol. 2001, 31, 233–242. [Google Scholar] [CrossRef]
- Akatsuka, A.; Ito, M.; Yamauchi, C.; Ochiai, A.; Yamamoto, K.; Matsumoto, N. Tumor cells of non-hematopoietic and hematopoietic origins express activation-induced C-type lectin, the ligand for killer cell lectin-like receptor F1. Int. Immunol. 2010, 22, 783–790. [Google Scholar] [CrossRef]
- Dennehy, K.M.; Klimosch, S.N.; Steinle, A.; Tsuji, T.; Matsuzaki, J.; Kelly, M.P.; Ramakrishna, V.; Vitale, L.; He, L.-Z.; Keler, T.; et al. Cutting Edge: NKp80 Uses an Atypical Hemi-ITAM To Trigger NK Cytotoxicity. J. Immunol. 2010, 186, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Eissmann, P.; Beauchamp, L.; Wooters, J.; Tilton, J.C.; Long, E.O.; Watzl, C. Molecular basis for positive and negative signaling by the natural killer cell receptor 2B4 (CD244). Blood 2005, 105, 4722–4729. [Google Scholar] [CrossRef] [Green Version]
- Flaig, R.M.; Stark, S.; Watzl, C. Cutting edge: NTB-A activates NK cells via homophilic interaction. J. Immunol. 2004, 172, 6524–6527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, S.; Watzl, C. 2B4 (CD244), NTB-A and CRACC (CS1) stimulate cytotoxicity but no proliferation in human NK cells. Int. Immunol. 2006, 18, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Ebert, L.M.; Meuter, S.; Moser, B. Homing and function of human skin gammadelta T cells and NK cells: Relevance for tumor surveillance. J. Immunol. 2006, 176, 4331–4336. [Google Scholar] [CrossRef] [PubMed]
- Luci, C.; Reynders, A.; Ivanov, I.I.; Cognet, C.; Chiche, L.; Chasson, L.; Hardwigsen, J.; Anguiano, E.; Banchereau, J.; Chaussabel, D.; et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat. Immunol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- de Andrade, L.F.; Lu, Y.; Luoma, A.; Ito, Y.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Yuan, G.-C.; Wucherpfennig, K.W. Discovery of specialized NK cell populations infiltrating human melanoma metastases. JCI Insight 2019, 4, e133103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casado, J.G.; Pawelec, G.; Morgado, S.; Sanchez-Correa, B.; Delgado, E.; Gayoso, I.; Duran, E.; Solana, R.; Tarazona, R. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol. Immunother. 2009, 58, 1517–1526. [Google Scholar] [CrossRef]
- Cagnano, E.; Hershkovitz, O.; Zilka, A.; Bar-Ilan, A.; Golder, A.; Sion-Vardy, N.; Bogdanov-Berezovsky, A.; Mandelboim, O.; Benharroch, D.; Porgador, A. Expression of Ligands to NKp46 in Benign and Malignant Melanocytes. J. Investig. Dermatol. 2008, 128, 972–979. [Google Scholar] [CrossRef] [Green Version]
- Byrd, A.; Hoffmann, S.C.; Jarahian, M.; Momburg, F.; Watzl, C. Expression Analysis of the Ligands for the Natural Killer Cell Receptors NKp30 and NKp44. PLoS ONE 2007, 2, e1339. [Google Scholar] [CrossRef]
- De Jonge, K.; Ebering, A.; Nassiri, S.; Hajjami, H.M.-E.; Ouertatani-Sakouhi, H.; Baumgaertner, P.; Speiser, D.E. Circulating CD56bright NK cells inversely correlate with survival of melanoma patients. Sci. Rep. 2019, 9, 4487. [Google Scholar] [CrossRef] [Green Version]
- Fregni, G.; Messaoudene, M.; Fourmentraux-Neves, E.; Mazouz-Dorval, S.; Chanal, J.; Maubec, E.; Marinho, E.; Scheer-Senyarich, I.; Cremer, I.; Avril, M.-F.; et al. Phenotypic and Functional Characteristics of Blood Natural Killer Cells from Melanoma Patients at Different Clinical Stages. PLoS ONE 2013, 8, e76928. [Google Scholar] [CrossRef]
- de Coaña, Y.P.; Wolodarski, M.; Àvila, I.V.D.H.; Nakajima, T.; Rentouli, S.; Lundqvist, A.; Masucci, G.; Hansson, J.; Kiessling, R. PD-1 checkpoint blockade in advanced melanoma patients: NK cells, monocytic subsets and host PD-L1 expression as predictive biomarker candidates. OncoImmunology 2020, 9, 1786888. [Google Scholar] [CrossRef]
- Cursons, J.; Souza-Fonseca-Guimaraes, F.; Foroutan, M.; Anderson, A.; Hollande, F.; Hediyeh-Zadeh, S.; Behren, A.; Huntington, N.D.; Davis, M.J. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol. Res. 2019, 7, 1162–1174. [Google Scholar] [CrossRef] [Green Version]
- Hersey, P.; Edwards, A.; McCarthy, W.H. Tumour-related changes in natural killer cell activity in melanoma patients. Influence of stage of disease, tumour thickness and age of patients. Int. J. Cancer 1980, 25, 187–194. [Google Scholar] [CrossRef]
- Konjević, G.; Martinović, K.M.; Vuletić, A.; Jović, V.; Jurisic, V.; Babović, N.; Spužić, I. Low expression of CD161 and NKG2D activating NK receptor is associated with impaired NK cell cytotoxicity in metastatic melanoma patients. Clin. Exp. Metastasis 2007, 24, 1–11. [Google Scholar] [CrossRef]
- Konjevic, G.; Martinović, K.M.; Jurišić, V.; Babović, N.; Spužić, I. Biomarkers of suppressed natural killer (NK) cell function in metastatic melanoma: Decreased NKG2D and increased CD158a receptors on CD3–CD16+ NK cells. Biomarkers 2009, 14, 258–270. [Google Scholar] [CrossRef]
- Pietra, G.; Manzini, C.; Rivara, S.; Vitale, M.; Cantoni, C.; Petretto, A.; Balsamo, M.; Conte, R.; Benelli, R.; Minghelli, S.; et al. Melanoma Cells Inhibit Natural Killer Cell Function by Modulating the Expression of Activating Receptors and Cytolytic Activity. Cancer Res. 2012, 72, 1407–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-Cell Exhaustion in Advanced Melanoma by Tim-3 Blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschen, A.; Sucker, A.; Hill, B.; Moll, I.; Zapatka, M.; Nguyen, X.D.; Sim, G.C.; Gutmann, I.; Hassel, J.; Becker, J.C.; et al. Differential Clinical Significance of Individual NKG2D Ligands in Melanoma: Soluble ULBP2 as an Indicator of Poor Prognosis Superior to S100B. Clin. Cancer Res. 2009, 15, 5208–5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlecker, E.; Fiegler, N.; Arnold, A.; Altevogt, P.; Rose-John, S.; Moldenhauer, G.; Sucker, A.; Paschen, A.; von Strandmann, E.P.; Textor, S.; et al. Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell-activating receptor NKp30. Cancer Res. 2014, 74, 3429–3440. [Google Scholar] [CrossRef] [Green Version]
- De Andrade, L.F.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B., Jr.; Harvey, C.J.; Kobold, S.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell–driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskus, W.; Mitsiades, N. B-Raf Inhibition in the Clinic: Present and Future. Annu. Rev. Med. 2016, 67, 29–43. [Google Scholar] [CrossRef]
- López-Cobo, S.; Pieper, N.; Campos-Silva, C.; García-Cuesta, E.M.; Reyburn, H.T.; Paschen, A.; Valés-Gómez, M. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. OncoImmunology 2017, 7, e1392426. [Google Scholar] [CrossRef]
- Murray, S.; Lundqvist, A. Targeting the tumor microenvironment to improve natural killer cell-based immunotherapies: On being in the right place at the right time, with resilience. Hum. Vaccines Immunother. 2015, 12, 607–611. [Google Scholar] [CrossRef] [Green Version]
- Ziani, L.; Ben Safta-Saadoun, T.; Gourbeix, J.; Cavalcanti, A.; Robert, C.; Favre, G.; Chouaib, S.; Thiery, J. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget 2017, 8, 19780–19794. [Google Scholar] [CrossRef] [Green Version]
- Balsamo, M.; Scordamaglia, F.; Pietra, G.; Manzini, C.; Cantoni, C.; Boitano, M.; Queirolo, P.; Vermi, W.; Facchetti, F.; Moretta, A.; et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 20847–20852. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Diergaarde, B.; Ferrone, S.; Kirkwood, J.M.; Whiteside, T.L. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Kok, N.; van der Vliet, H.; Verheul, H.; De Gruijl, T.D.; Spanholtz, J. The Rise of Allogeneic Natural Killer Cells As a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front. Immunol. 2017, 8, 631. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Riley, J.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive Transfer of Autologous Natural Killer Cells Leads to High Levels of Circulating Natural Killer Cells but Does Not Mediate Tumor Regression. Clin. Cancer Res. 2011, 17, 6287–6297. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; DeFor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Wu, Z.; Niu, L.; Chen, J.; Ma, Y.; Zhang, M. Preparation of highly activated natural killer cells for advanced lung cancer therapy. OncoTargets Ther. 2019, 12, 5077–5086. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2020, 18, 85–100. [Google Scholar] [CrossRef]
- Hallett, W.H.; Ames, E.; Motarjemi, M.; Barao, I.; Shanker, A.; Tamang, D.L.; Sayers, T.J.; Hudig, D.; Murphy, W.J. Sensitization of Tumor Cells to NK Cell-Mediated Killing by Proteasome Inhibition. J. Immunol. 2007, 180, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Mgrditchian, T.; Arakelian, T.; Paggetti, J.; Noman, M.Z.; Viry, E.; Moussay, E.; Van Moer, K.; Kreis, S.; Guerin, C.; Buart, S.; et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc. Natl. Acad. Sci. USA 2017, 114, E9271–E9279. [Google Scholar] [CrossRef] [Green Version]
- Wennerberg, E.; Kremer, V.; Childs, R.; Lundqvist, A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol. Immunother. 2014, 64, 225–235. [Google Scholar] [CrossRef]
- Ng, Y.Y.; Tay, J.C.; Wang, S. CXCR1 Expression to Improve Anti-Cancer Efficacy of Intravenously Injected CAR-NK Cells in Mice with Peritoneal Xenografts. Mol. Ther. Oncolytics 2019, 16, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Otegbeye, F.; Ojo, E.; Moreton, S.; Mackowski, N.; Lee, D.A.; de Lima, M.; Wald, D.N. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE 2018, 13, e0191358. [Google Scholar]
- Pekar, L.; Klausz, K.; Busch, M.; Valldorf, B.; Kolmar, H.; Wesch, D.; Oberg, H.-H.; Krohn, S.; Boje, A.S.; Gehlert, C.L.; et al. Affinity Maturation of B7-H6 Translates into Enhanced NK Cell–Mediated Tumor Cell Lysis and Improved Proinflammatory Cytokine Release of Bispecific Immunoligands via NKp30 Engagement. J. Immunol. 2020, 206, 225–236. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef] [PubMed]
- Sayitoglu, E.C.; Georgoudaki, A.-M.; Chrobok, M.; Ozkazanc, D.; Josey, B.J.; Arif, M.; Kusser, K.; Hartman, M.; Chinn, T.M.; Potens, R.; et al. Boosting Natural Killer Cell-Mediated Targeting of Sarcoma Through DNAM-1 and NKG2D. Front. Immunol. 2020, 11, 40. [Google Scholar] [CrossRef]
- Simon, B.; Uslu, U. CAR-T cell therapy in melanoma: A future success story? Exp. Dermatol. 2018, 27, 1315–1321. [Google Scholar] [CrossRef] [Green Version]
- Campoli, M.R.; Chang, C.-C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Wiesinger, M.; März, J.; Wistuba-Hamprecht, K.; Weide, B.; Schuler-Thurner, B.; Schuler, G.; Dörrie, J.; Uslu, U. The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma. Int. J. Mol. Sci. 2018, 19, 2365. [Google Scholar] [CrossRef] [Green Version]
- Carmo-Fonseca, M.; Pfeifer, K.; Schröder, H.C.; Vaz, M.F.; Fonseca, J.E.; Müller, W.E.; Bachmann, M. Identification of La ribonucleoproteins as a component of interchromatin granules. Exp. Cell Res. 1989, 185, 73–85. [Google Scholar] [CrossRef]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.; von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M.P. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef] [Green Version]
- Mitwasi, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Jureczek, J.; Loureiro, L.R.; Bergmann, R.; Máthé, D.; Hegedüs, N.; et al. "UniCAR"-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci. Rep. 2020, 10, 2141. [Google Scholar] [CrossRef] [Green Version]
- Grote, S.; Ureña-Bailén, G.; Chan, K.C.; Baden, C.; Mezger, M.; Handgretinger, R.; Schleicher, S. In Vitro Evaluation of CD276-CAR NK-92 Functionality, Migration and Invasion Potential in the Presence of Immune Inhibitory Factors of the Tumor Microenvironment. Cells 2021, 10, 1020. [Google Scholar] [CrossRef] [PubMed]
- Parlar, A.; Sayitoglu, E.C.; Ozkazanc, D.; Georgoudaki, A.M.; Pamukcu, C.; Aras, M.; Josey, B.J.; Chrobok, M.; Branecki, S.; Zahedimaram, P.; et al. Engineering antigen-specific natural killer cell lines against the melanoma-associated antigen tyrosinase via TCR gene transfer. Eur. J. Immunol. 2019, 49, 1278–1290. [Google Scholar]
- Zhang, G.; Liu, R.; Zhu, X.; Wang, L.; Ma, J.; Han, H.; Wang, X.; Zhang, G.; He, W.; Wang, W.; et al. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol. Cell Biol. 2013, 91, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Mensali, N.; Dillard, P.; Hebeisen, M.; Lorenz, S.; Theodossiou, T.; Myhre, M.R.; Fåne, A.; Gaudernack, G.; Kvalheim, G.; Myklebust, J.H.; et al. NK cells specifically TCR-dressed to kill cancer cells. EBioMedicine 2019, 40, 106–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef] [Green Version]
- Mingari, M.C.; Pietra, G.; Moretta, L. Immune Checkpoint Inhibitors: Anti-NKG2A Antibodies on Board. Trends Immunol. 2019, 40, 83–85. [Google Scholar] [CrossRef]
- Tremante, E.; Ginebri, A.; Lo Monaco, E.; Benassi, B.; Frascione, P.; Grammatico, P.; Cappellacci, S.; Catricalà, C.; Arcelli, D.; Natali, P.G.; et al. A melanoma immune response signature including Human Leukocyte Antigen-E. Pigment Cell Melanoma Res. 2014, 27, 103–112. [Google Scholar] [CrossRef]
- Kim, K.S.; Kim, D.H.; Kim, D.H. Recent Advances to Augment NK Cell Cancer Immunotherapy Using Nanoparticles. Pharmaceutics 2021, 13, 525. [Google Scholar] [CrossRef]
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Licona Limon, P.; et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| NK Cell Source | Study (NCT Number/Reference) and Clinical Phase | Study Title | Start Date-End Date | Patient Inclusion | Cell Culture Method | Infusion Dose | Pre-Treatment | Outcome | Academic/Industry |
|---|---|---|---|---|---|---|---|---|---|
| Autologous NK cells | Parkhurst et al. [104], NCT00328861, phase II | Natural Killer Cells Plus IL-2 Following Chemotherapy to Treat Advanced Melanoma or Kidney Cancer | May 2006, April 2009 | 7, progressive stage IV melanoma | 3-week culture period of CD3+ depleted cells using irradiated PBMCs as feeder cells | 4.7 × 1010 (±2.1 × 1010) cells, +high dose I.V. IL-2 (720,000 IU/kg/dose) | 2 days of cyclophosphamide (60 mg/kg) followed by 5 days of fludarabine (25 mg/m2) | no objective clinical responses | National Cancer Institute, Bethesda, Maryland |
| NCT00720785, phase I | Natural Killer Cells and Bortezomib to Treat Cancer | Recruiting in Sep 2020 | Malignant melanoma | Ex vivo expanded NK cells after apheresis | unknown | unknown | NA | National Cancer Institute, Bethesda, Maryland | |
| Allogeneic NK cells | Miller et al. [105], phase I | - | 2005 | 10, Metastatic melanoma | Depletion of CD3+ cells from PBMCs of haploidentical donors, NK cells were cultured overnight in medium containing IL-2 | 1 × 105, 1 × 106, 1 × 107, or 2 × 107 cells/kg of recipient body weight, +IL-2 injection for 14 days (1.75 × 106 IU/m2) | 48 hours prior to infusion: 750 mg/m2 intravenous cyclophosphamide and 1000 mg/m2 methylprednisolone | 4 patients with stable disease. After second infusion, disease progression was found within 4 to 6 weeks | University of Minnesota Cancer Center, Minneapolis |
| NCT00846833, phase I/II | Haploidentical NK Cell Infusion in Malignant Melanoma | Feb 2009–April 2012 | Confirmed metastatic or relapsed malignant melanoma and who received prior chemotherapy or immunotherapy | Depletion of CD3+ cells from PBMCs of haploidentical donors | unknown | Cyclophosphamide | unknown | Seoul National University Hospital Seoul, Korea | |
| NCT03420963, phase I | Donor Natural Killer Cells, Cyclophosphamide, and Etoposide in Treating Children and Young Adults With Relapsed or Refractory Solid Tumors | Feb 2018–recruiting | Recurrent Cutaneous Melanoma, Refractory Cutaneous Melanoma | Cord blood derived expanded allogeneic NK cells | unknown | Cyclophosphamide and etoposide | NA | M. D. Anderson Cancer CenterHouston, Texas, United States | |
| NCT03841110, phase I | FT500 as Monotherapy and in Combination With Immune Checkpoint Inhibitors in Subjects With Advanced Solid Tumors | Feb 2019–recruiting | Melanoma | NK cell derived from a clonal master iPSC line | 3 infusions, once weekly | Fludarabine/ Cyclophosphamide | NA | Fate therapeutics, San Diego, United states | |
| NCT03319459, phase I | FATE-NK100 as Monotherapy and in Combination With Monoclonal Antibody in Subjects With Advanced Solid Tumors | Jan 2018–recruiting | Melanoma | Allogeneic donor derived NK cells | unknown | unknown | NA | Fate therapeutics, San Diego, United states | |
| NCT03007823, phase I/II | High-activity Natural Killer Immunotherapy for Small Metastases of Melanoma | Dec 2016–Jun 2019 | Small metastasis of melanoma | 14 days of culture of allogeneic NK cells using a human NK cell in vitro culture kit (HANK bioengineering) [106] | 8–10 ×109 cells per infusion, 3 times infusion | unknown | unknown | Fuda Cancer Hospital, Guangzhou, China | |
| NK-92 cell line | Arai et al. [107], phase I | - | April 2002–June 2004 | malignant melanoma refractory with failed standard therapy | 3 weeks of ex vivo culture of NK-92 cell line with IL-2. Irradiation before infusion | 3 × 109 cells/m2 | Diphenhydramine prior to infusion | Minor response | Stanford University, Stanford, California, United States |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Vliet, A.A.; Georgoudaki, A.-M.; Raimo, M.; de Gruijl, T.D.; Spanholtz, J. Adoptive NK Cell Therapy: A Promising Treatment Prospect for Metastatic Melanoma. Cancers 2021, 13, 4722. https://doi.org/10.3390/cancers13184722
van Vliet AA, Georgoudaki A-M, Raimo M, de Gruijl TD, Spanholtz J. Adoptive NK Cell Therapy: A Promising Treatment Prospect for Metastatic Melanoma. Cancers. 2021; 13(18):4722. https://doi.org/10.3390/cancers13184722
Chicago/Turabian Stylevan Vliet, Amanda A., Anna-Maria Georgoudaki, Monica Raimo, Tanja D. de Gruijl, and Jan Spanholtz. 2021. "Adoptive NK Cell Therapy: A Promising Treatment Prospect for Metastatic Melanoma" Cancers 13, no. 18: 4722. https://doi.org/10.3390/cancers13184722
APA Stylevan Vliet, A. A., Georgoudaki, A.-M., Raimo, M., de Gruijl, T. D., & Spanholtz, J. (2021). Adoptive NK Cell Therapy: A Promising Treatment Prospect for Metastatic Melanoma. Cancers, 13(18), 4722. https://doi.org/10.3390/cancers13184722