
Prostate Cancer—Focus on Cholesterol

 ,
,
Simple Summary
Abstract
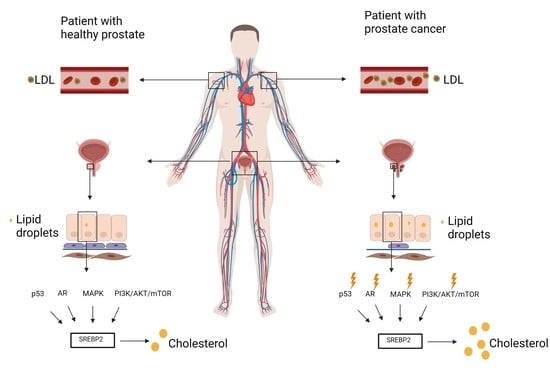
1. Introduction
2. Prostate Metabolism
3. Cholesterol Function and Prostate Cell Supply
4. Regulation of Cholesterol Homeostasis
5. Cholesterol Profile in Blood
6. Cholesterol Profile in the Prostate Tissue
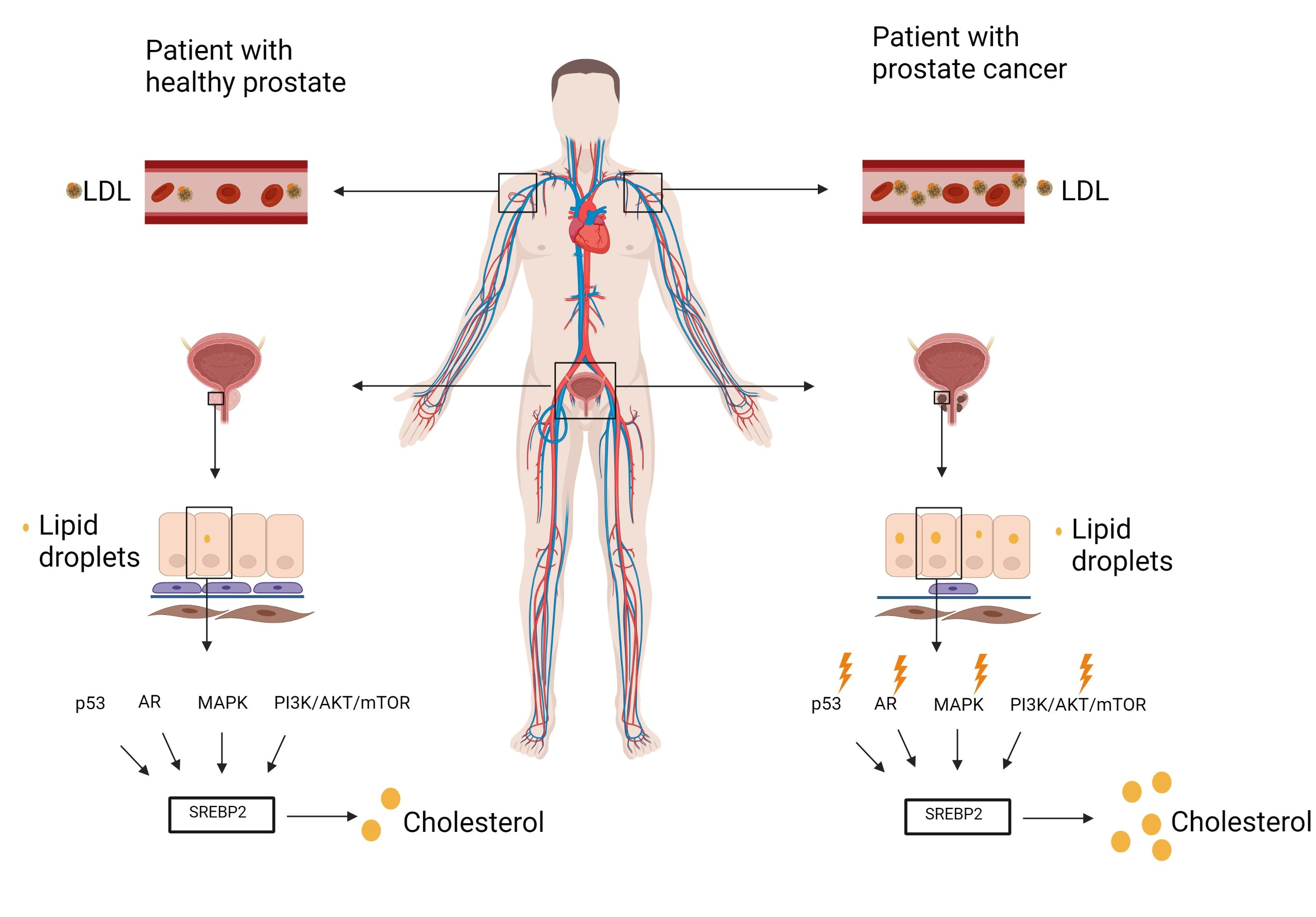
7. Dysregulated Signaling Pathways in Prostate Cancer
7.1. P53 and SREBP2
7.2. AR (Androgen Receptor)
7.3. PI3K/AKT/MTOR
7.4. MAPK
8. Acidity
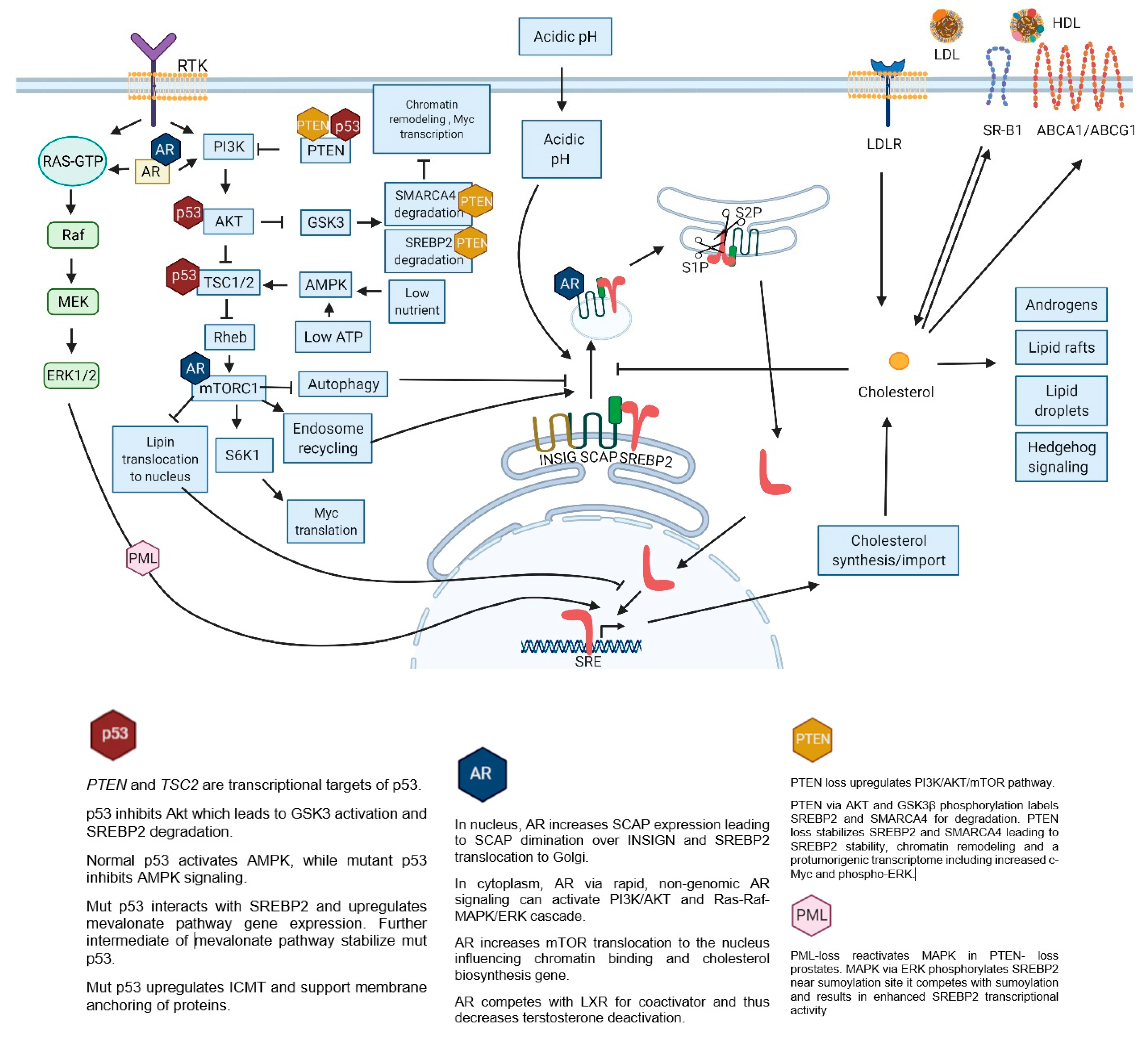
9. SREBP2 Targets
10. Cholesterol-Lowering Drugs and HDL Particles
11. Perspective
12. Conclusions
- increased membrane/lipid rafts synthesis and protein prenylation which alter membrane composition and cell signaling
- mut p53 stabilization and further mevalonate pathway promotion
- nucleotide synthesis, mitochondrial electron transport, protein anchoring and stabilization, ferroptosis inhibition
- intracellular androgen synthesis
- SREBP2 interaction with Myc promoter
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette transporter A1 |
| ABCG1 | ATP-binding cassette transporter G |
| ACLY | ATP citrate lyase |
| AMPK | AMP-activated protein kinase |
| AR | Androgen receptor |
| ARE | Androgen responsive element |
| BCR | Biochemical recurrence |
| BPH | Benign prostatic hyperplasia |
| COPII | Coatomer II |
| CRPC | Castrate-resistant prostate cancer |
| DHT | Dihydrotestosterone |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular signal-regulated kinase |
| FA | Fatty acid |
| FDG-PET | Fluorodeoxyglucose-positron emission tomography |
| FPP | Farnesyl diphosphate |
| GGPP | Geranyl-geranyl-pyrophosphate |
| GSK3 | Glycogen synthase kinase 3 |
| HDL | High-density lipoprotein |
| HMG-CoA | 3-hydroxy-3methylglutaryl-CoA |
| INSIG | Insulin-induced gene |
| ICMT | Isoprenylcysteine carboxyl methyltransferase |
| LDL | Low-density lipoprotein |
| LDLR | Low-density lipoprotein receptor |
| LRP8 | Lipoprotein receptor-related proteins 8 |
| LXR | Liver X receptor |
| MAPK | Mitogen-activated protein kinase |
| mTOR | Mechanistic target of rapamycin |
| PC | Prostate cancer |
| PSA | Prostate-specific antigen |
| ROS | Reactive oxygen species |
| S1P | Site-1 protease |
| S2P | Site-2 protease |
| SCAP | SREBP cleavage-activating protein |
| SHBG | Sex hormone-binding globulin |
| SQLE | Squalene monooxygenase |
| SR-B1 | Scavenger receptor class B member 1 |
| SRE | Sterol response element |
| SREBP2 | Sterol regulatory element–binding protein-2 |
References
- Krušlin, B.; Škara, L.; Vodopić, T.; Vrhovec, B.; Murgić, J.; Štimac, G.; Fröbe, A.; Lež, C.; Ulamec, M.; Gall-Trošelj, K. Genetics of Prostate Carcinoma. Acta Med. Acad. 2021, 50, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Abramovic, I.; Vrhovec, B.; Skara, L.; Vrtaric, A.; Gabaj, N.N.; Kulis, T.; Stimac, G.; Ljiljak, D.; Ruzic, B.; Kastelan, Z.; et al. MiR-182-5p and miR-375-3p Have Higher Performance Than PSA in Discriminating Prostate Cancer from Benign Prostate Hyperplasia. Cancers 2021, 13, 2068. [Google Scholar] [CrossRef]
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52. Available online: http://www.sciencedirect.com/science/article/pii/S0302283819306190 (accessed on 5 September 2021). [CrossRef]
- Adamaki, M.; Zoumpourlis, V. Immunotherapy as a Precision Medicine Tool for the Treatment of Prostate Cancer. Cancers 2021, 13, 173. [Google Scholar] [CrossRef]
- Milliron, B.; Bruneau, M.; Obeid, E.; Gross, L.; Bealin, L.; Smaltz, C.; Giri, V.N. Diet assessment among men undergoing genetic counseling and genetic testing for inherited prostate cancer: Exploring a teachable moment to support diet intervention. Prostate 2019, 79, 778–783. [Google Scholar] [CrossRef]
- Hager, M.H.; Solomon, K.R.; Freeman, M.R. The role of cholesterol in prostate cancer. Curr. Opin. Clin. Nutr. Metab. Care. 2006, 9, 379–385. [Google Scholar] [CrossRef]
- Moon, H.; Ruelcke, J.; Choi, E.; Sharpe, L.; Nassar, Z.; Bielefeldt-Ohmann, H.; Parat, M.-O.; Shah, A.; Francois, M.; Inder, K.L.; et al. Diet-Induced Hypercholesterolemia Promotes Androgen-Independent Prostate Cancer Metastasis via IQGAP1 and Caveolin-1. Oncotarget 2015, 6, 7438–7453. [Google Scholar] [CrossRef]
- Whittemore, A.S.; Kolonel, L.N.; Wu, A.H.; John, E.M.; Gallagher, R.P.; Howe, G.R.; Burch, J.D.; Hankin, J.; Dreon, D.M.; West, D.W.; et al. Prostate Cancer in Relation to Diet, Physical Activity, and Body Size in Blacks, Whites, and Asians in the United States and Canada. J. Natl. Cancer Inst. 1995, 87, 652–661. [Google Scholar] [CrossRef]
- Buszewska-Forajta, M.; Pomastowski, P.; Monedeiro, F.; Walczak-Skierska, J.; Markuszewski, M.; Matuszewski, M.; Markuszewski, M.J.; Buszewski, B. Lipidomics as a Diagnostic Tool for Prostate Cancer. Cancers 2021, 13, 2000. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, V.; Karunasinghe, N.; Jabed, A.; Pallati, R.; Kao, C.H.-J.; Wang, A.; Marlow, G.; Ferguson, L.R. Prostate Cancer: Is It a Battle Lost to Age? Geriatrics 2016, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Pelton, K.; Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. Curr. Opin. Pharmacol. 2012, 12, 751–759. [Google Scholar] [CrossRef]
- Singh, G.; Sankanagoudar, S.; Dogra, P.; Chandra, N.C. Interlink between cholesterol & cell cycle in prostate carcinoma. Indian J. Med. Res. 2017, 146 (Suppl. 2), S38–S44. [Google Scholar] [PubMed]
- Solomon, K.R.; Pelton, K.; Boucher, K.; Joo, J.; Tully, C.; Zurakowski, D.; Schaffner, C.P.; Kim, J.; Freeman, M.R. Ezetimibe Is an Inhibitor of Tumor Angiogenesis. Am. J. Pathol. 2009, 174, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Uo, T.; Sprenger, C.C.; Plymate, S.R. Androgen Receptor Signaling and Metabolic and Cellular Plasticity during Progression to Castration Resistant Prostate Cancer. Front. Oncol. 2020, 10, 2275. [Google Scholar] [CrossRef] [PubMed]
- Mah, C.Y.; Nassar, Z.D.; Swinnen, J.V.; Butler, L.M. Lipogenic Effects of Androgen Signaling in Normal and Malignant Prostate. Asian J. Urol. 2020, 7, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Franklin, R.B. A Comprehensive Review of the Role of Zinc in Normal Prostate Function and Metabolism; and Its Implications in Prostate Cancer. Arch. Biochem. Biophys. 2016, 611, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Feng, P.; Milon, B.; Tan, M.; Franklin, R.B. Role of zinc in the pathogenesis and treatment of prostate cancer: Critical issues to resolve. Prostate Cancer Prostatic Dis. 2004, 7, 111–117. [Google Scholar] [CrossRef]
- Bader, D.A.; McGuire, S.E. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat. Rev. Urol. 2020, 17, 214–231. [Google Scholar] [CrossRef]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo Lipogenesis Targets Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 2018, 116, 631–640. [Google Scholar] [CrossRef]
- Bertilsson, H.; Tessem, M.-B.; Flatberg, A.; Viset, T.; Gribbestad, I.; Angelsen, A.; Halgunset, J. Changes in Gene Transcription Underlying the Aberrant Citrate and Choline Metabolism in Human Prostate Cancer Samples. Clin. Cancer Res. 2012, 18, 3261–3269. [Google Scholar] [CrossRef]
- Gonzalez-Menendez, P.; Hevia, D.; Mayo, J.C.; Sainz, R.M. The dark side of glucose transporters in prostate cancer: Are they a new feature to characterize carcinomas? Int. J. Cancer 2018, 142, 2414–2424. [Google Scholar] [CrossRef]
- Krycer, J.R.; Brown, A.J. Cholesterol Accumulation in Prostate Cancer: A Classic Observation from a Modern Perspective. Biochim. Biophys. Acta-Rev. Cancer 2013, 1835, 219–229. [Google Scholar]
- Poulose, N.; Mills, I.G.; Steele, R.E. The impact of transcription on metabolism in prostate and breast cancers. Endocr. Relat. Cancer. 2018, 2 5, R435–R452. [Google Scholar] [CrossRef]
- Yupeng, L.; Yuxue, Z.; Pengfei, L.; Cheng, C.; Yashuang, Z.; Dapeng, L.; Chen, D. Cholesterol Levels in Blood and the Risk of Prostate Cancer: A Meta-analysis of 14 Prospective Studies. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1086–1093. [Google Scholar] [CrossRef]
- Huang, B.; Song, B.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef]
- Likus, W.; Siemianowicz, K.; Bieńk, K.; Pakuła, M.; Pathak, H.; Dutta, C.; Wang, Q.; Shojaei, S.; Assaraf, Y.G.; Ghavami, S.; et al. Could drugs inhibiting the mevalonate pathway also target cancer stem cells? Drug Resist. Updat. Rev. Comment Antimicrob. Anticancer Chemother. 2016, 25, 13–25. [Google Scholar] [CrossRef]
- Hryniewicz-Jankowska, A.; Augoff, K.; Sikorski, A.F. Highlight article: The role of cholesterol and cholesterol-driven membrane raft domains in prostate cancer. Exp. Biol. Med. (Maywood) 2019, 244, 1053–1061. [Google Scholar] [CrossRef]
- Li, H.; Feng, Z.; He, M.-L. Lipid Metabolism Alteration Contributes to and Maintains the Properties of Cancer Stem Cells. Theranostics 2020, 10, 7053–7069. [Google Scholar] [PubMed]
- Matsushita, Y.; Nakagawa, H.; Koike, K. Lipid Metabolism in Oncology: Why It Matters, How to Research, and How to Treat. Cancers 2021, 13, 474. [Google Scholar] [CrossRef]
- Brown, A.J. Cholesterol, Statins and Cancer. Clin. Exp. Pharmacol. Physiol. 2007, 34, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zhang, W.; Li, S.; Yang, H. The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 2019, 9, 219–227. [Google Scholar]
- Luo, J.; Jiang, L.-Y.; Yang, H.; Song, B.-L. Intracellular Cholesterol Transport by Sterol Transfer Proteins at Membrane Contact Sites. Trends Biochem. Sci. 2019, 44, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.; Blanco, G. Chapter 15-Lipid Metabolism; Blanco, A., Blanco, G., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 325–365. Available online: http://www.sciencedirect.com/science/article/pii/B978012803550400015X (accessed on 5 September 2021).
- Shapiro, M.D.; Tavori, H.; Fazio, S. PCSK9: From Basic Science Discoveries to Clinical Trials. Circ. Res. 2018, 122, 1420–1438. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.-J.; Azhar, S.; Kraemer, F.B. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Traughber, C.A.; Opoku, E.; Brubaker, G.; Major, J.; Lu, H.; Lorkowski, S.W.; Neumann, C.; Hardaway, A.; Chung, Y.-M.; Gulshan, K.; et al. Uptake of high-density lipoprotein by scavenger receptor class B type 1 is associated with prostate cancer proliferation and tumor progression in mice. J. Biol. Chem. 2020, 295, 8252–8261. [Google Scholar] [CrossRef]
- Cha, J.-Y.; Lee, H.-J. Targeting Lipid Metabolic Reprogramming as Anticancer Therapeutics. J. Cancer Prev. 2016, 21, 209–215. [Google Scholar] [CrossRef]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth Under Metabolic Stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef]
- Giacomini, I.; Gianfanti, F.; Desbats, M.A.; Orso, G.; Berretta, M.; Prayer-Galetti, T.; Ragazzi, E.; Cocetta, V. Cholesterol Metabolic Reprogramming in Cancer and Its Pharmacological Modulation as Therapeutic Strategy. Front. Oncol. 2021, 11, 1946. [Google Scholar] [CrossRef]
- Afonso, M.S.; Machado, R.M.; Lavrador, M.S.; Quintao, E.C.R.; Moore, K.J.; Lottenberg, A.M. Molecular Pathways Underlying Cholesterol Homeostasis. Nutrients 2018, 10, 760. [Google Scholar]
- Gesto, D.S.; Pereira, C.M.S.; Cerqueira, N.M.F.S.; Sousa, S.F. An Atomic-Level Perspective of HMG-CoA-Reductase: The Target Enzyme to Treat Hypercholesterolemia. Molecules 2020, 25, 3891. [Google Scholar] [CrossRef]
- Alioui, A.; Celhay, O.; Baron, S.; Lobaccaro, J.-M.A. Lipids and Prostate Cancer Adenocarcinoma. Clin. Lipidol. 2014, 9, 643–655. [Google Scholar] [CrossRef]
- Rye, M.B.; Bertilsson, H.; Andersen, M.K.; Rise, K.; Bathen, T.F.; Drabløs, F.; Tessem, M.-B. Cholesterol synthesis pathway genes in prostate cancer are transcriptionally downregulated when tissue confounding is minimized. BMC Cancer 2018, 18, 478. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q.-H. Regulation of intestinal cholesterol absorption. Annu. Rev. Physiol. 2007, 69, 221–248. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Mangelsdorf, D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 2000, 16, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Ru, P.; Geng, F.; Liu, J.; Yoo, J.Y.; Wu, X.; Cheng, X.; Euthine, V.; Hu, P.; Guo, J.Y.; et al. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell 2015, 28, 569–581. [Google Scholar] [CrossRef]
- Xue, L.; Qi, H.; Zhang, H.; Ding, L.; Huang, Q.; Zhao, D.; Wu, B.J.; Li, X. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front. Oncol. 2020, 10, 1510. [Google Scholar] [CrossRef] [PubMed]
- Rawson, R.B.; DeBose-Boyd, R.; Goldstein, J.L.; Brown, M.S. Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J. Biol. Chem. 1999, 274, 28549–28556. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, Y.; Goldstein, J.L.; Brown, M.S.; Radhakrishnan, A. Cholesterol-induced conformational changes in the sterol-sensing domain of the Scap protein suggest feedback mechanism to control cholesterol synthesis. J. Biol. Chem. 2017, 292, 8729–8737. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Goldstein, J.L.; McDonald, J.G.; Brown, M.S. Switch-like Control of SREBP-2 Transport Triggered by Small Changes in ER Cholesterol: A Delicate Balance. Cell Metab. 2008, 8, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.N.; Song, B.; DeBose-Boyd, R.A.; Ye, J. Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J. Biol. Chem. 2006, 281, 39308–39315. [Google Scholar] [CrossRef] [PubMed]
- Xiao-Ying, D.; Sheng-Qiu, T.; Jin-Ding, C. Dual functions of Insig proteins in cholesterol homeostasis. Lipids. Health Dis. 2012, 11, 173. [Google Scholar]
- Kuan, Y.-C.; Takahashi, Y.; Maruyama, T.; Shimizu, M.; Yamauchi, Y.; Sato, R. Ring Finger Protein 5 Activates Sterol Regulatory Element–Binding Protein 2 (SREBP2) to Promote Cholesterol Biosynthesis via Inducing Polyubiquitination of SREBP Chaperone SCAP. J. Biol. Chem. 2020, 295, 3918–3928. [Google Scholar] [CrossRef]
- Fan, Z.; Kong, M.; Li, M.; Hong, W.; Fan, X.; Xu, Y. Brahma Related Gene 1 (Brg1) Regulates Cellular Cholesterol Synthesis by Acting as a Co-factor for SREBP2. Front. cell Dev. Biol. 2020, 8, 259. [Google Scholar] [CrossRef]
- Matsuzaka, T.; Shimano, H. Insulin-dependent and -independent regulation of sterol regulatory element-binding protein-1c. J. Diabetes Investig. 2013, 4, 411–412. [Google Scholar] [CrossRef]
- Sundqvist, A.; Bengoechea-Alonso, M.T.; Ye, X.; Lukiyanchuk, V.; Jin, J.; Harper, J.W.; Harper, J.; Ericsson, J. Control of Lipid Metabolism by Phosphorylation-Dependent Degradation of the SREBP Family of Transcription Factors by SCFFbw7. Cell Metab. 2005, 1, 379–391. [Google Scholar] [CrossRef]
- Giandomenico, V.; Simonsson, M.; Grönroos, E.; Ericsson, J. Coactivator-Dependent Acetylation Stabilizes Members of the SREBP Family of Transcription Factors. Mol. Cell Biol. 2003, 23, 2587–2599. [Google Scholar] [CrossRef]
- Arito, M.; Horiba, T.; Hachimura, S.; Inoue, J.; Sato, R. Growth factor-induced phosphorylation of sterol regulatory element-binding proteins inhibits sumoylation, thereby stimulating the expression of their target genes, low density lipoprotein uptake, and lipid synthesis. J. Biol. Chem. 2008, 283, 15224–15231. [Google Scholar] [CrossRef]
- Elhanati, S.; Kanfi, Y.; Varvak, A.; Roichman, A.; Carmel-Gross, I.; Barth, S.; Gibor, G.; Cohen, H.Y. Multiple Regulatory Layers of SREBP1/2 by SIRT6. Cell Rep. 2013, 4, 905–912. [Google Scholar] [CrossRef]
- Dufour, J.; Viennois, E.; De Boussac, H.; Baron, S.; Lobaccaro, J.-M. Oxysterol receptors, AKT and prostate cancer. Curr. Opin. Pharmacol. 2012, 12, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Ikeda, Y.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Sterol-Regulated Transport of SREBPs from Endoplasmic Reticulum to Golgi: Oxysterols Block Transport by Binding to INSIG. Proc. Natl. Acad. Sci. USA 2007, 104, 6511–6518. [Google Scholar] [CrossRef]
- Scotti, E.; Calamai, M.; Goulbourne, C.N.; Zhang, L.; Hong, C.; Lin, R.R.; Choi, J.; Pilch, P.F.; Fong, L.G.; Zou, P.; et al. IDOL Stimulates Clathrin-Independent Endocytosis and Multivesicular Body-Mediated Lysosomal Degradation of the Low-Density Lipoprotein Receptor. Mol. Cell. Biol. 2013, 33, 1503–1514. [Google Scholar] [CrossRef]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef]
- Wang, Y.; Rogers, P.M.; Su, C.; Varga, G.; Stayrook, K.R.; Burris, T.P. Regulation of Cholesterologenesis by the Oxysterol Receptor, LXRalpha. J. Biol. Chem. 2008, 283, 26332–26339. [Google Scholar] [CrossRef]
- Mok, E.H.K.; Lee, T.K.W. The Pivotal Role of the Dysregulation of Cholesterol Homeostasis in Cancer: Implications for Therapeutic Targets. Cancers 2020, 12, 1410. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, H.; Coates, H.W.; Chua, N.K.; Hashimoto, Y.; Brown, A.J.; Ohgane, K. A Key Mammalian Cholesterol Synthesis Enzyme, Squalene Monooxygenase, is Allosterically Stabilized by Its Substrate. Proc. Natl. Acad. Sci. USA 2020, 117, 7150–7158. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.; Verrijdt, G.; Organe, S.; Claessens, F.; Heyns, W.; Verhoeven, G.; Swinnen, J. Identification of an Androgen Response Element in Intron 8 of the Sterol Regulatory Element-binding Protein Cleavage-activating Protein Gene Allowing Direct Regulation by the Androgen Receptor. J. Biol. Chem. 2004, 279, 30880–30887. [Google Scholar] [CrossRef] [PubMed]
- Krycer, J.R.; Brown, A.J. Cross-talk between the androgen receptor and the liver X receptor: Implications for cholesterol homeostasis. J. Biol. Chem. 2011, 286, 20637–20647. [Google Scholar] [CrossRef] [PubMed]
- Runge-Morris, M.; Kocarek, T.A.; Falany, C.N. Regulation of the Cytosolic Sulfotransferases by Nuclear Receptors. Drug Metab Rev. 2013, 45, 15–33. [Google Scholar] [CrossRef]
- Lee, J.H.; Gong, H.; Khadem, S.; Lu, Y.; Gao, X.; Li, S.; Zhang, J.; Xie, W. Androgen Deprivation by Activating the Liver X Receptor. Endocrinology 2008, 149, 3778–3788. [Google Scholar] [PubMed]
- Lee, H.J.; Li, J.; Vickman, R.E.; Li, J.; Liu, R.; Durkes, A.C.; Elzey, B.D.; Yue, S.; Liu, X.; Ratliff, T.L.; et al. Cholesterol Esterification Inhibition Suppresses Prostate Cancer Metastasis by Impairing the Wnt/β-catenin Pathway. Mol. Cancer Res. 2018, 16, 974–985. [Google Scholar] [CrossRef]
- Asare, G.A.; Owusu-Boateng, E.; Asiedu, B.; Amoah, B.Y.; Essendoh, E.; Otoo, R.Y. Oxidised low-density lipoprotein, a possible distinguishing lipid profile biomolecule between prostate cancer and benign prostatic hyperplasia. Andrologia. 2019, 51, e13321. [Google Scholar] [CrossRef]
- Kong, Y.; Cheng, L.; Mao, F.; Zhang, Z.; Zhang, Y.; Farah, E.; Bosler, J.; Bai, Y.; Ahmad, N.; Kuang, S.; et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J. Biol. Chem. 2018, 293, 14328–14341. [Google Scholar] [CrossRef] [PubMed]
- Jamnagerwalla, J.; Howard, L.E.; Allott, E.H.; Vidal, A.C.; Moreira, D.M.; Castro-Santamaria, R.; Andriole, G.L.; Freeman, M.R.; Freedland, S.J. Serum cholesterol and risk of high-grade prostate cancer: Results from the REDUCE study. Prostate Cancer Prostatic Dis. 2018, 21, 252–259. [Google Scholar] [CrossRef]
- Zapata, D.; Howard, L.E.; Allott, E.H.; Hamilton, R.J.; Goldberg, K.; Freedland, S.J. Is PSA related to serum cholesterol and does the relationship differ between black and white men? Prostate 2015, 75, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Sankanagoudar, S.; Dogra, P.N.; Mathur, S.R.; Chandra, N.C. A study on lipid profile in prostate carcinoma patients admitted in AIIMS, New Delhi. J. Biomed Pharm. Res. 2014, 3, 49–51. [Google Scholar]
- Henriksson, P.; Ericsson, S.; Stege, R.; Eriksson, M.; Rudling, M.; Berglund, L.; Angelin, B. Hypocholesterolaemia and Increased Elimination of Low-Density Lipoproteins in Metastatic Cancer of the Prostate. Lancet 1989, 334, 1178–1180. [Google Scholar] [CrossRef]
- Tatidis, L.; Gruber, A.; Vitols, S. Decreased feedback regulation of low density lipoprotein receptor activity by sterols in leukemic cells from patients with acute myelogenous leukemia. J. Lipid. Res. 1997, 38, 2436–2445. [Google Scholar] [CrossRef]
- Gonçalves, R.P.; Rodrigues, D.G.; Maranhão, R.C. Uptake of High Density Lipoprotein (HDL) Cholesteryl Esters by Human Acute Leukemia Cells. Leuk. Res. 2005, 29, 955–959. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Zuber, V.; Thompson, W.K.; Schork, A.J.; Bettella, F.; Djurovic, S.; Desikan, R.S.; Mills, I.; Dale, A.M.; The PRACTICAL Consortium; et al. Shared common variants in prostate cancer and blood lipids. Int. J. Epidemiol. 2014, 43, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; Casaburi, I.; Avena, P.; Trotta, F.; De Luca, A.; Rago, V.; Pezzi, V.; Sirianni, R. Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front. Endocrinol. 2019, 9, 807. [Google Scholar] [CrossRef]
- Li, X.; Wu, J.B.; Li, Q.; Shigemura, K.; Chung, L.W.K.; Huang, W.-C. SREBP-2 Promotes Stem Cell-Like Properties and Metastasis by Transcriptional Activation of c-Myc in Prostate Cancer. Oncotarget 2016, 7, 12869–12884. Available online: https://pubmed.ncbi.nlm.nih.gov/26883200 (accessed on 5 September 2021). [CrossRef]
- Longo, J.; Mullen, P.J.; Yu, R.; van Leeuwen, J.E.; Masoomian, M.; Woon, D.T.; Wang, Y.; Chen, E.X.; Hamilton, R.J.; Sweet, J.M.; et al. An Actionable Sterol-Regulated Feedback Loop Modulates Statin Sensitivity in Prostate Cancer. Mol. Metab. 2019, 25, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Taylor, M.; Robinet, P.; Smith, J.D.; Schweitzer, J.; Sehayek, E.; Falzarano, S.M.; Magi-Galluzzi, C.; Klein, E.A.; Ting, A.H. Dysregulation of Cholesterol Homeostasis in Human Prostate Cancer through Loss of ABCA1. Cancer Res. 2012, 73, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Schörghofer, D.; Kinslechner, K.; Preitschopf, A.; Schütz, B.; Röhrl, C.; Hengstschläger, M.; Stangl, H.; Mikula, M. The HDL receptor SR-BI is associated with human prostate cancer progression and plays a possible role in establishing androgen independence. Reprod. Biol. Endocrinol. 2015, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Thysell, E.; Surowiec, I.; Hörnberg, E.; Crnalic, S.; Widmark, A.; Johansson, A.I.; Stattin, P.; Bergh, A.; Moritz, T.; Antti, H.; et al. Metabolomic Characterization of Human Prostate Cancer Bone Metastases Reveals Increased Levels of Cholesterol. PLoS ONE 2010, 5, e14175. [Google Scholar] [CrossRef]
- Chen, Y.; Hughes-Fulford, M. Human prostate cancer cells lack feedback regulation of low-density lipoprotein receptor and its regulator, SREBP2. Int. J. Cancer 2001, 91, 41–45. [Google Scholar] [CrossRef]
- Ossoliński, K.; Nizioł, J.; Arendowski, A.; Ossolińska, A.; Ossoliński, T.; Kucharz, J.; Wiechno, P.; Ruman, T. Mass spectrometry-based metabolomic profiling of prostate cancer-a pilot study. J. Cancer Metastasis Treat. 2019, 2019. [Google Scholar] [CrossRef]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Wilson, S.; Qi, J.; Filipp, F.V. Refinement of the androgen response element based on ChIP-Seq in androgen-insensitive and androgen-responsive prostate cancer cell lines. Sci. Rep. 2016, 6, 32611. [Google Scholar] [CrossRef]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen Receptor Gene Expression in Prostate Cancer Is Directly Suppressed by the Androgen Receptor Through Recruitment of Lysine-Specific Demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef]
- Xiao, Q.; Sun, Y.; Dobi, A.; Srivastava, S.; Wang, W.; Srivastava, S.; Ji, Y.; Hou, J.; Zhao, G.-P.; Li, Y.; et al. Systematic analysis reveals molecular characteristics of ERG-negative prostate cancer. Sci. Rep. 2018, 8, 12868. [Google Scholar] [CrossRef] [PubMed]
- Khemlina, G.; Ikeda, S.; Kurzrock, R. Molecular Landscape of Prostate Cancer: Implications for Current Clinical Trials. Cancer Treat. Rev. 2015, 41, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Agell, L.; Hernández, S.; Salido, M.; De Muga, S.; Juanpere, N.; Arumi-Uria, M.; Menendez, S.; Lorenzo, M.; Lorente, J.A.; Serrano, S.; et al. PI3K signaling pathway is activated by PIK3CA mRNA overexpression and copy gain in prostate tumors, but PIK3CA, BRAF, KRAS and AKT1 mutations are infrequent events. Mod. Pathol. 2010, 24, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Huang, J.; Homma, T.; Kita, D.; Klocker, H.; Schafer, G.; Boyle, P.; Ohgaki, H. Genetic alterations in the PI3K pathway in prostate cancer. Anticancer Res. 2009, 29, 1739–1743. [Google Scholar]
- Li, X.; Wu, J.B.; Chung, L.W.K.; Huang, W.-C. Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget 2015, 6, 41018–41032. [Google Scholar] [CrossRef]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Borini Etichetti, C.M.; Arel Zalazar, E.; Cocordano, N.; Girardini, J. Beyond the Mevalonate Pathway: Control of Post-Prenylation Processing by Mutant p53. Front. Oncol. 2020, 10, 2389. [Google Scholar] [CrossRef]
- Flöter, J.; Kaymak, I.; Schulze, A. Regulation of Metabolic Activity by p53. Metabolites 2017, 7, 21. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.-J.; et al. AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.-X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.; Jasser, S.A.; et al. Gain-of-Function Mutant p53 Promotes Cell Growth and Cancer Cell Metabolism via Inhibition of AMPK Activation. Mol. Cell 2014, 54, 960–974. [Google Scholar]
- Kaymak, I.; Maier, C.R.; Schmitz, W.; Campbell, A.D.; Dankworth, B.; Ade, C.P.; Walz, S.; Paauwe, M.; Kalogirou, C.; Marouf, H.; et al. Mevalonate Pathway Provides Ubiquinone to Maintain Pyrimidine Synthesis and Survival in p53-Deficient Cancer Cells Exposed to Metabolic Stress. Cancer Res. 2019, 80, 189–203. [Google Scholar] [CrossRef]
- Kallio, H.M.L.; Hieta, R.; Latonen, L.; Brofeldt, A.; Annala, M.; Kivinummi, K.; Tammela, T.L.; Nykter, M.; Isaacs, W.B.; Lilja, H.G.; et al. Constitutively active androgen receptor splice variants AR-V3, AR-V7 and AR-V9 are co-expressed in castration-resistant prostate cancer metastases. Br. J. Cancer 2018, 119, 347–356. [Google Scholar] [CrossRef]
- Marchetti, P.M.; Barth, J.H. Clinical Biochemistry of Dihydrotestosterone. Ann. Clin. Biochem. 2013, 50, 95–107. [Google Scholar] [CrossRef]
- Holt, S.K.; Karyadi, D.M.; Kwon, E.M.; Stanford, J.L.; Nelson, P.S.; Ostrander, E.A. Association of Megalin Genetic Polymorphisms with Prostate Cancer Risk and Prognosis. Clin. Cancer Res. 2008, 14, 3823–3831. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Pham, T.; McWhinney, B.C.; Ungerer, J.; Pretorius, C.; Richard, D.J.; Mortimer, R.H.; D’Emden, M.; Richard, K. Sex Hormone Binding Globulin Modifies Testosterone Action and Metabolism in Prostate Cancer Cells. Int. J. Endocrinol. 2016, 2016, 6437585. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer (Review). Oncol. Lett. 2018, 15, 6063–6076. [Google Scholar] [CrossRef] [PubMed]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Research 2019, 8, 998. [Google Scholar] [CrossRef] [PubMed]
- Valvezan, A.J.; Manning, B.D. Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat. Metab. 2019, 1, 321–333. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.; Finck, B.N.; et al. mTOR Complex 1 Regulates Lipin 1 Localization to Control the SREBP Pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef]
- Ricoult, S.J.H.; Manning, B.D. The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep. 2013, 14, 242–251. [Google Scholar] [CrossRef]
- Yoon, M.-S. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef]
- Audet-Walsh, É.; Dufour, C.R.; Yee, T.; Zouanat, F.Z.; Yan, M.; Kalloghlian, G.; Vernier, M.; Caron, M.; Bourque, G.; Scarlata, E.; et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev. 2017, 31, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Eid, W.; Dauner, K.; Courtney, K.C.; Gagnon, A.; Parks, R.J.; Sorisky, A.; Zha, X. mTORC1 Activates SREBP-2 by Suppressing Cholesterol Trafficking to Lysosomes in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2017, 114, 7999–8004. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and Its Impact on Gene Expression at a Glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [PubMed]
- Bitting, R.L.; Armstrong, A.J. Targeting the PI3K/Akt/mTOR Pathway in Castration-Resistant Prostate Cancer. Endocr. Relat. Cancer 2013, 20, R83–R99. [Google Scholar] [CrossRef]
- Lokody, I.B.; Francis, J.C.; Gardiner, J.R.; Erler, J.T.; Swain, A. Pten Regulates Epithelial Cytodifferentiation during Prostate Development. PLoS ONE 2015, 10, e0129470. [Google Scholar] [CrossRef]
- Jillson, L.K.; Yette, G.A.; Laajala, T.D.; Tilley, W.D.; Costello, J.C.; Cramer, S.D. Androgen Receptor Signaling in Prostate Cancer Genomic Subtypes. Cancers 2021, 13, 3272. [Google Scholar] [CrossRef]
- Ding, Y.; Li, N.; Dong, B.; Guo, W.; Wei, H.; Chen, Q.; Yuan, H.; Han, Y.; Chang, H.; Kan, S.; et al. Chromatin Remodeling ATPase BRG1 and PTEN are Synthetic Lethal in Prostate Cancer. J. Clin. Investig. 2019, 129, 759–773. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.-S.; Lee, Y.-R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Tang, Y.; Peng, X.; Cai, X.; Wa, Q.; Ren, D.; Li, Q.; Luo, J.; Li, L.; Zou, X.; et al. Acidic extracellular pH promotes prostate cancer bone metastasis by enhancing PC-3 stem cell characteristics, cell invasiveness and VEGF-induced vasculogenesis of BM-EPCs. Oncol. Rep. 2016, 36, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Logozzi, M.; Angelini, D.F.; Iessi, E.; Mizzoni, D.; Di Raimo, R.; Federici, C.; Lugini, L.; Borsellino, G.; Gentilucci, A.; Pierella, F.; et al. Increased PSA expression on prostate cancer exosomes in in vitro condition and in cancer patients. Cancer Lett. 2017, 403, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Thews, O.; Gassner, B.; Kelleher, D.K.; Schwerd, G.; Gekle, M. Impact of Extracellular Acidity on the Activity of P-Glycoprotein and the Cytotoxicity of Chemotherapeutic Drugs. Neoplasia 2006, 8, 143–152. [Google Scholar] [CrossRef]
- Kondo, A.; Yamamoto, S.; Nakaki, R.; Shimamura, T.; Hamakubo, T.; Sakai, J.; Kodama, T.; Yoshida, T.; Aburatani, H.; Osawa, T. Extracellular Acidic pH Activates the Sterol Regulatory Element-Binding Protein 2 to Promote Tumor Progression. Cell Rep. 2017, 18, 2228–2242. [Google Scholar] [CrossRef]
- Leon, C.G.; Locke, J.A.; Adomat, H.H.; Etinger, S.L.; Twiddy, A.L.; Neumann, R.D.; Nelson, C.C.; Guns, E.S.; Wasan, K.M. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model. Prostate 2009, 70, 390–400. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nat. Cell Biol. 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Khutornenko, A.A.; Roudko, V.V.; Chernyak, B.V.; Vartapetian, A.B.; Chumakov, P.M.; Evstafieva, A.G. Pyrimidine Biosynthesis Links Mitochondrial Respiration to the p53 Pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12828–12833. [Google Scholar] [CrossRef]
- Cheng, C.; Guo, J.Y.; Geng, F.; Wu, X.; Cheng, X.; Li, Q.; Guo, D. Analysis of SCAP N-glycosylation and Trafficking in Human Cells. J. Vis. Exp. 2016, 117, e54709. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Gabitova, L.; Gorin, A.; Astsaturov, I. Molecular pathways: Sterols and receptor signaling in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 28–34. [Google Scholar] [CrossRef]
- Zhang, Z.; Hou, X.; Shao, C.; Li, J.; Cheng, J.-X.; Kuang, S.; Ahmad, N.; Ratliff, T.; Liu, X. Plk1 Inhibition Enhances the Efficacy of Androgen Signaling Blockade in Castration-Resistant Prostate Cancer. Cancer Res. 2014, 74, 6635–6647. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.-T.; Hu, P.; Huang, W.-C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 2014, 13, 855–866. [Google Scholar] [CrossRef]
- Csibi, A.; Lee, G.; Yoon, S.-O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.-M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 Pathway Regulates Glutamine Metabolism through the eIF4B-Dependent Control of c-Myc Translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef]
- Karlic, H.; Varga, F. Mevalonate Pathway. In Encyclopedia of Cancer, 3rd ed.; Boffetta, P., Hainaut, P.B.T., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 445–457. Available online: http://www.sciencedirect.com/science/article/pii/B9780128012383650006 (accessed on 5 September 2021).
- Wang, K.; Gerke, T.A.; Chen, X.; Prosperi, M. Association of statin use with risk of Gleason score-specific prostate cancer: A hospital-based cohort study. Cancer Med. 2019, 8, 7399–7407. [Google Scholar] [CrossRef] [PubMed]
- Harshman, L.C.; Wang, X.; Nakabayashi, M.; Xie, W.; Valenca, L.; Werner, L.; Yu, Y.; Kantoff, A.M.; Sweeney, C.J.; Mucci, L.A.; et al. Statin Use at the Time of Initiation of Androgen Deprivation Therapy and Time to Progression in Patients With Hormone-Sensitive Prostate Cancer. JAMA Oncol. 2015, 1, 495–504. [Google Scholar] [CrossRef]
- Gutt, R.; Tonlaar, N.; Kunnavakkam, R.; Karrison, T.; Weichselbaum, R.R.; Liauw, S.L. Statin Use and Risk of Prostate Cancer Recurrence in Men Treated With Radiation Therapy. J. Clin. Oncol. 2010, 28, 2653–2659. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.J.; Banez, L.L.; Aronson, W.J.; Terris, M.; Platz, E.A.; Kane, C.J.; Presti, J.C.; Amling, C.L.; Freedland, S.J. Statin medication use and the risk of biochemical recurrence after radical prostatectomy. Cancer 2010, 116, 3389–3398. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Riviere, P.; Luterstein, E.; Nalawade, V.; Vitzthum, L.; Sarkar, R.R.; Bryant, A.K.; Einck, J.P.; Mundt, A.J.; Murphy, J.D.; et al. Associations among statins, preventive care, and prostate cancer mortality. Prostate Cancer Prostatic Dis. 2020, 23, 475–485. [Google Scholar] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 Controls the Fate of Misfolded Mutant p53 Through the Mevalonate Pathway. Nature 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.; Deplazes, E.; Cranfield, C.G.; Garcia, A. The Role of Structure and Biophysical Properties in the Pleiotropic Effects of Statins. Int. J. Mol. Sci. 2020, 21, 8745. [Google Scholar] [CrossRef]
- Gholkar, A.; Cheung, K.; Williams, K.J.; Lo, Y.-C.; Hamideh, S.A.; Nnebe, C.; Khuu, C.; Bensinger, S.J.; Torres, J.Z. Fatostatin Inhibits Cancer Cell Proliferation by Affecting Mitotic Microtubule Spindle Assembly and Cell Division. J. Biol. Chem. 2016, 291, 17001–17008. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Inoue, J.; Shimizu, M.; Sato, R. Xanthohumol Improves Diet-induced Obesity and Fatty Liver by Suppressing Sterol Regulatory Element-binding Protein (SREBP) Activation. J. Biol. Chem. 2015, 290, 20565–20579. [Google Scholar] [CrossRef]
- Tang, J.-J.; Li, J.-G.; Qi, W.; Qiu, W.-W.; Li, P.-S.; Li, B.-L.; Song, B.-L. Inhibition of SREBP by a Small Molecule, Betulin, Improves Hyperlipidemia and Insulin Resistance and Reduces Atherosclerotic Plaques. Cell Metab. 2011, 13, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Deeb, D.; Gao, X.; Jiang, H.; Arbab, A.S.; Dulchavsky, S.A.; Gautam, S.C. Growth inhibitory and apoptosis-inducing effects of xanthohumol, a prenylated chalone present in hops, in human prostate cancer cells. Anticancer Res. 2010, 30, 3333–3339. [Google Scholar] [PubMed]
- Venè, R.; Benelli, R.; Minghelli, S.; Astigiano, S.; Tosetti, F.; Ferrari, N. Xanthohumol impairs human prostate cancer cell growth and invasion and diminishes the incidence and progression of advanced tumors in TRAMP mice. Mol. Med. 2012, 18, 1292–1302. [Google Scholar] [CrossRef]
- Reiner, T.; Parrondo, R.; de Las Pozas, A.; Palenzuela, D.; Perez-Stable, C. Betulinic acid selectively increases protein degradation and enhances prostate cancer-specific apoptosis: Possible role for inhibition of deubiquitinase activity. PLoS ONE 2013, 8, e56234. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Pandyra, A.A.; Mullen, P.J.; Goard, C.A.; Ericson, E.; Sharma, P.; Kalkat, M.; Yu, R.; Pong, J.T.; Brown, K.; Hart, T.; et al. Genome-wide RNAi analysis reveals that simultaneous inhibition of specific mevalonate pathway genes potentiates tumor cell death. Oncotarget 2015, 6, 26909–26921. [Google Scholar] [CrossRef]
- Zhu, M.-L.; Horbinski, C.M.; Garzotto, M.; Qian, D.Z.; Beer, T.M.; Kyprianou, N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010, 70, 7992–8002. [Google Scholar] [CrossRef] [PubMed]
- Meor Anuar Shuhaili, M.F.R.; Samsudin, I.N.; Stanslas, J.; Hasan, S.; Thambiah, S.C. Effects of Different Types of Statins on Lipid Profile: A Perspective on Asians. Int. J. Endocrinol. Metab. 2017, 15, e43319. [Google Scholar] [CrossRef]
- Menter, D.G.; Ramsauer, V.P.; Harirforoosh, S.; Chakraborty, K.; Yang, P.; Hsi, L.; Newman, R.A.; Krishnan, K. Differential Effects of Pravastatin and Simvastatin on the Growth of Tumor Cells from Different Organ Sites. PLoS ONE 2011, 6, e28813. [Google Scholar] [CrossRef]
- Caro-Maldonado, A.; Camacho, L.; Zabala-Letona, A.; Torrano, V.; Fernández-Ruiz, S.; Zamacola-Bascaran, K.; Arreal, L.; Valcárcel-Jiménez, L.; Martín-Martín, N.; Flores, J.M.; et al. Low-dose statin treatment increases prostate cancer aggressiveness. Oncotarget 2017, 9, 1494–1504. Available online: https://pubmed.ncbi.nlm.nih.gov/29416709 (accessed on 5 September 2021). [CrossRef]
- Murtola, T.J.; Syvälä, H.; Tolonen, T.; Helminen, M.; Riikonen, J.; Koskimäki, J.; Pakarainen, T.; Kaipia, A.; Isotalo, T.; Kujala, P.; et al. Atorvastatin Versus Placebo for Prostate Cancer Before Radical Prostatectomy-A Randomized, Double-blind, Placebo-controlled Clinical Trial. Eur. Urol. 2018, 74, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Longo, J.; Hamilton, R.J.; Masoomian, M.; Khurram, N.; Branchard, E.; Mullen, P.J.; Elbaz, M.; Hersey, K.; Chadwick, D.; Ghai, S.; et al. A pilot window-of-opportunity study of preoperative fluvastatin in localized prostate cancer. Prostate Cancer Prostatic Dis. 2020, 23, 630–637. [Google Scholar] [CrossRef]
- Rajora, M.A.; Zheng, G. Targeting SR-BI for Cancer Diagnostics, Imaging and Therapy. Front. Pharmacol. 2016, 7, 326. [Google Scholar] [CrossRef]
- Sarhadi, S.; Ganjali, S.; Pirro, M.; Sahebkar, A. The role of high-density lipoproteins in antitumor drug delivery. IUBMB Life 2019, 71, 1442–1452. [Google Scholar] [CrossRef]
- McMahon, K.M.; Plebanek, M.P.; Thaxton, C.S. Properties of Native High-Density Lipoproteins Inspire Synthesis of Actively Targeted In Vivo siRNA Delivery Vehicles. Adv. Funct. Mater. 2016, 26, 7824–7835. [Google Scholar] [CrossRef]
- Sabnis, S.; Sabnis, N.A.; Raut, S.; Lacko, A.G. Superparamagnetic reconstituted high-density lipoprotein nanocarriers for magnetically guided drug delivery. Int. J. Nanomed. 2017, 12, 1453–1464. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Maitland, N.J. Getting closer to prostate cancer in patients—What scientists should want from clinicians. J. Cancer Metastasis Treat. 2017, 3, 262–270. [Google Scholar] [CrossRef]
- Latonen, L.; Afyounian, E.; Jylhä, A.; Nättinen, J.; Aapola, U.; Annala, M.; Kivinummi, K.K.; Tammela, T.T.L.; Beuerman, R.W.; Uusitalo, H.; et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat. Commun. 2018, 9, 1176. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Function | Reference |
|---|---|---|---|
| Statins (in general) | HMGCR | Reduces HMG-CoA to mevalonate | [143] |
| Atorvastatin | SLCO2B1 | Androgen transporting gene (uptake) | [139] |
| DNAJA1 | Cochaperone protein (protects mut p53 form degradation) | [143] | |
| Pravastatin | SLCO2B1 | Androgen transporting gene (uptake) | [139] |
| Fatostatin | SCAP | SREBP2 and SREBP1 maturation | [145] |
| Tubulin | Maintenance of microtubule organization | [145] | |
| Lovastatin | DNAJA1 | Cochaperone protein (protects mut p53 form degradation) | [143] |
| Mevastatin | DNAJA1 | Cochaperone protein (protects mut p53 form degradation) | [143] |
| Betulin | SCAP-INSIGN interaction | SCAP/SREBP translocation | [147] |
| Xanthohumol | Sec23/24 (COPII vesicle) | SCAP/SREBP translocation | [146] |
| Combination therapy | |||
| Statins + Dipyridamole (antiplatelet agent) | Phosphodiesterase | Hydrolysis of cyclic nucleotides | [84,152] |
| Simvastin + enzalutamide (antiandrogen) | AR | Activation of AR signaling pathway | [74] |
| Fatostatin + Docetaxel (chemotherapeutic agent) | Tubulin | Maintenance of microtubule organization | [98,153] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Škara, L.; Huđek Turković, A.; Pezelj, I.; Vrtarić, A.; Sinčić, N.; Krušlin, B.; Ulamec, M. Prostate Cancer—Focus on Cholesterol. Cancers 2021, 13, 4696. https://doi.org/10.3390/cancers13184696
Škara L, Huđek Turković A, Pezelj I, Vrtarić A, Sinčić N, Krušlin B, Ulamec M. Prostate Cancer—Focus on Cholesterol. Cancers. 2021; 13(18):4696. https://doi.org/10.3390/cancers13184696
Chicago/Turabian StyleŠkara, Lucija, Ana Huđek Turković, Ivan Pezelj, Alen Vrtarić, Nino Sinčić, Božo Krušlin, and Monika Ulamec. 2021. "Prostate Cancer—Focus on Cholesterol" Cancers 13, no. 18: 4696. https://doi.org/10.3390/cancers13184696
APA StyleŠkara, L., Huđek Turković, A., Pezelj, I., Vrtarić, A., Sinčić, N., Krušlin, B., & Ulamec, M. (2021). Prostate Cancer—Focus on Cholesterol. Cancers, 13(18), 4696. https://doi.org/10.3390/cancers13184696