
Crosstalk between PRLR and EGFR/HER2 Signaling Pathways in Breast Cancer


{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
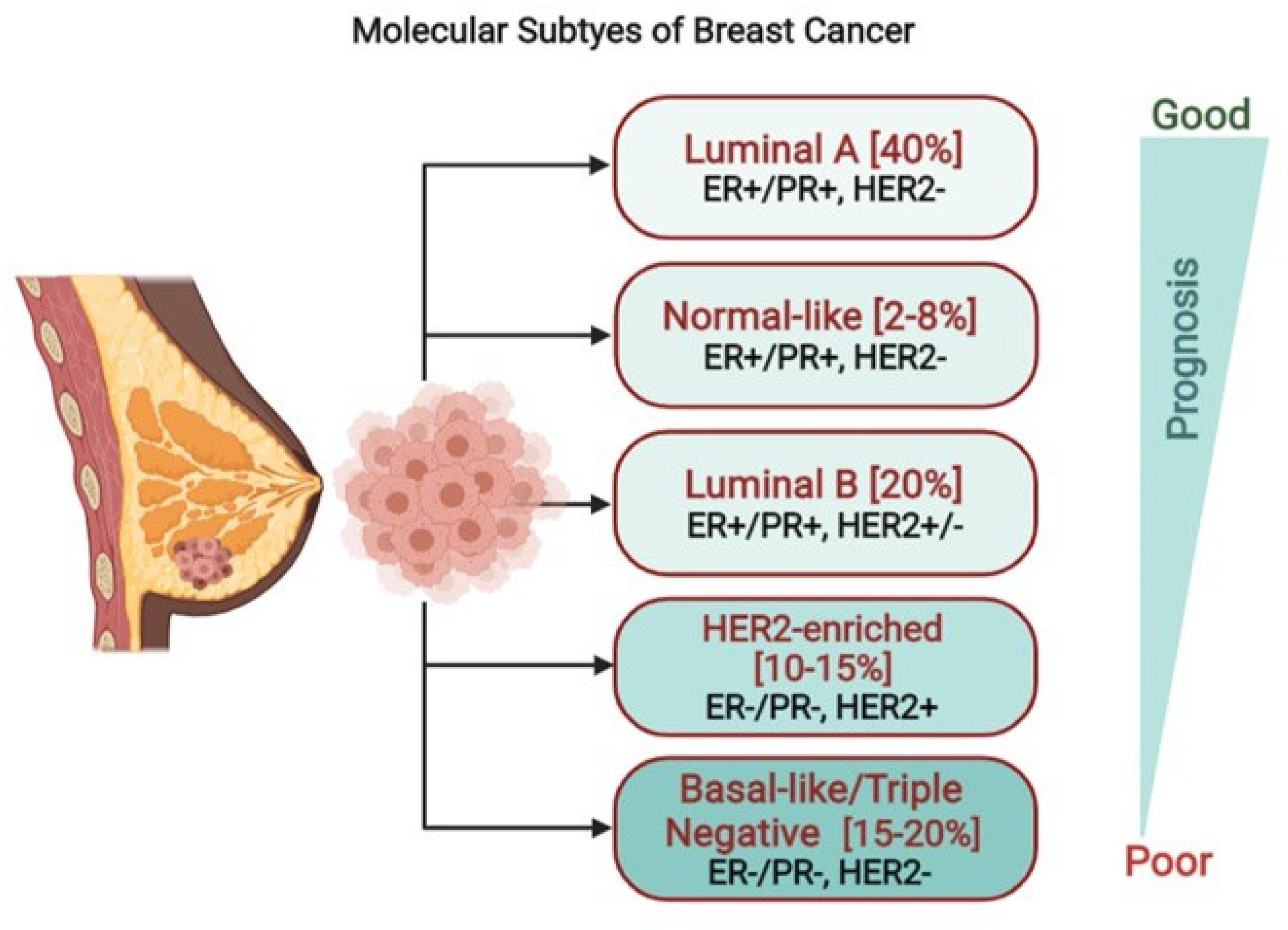
1. Introduction
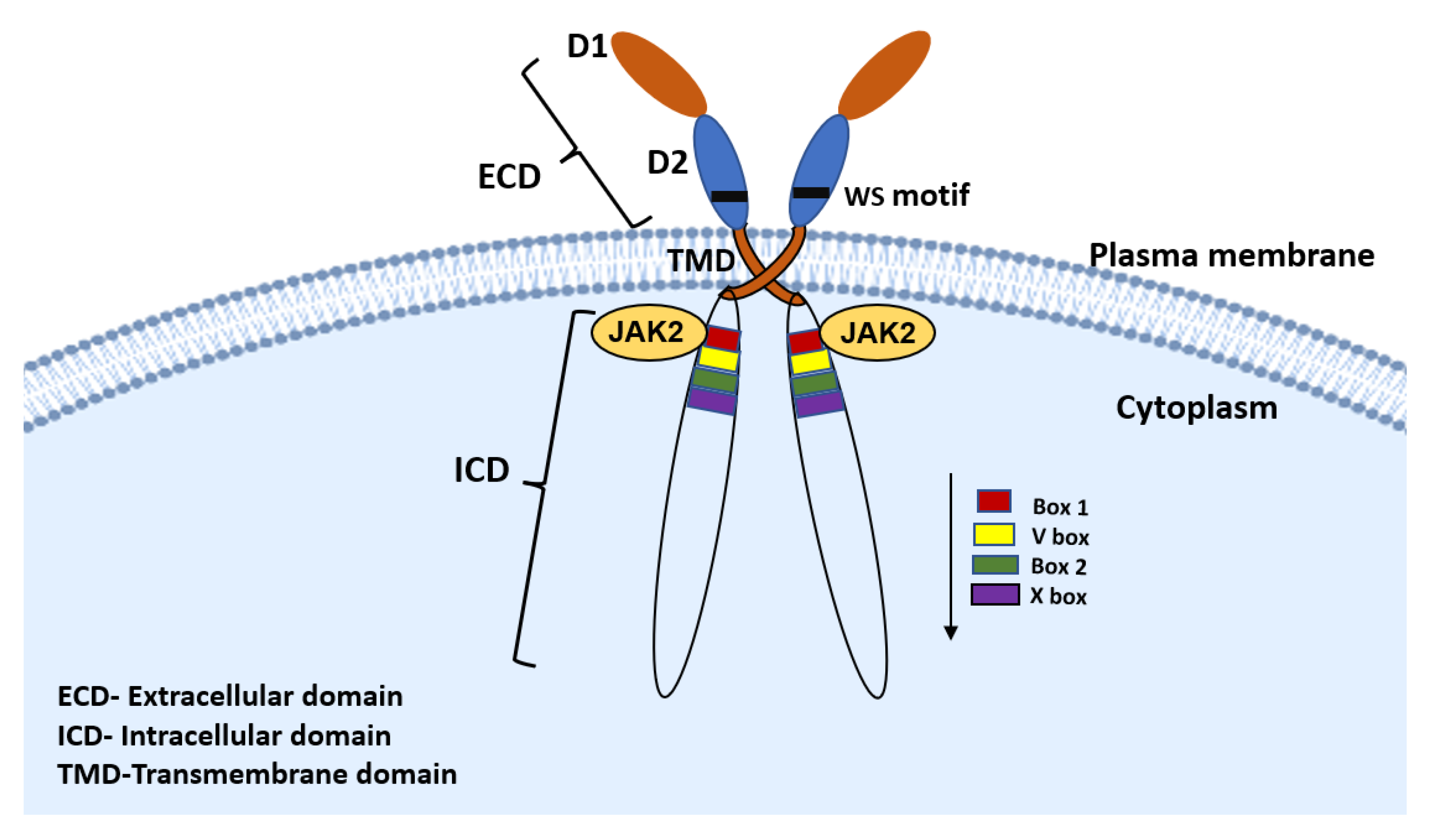
2. PRLR Signaling Pathway
3. ERBB Signaling Pathway
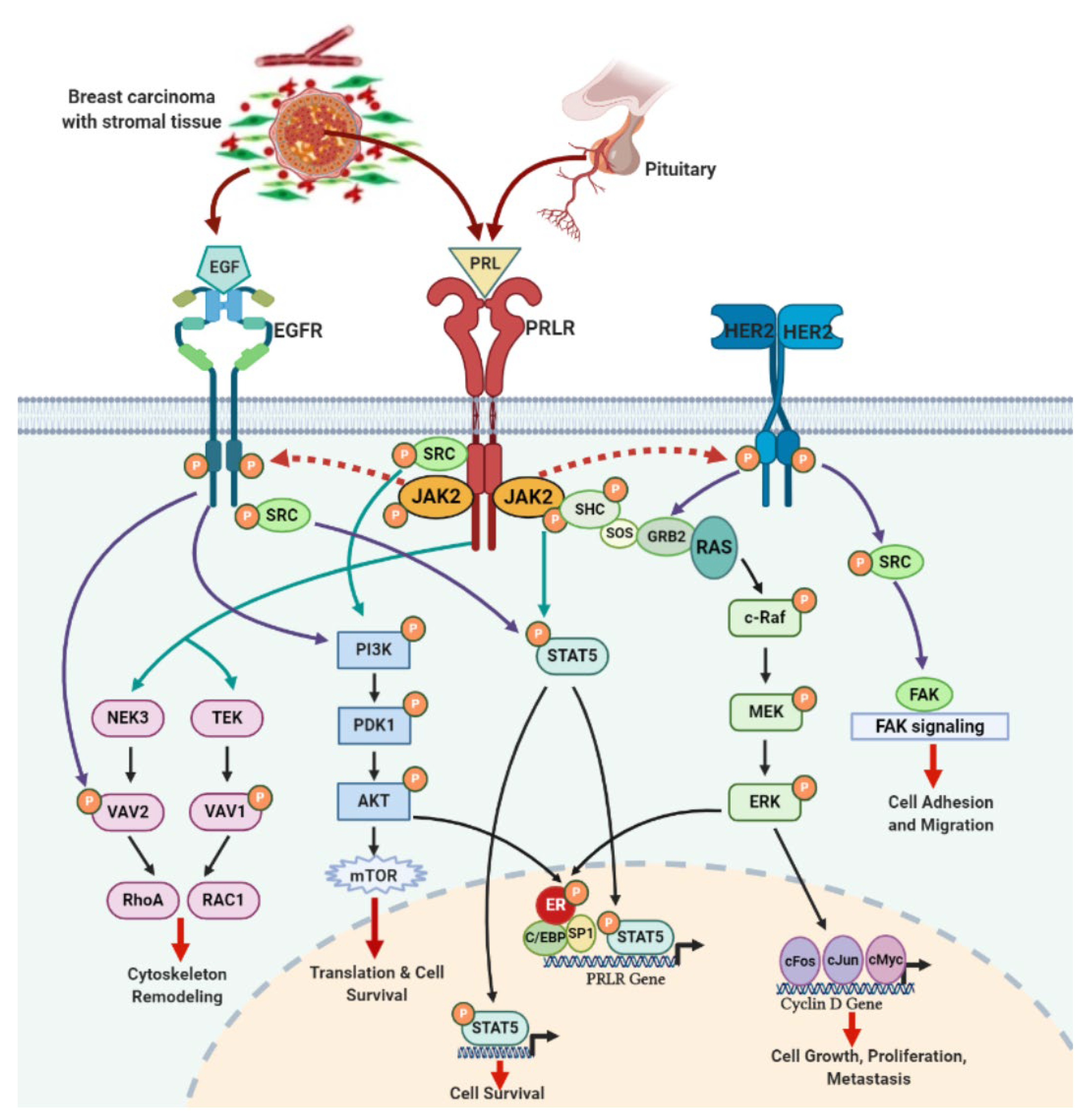
4. PRLR and EGFR Signaling Crosstalk in Breast Cancer
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R. Molecular mechanisms of Trastuzumab-based treatment in HER2-overexpressing breast cancer. ISRN Oncol. 2012, 2012, 428062. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanna, M.P.; Stern, D.F.; Edgerton, S.M.; Whalen, S.G.; Moore, D.; Thor, A.D. Relationship of epidermal growth factor receptor expression to ErbB-2 signaling activity and prognosis in breast cancer patients. J. Clin. Oncol. 2005, 23, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Nahta, R.; Hung, M.C.; Esteva, F.J. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 2004, 64, 2343–2346. [Google Scholar] [CrossRef]
- Orphanos, G.; Kountourakis, P. Targeting the HER2 receptor in metastatic breast cancer. Hematol. Oncol. Stem Cell Ther. 2012, 5, 127–137. [Google Scholar] [CrossRef]
- Ferreira, M.; Mesquita, M.; Quaresma, M.; Andre, S. Prolactin receptor expression in gynaecomastia and male breast carcinoma. Histopathology 2008, 53, 56–61. [Google Scholar] [CrossRef]
- Tworoger, S.S.; Hankinson, S.E. Prolactin and breast cancer etiology: An epidemiologic perspective. J. Mammary Gland. Biol. Neoplasia 2008, 13, 41–53. [Google Scholar] [CrossRef]
- Tworoger, S.S.; Eliassen, A.H.; Zhang, X.; Qian, J.; Sluss, P.M.; Rosner, B.A.; Hankinson, S.E. A 20-year prospective study of plasma prolactin as a risk marker of breast cancer development. Cancer Res. 2013, 73, 4810–4819. [Google Scholar] [CrossRef]
- Tworoger, S.S.; Rice, M.S.; Rosner, B.A.; Feeney, Y.B.; Clevenger, C.V.; Hankinson, S.E. Bioactive prolactin levels and risk of breast cancer: A nested case-control study. Cancer Epidemiol. Biomark. Prev. 2015, 24, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Hachim, I.Y.; Hachim, M.Y.; Lopez, V.M.; Lebrun, J.J.; Ali, S. Prolactin Receptor Expression is an independent favorable prognostic marker in human breast cancer. Appl. Immunohistochem. Mol. Morphol. 2016, 24, 238–245. [Google Scholar] [CrossRef]
- Hachim, I.Y.; Shams, A.; Lebrun, J.J.; Ali, S. A favorable role of prolactin in human breast cancer reveals novel pathway based gene signatures indicative of tumor differentiation and favorable patient outcome: Prolactin-induced mammary differentiation program in breast cancer prognosis. Hum. Pathol. 2016, 53, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Sa-nguanraksa, D.; Thasripoo, C.; Samarnthai, N.; Kummalue, T.; Thumrongtaradol, T.; O-charoenrat, P. The role of Prolactin/Prolactin Receptor polymorphisms and expression in breast cancer susceptibility and outcome. Trans. Cancer Res. 2020, 9, 10. [Google Scholar] [CrossRef]
- Hachim, I.Y.; López-Ozuna, V.M.; Hachim, M.Y.; Lebrun, J.J.; Ali, S. Prolactin hormone exerts anti-tumorigenic effects in HER-2 overexpressing breast cancer cells through regulation of stemness. Stem. Cell Res. 2019, 40, 101538. [Google Scholar] [CrossRef] [PubMed]
- Hachim, M.Y.; Hachim, I.Y.; Talaat, I.M.; Yakout, N.M.; Hamoudi, R. M1 Polarization markers are upregulated in basal-like breast cancer molecular subtype and associated with favorable patient outcome. Front. Immunol. 2020, 11, 560074. [Google Scholar] [CrossRef]
- Shams, A.; Binothman, N.; Boudreault, J.; Wang, N.; Shams, F.; Hamam, D.; Tian, J.; Moamer, A.; Dai, M.; Lebrun, J.-J.; et al. Prolactin receptor-driven combined luminal and epithelial differentiation in breast cancer restricts plasticity, stemness, tumorigenesis and metastasis. Oncogenesis 2021, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- López-Ozuna, V.M.; Hachim, I.Y.; Hachim, M.Y.; Lebrun, J.J.; Ali, S. Prolactin pro-differentiation pathway in triple negative breast cancer: Impact on prognosis and potential therapy. Sci. Rep. 2016, 6, 30934. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, K.A.; Shea, M.P.; Schuler, L.A. Modeling prolactin actions in breast cancer in vivo: Insights from the NRL-PRL mouse. Adv. Exp. Med. Biol. 2015, 846, 201–220. [Google Scholar] [CrossRef] [PubMed]
- von Lintig, F.C.; Dreilinger, A.D.; Varki, N.M.; Wallace, A.M.; Casteel, D.E.; Boss, G.R. Ras activation in human breast cancer. Breast Cancer Res. Treat. 2000, 62, 51–62. [Google Scholar] [CrossRef]
- Wright, K.L.; Adams, J.R.; Liu, J.C.; Loch, A.J.; Wong, R.G.; Jo, C.E.B.; Beck, L.A.; Santhanam, D.R.; Weiss, L.; Mei, X.; et al. Ras signaling is a key determinant for metastatic dissemination and poor survival of luminal breast cancer patients. Cancer Res. 2015, 75, 4960–4972. [Google Scholar] [CrossRef]
- Campbell, K.M.; O’Leary, K.A.; Rugowski, D.E.; Mulligan, W.A.; Barnell, E.K.; Skidmore, Z.L.; Krysiak, K.; Griffith, M.; Schuler, L.A.; Griffith, O.L. A Spontaneous Aggressive ERα + Mammary Tumor Model Is Driven by Kras Activation. Cell Rep. 2019, 28, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Gutzman, J.H.; Miller, K.K.; Schuler, L.A. Endogenous human prolactin and not exogenous human prolactin induces estrogen receptor alpha and prolactin receptor expression and increases estrogen responsiveness in breast cancer cells. J. Steroid Biochem. Mol. Biol. 2004, 88, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Kavarthapu, R.; Dufau, M.L. Essential role of endogenous prolactin and CDK7 in estrogen-induced upregulation of the prolactin receptor in breast cancer cells. Oncotarget 2017, 8, 27353–27363. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gutzman, J.H.; Nikolai, S.E.; Rugowski, D.E.; Watters, J.J.; Schuler, L.A. Prolactin and Estrogen Enhance the Activity of Activating Protein 1 in Breast Cancer Cells: Role of Extracellularly Regulated Kinase 1/2-Mediated Signals to c-fos. Mol. Endocrinol. 2005, 19, 1765–1778. [Google Scholar] [CrossRef]
- Dong, J.; Tsai-Morris, C.H.; Dufau, M.L. A novel estradiol/estrogen receptor alpha-dependent transcriptional mechanism controls expression of the human prolactin receptor. J. Biol. Chem. 2006, 281, 18825–18836. [Google Scholar] [CrossRef] [PubMed]
- Ben-Jonathan, N.; LaPensee, C.R.; LaPensee, E.W. What Can We Learn from Rodents about Prolactin in Humans? Endocr. Rev. 2008, 29, 1–41. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, C.H. ERalpha and STAT5a cross-talk: Interaction through C-terminal portions of the proteins decreases STAT5a phosphorylation, nuclear translocation, and DNA-binding. FEBS Lett. 2004, 572, 238–244. [Google Scholar] [CrossRef]
- Schroeder, M.D.; Symowicz, J.; Schuler, L.A. Prolactin modulates cell cycle regulators in mammary tumor epithelial cells. Mol. Endocrinol. 2002, 16, 45–57. [Google Scholar] [CrossRef]
- Acosta, J.J.; Munoz, R.M.; Gonzalez, L.; Subtil-Rodriguez, A.; Dominguez-Caceres, M.A.; Garcia-Martinez, J.M.; Calcabrini, A.; Lazaro-Trueba, I.; Martín-Pérez, J. Src mediates prolactin-dependent proliferation of T47D and MCF7 cells via the activation of focal adhesion kinase/Erk1/2 and phosphatidylinositol 3-kinase pathways. Mol. Endocrinol. 2003, 17, 2268–2282. [Google Scholar] [CrossRef]
- Kavarthapu, R.; Tsai Morris, C.H.; Dufau, M.L. Prolactin induces up-regulation of its cognate receptor in breast cancer cells via transcriptional activation of its generic promoter by cross-talk between ERα and STAT5. Oncotarget 2014, 5, 9079–9091. [Google Scholar] [CrossRef]
- Lopez Vicchi, F.; Becu-Villalobos, D. Prolactin: The Bright and the Dark Side. Endocrinology 2017, 158, 1556–1559. [Google Scholar] [CrossRef]
- Neradugomma, N.K.; Subramaniam, D.; Tawfik, O.W.; Goffin, V.; Kumar, T.R.; Jensen, R.A.; Anant, S. Prolactin signaling enhances colon cancer stemness by modulating Notch signaling in a Jak2-STAT3/ERK manner. Carcinogenesis 2014, 35, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Levina, V.V.; Nolen, B.; Su, Y.Y.; Godwin, A.K.; Fishman, D.; Liu, J.; Mor, G.; Maxwell, L.G.; Herberman, R.B.; Szczepanski, M.J.; et al. Biological significance of prolactin in gynecologic cancers. Cancer Res. 2009, 69, 5226–5233. [Google Scholar] [CrossRef] [PubMed]
- Dagil, R.; Knudsen, M.J.; Olsen, J.G.; O’Shea, C.; Franzmann, M.; Goffin, V.; Teilum, K.; Breinholt, J.; Kragelund, B.B. The WSXWS motif in cytokine receptors is a molecular switch involved in receptor activation: Insight from structures of the prolactin receptor. Structure 2012, 20, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Bole-Feysot, C.; Goffin, V.; Edery, M.; Binart, N.; Kelly, P.A. Prolactin (PRL) and its receptor: Actions, signal transduction pathways and phenotypes observed in prl receptor knockout mice. Endocrinol. Rev. 1998, 19, 225–268. [Google Scholar] [CrossRef] [PubMed]
- Aksamitiene, E.; Achanta, S.; Kolch, W.; Kholodenko, B.N.; Hoek, J.B.; Kiyatkin, A. Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cell Signal. 2011, 23, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Zambrano, A.; Lazaro-Trueba, I.; Lopéz, E.; González, J.J.; Martín-Pérez, J.; Aranda, A. Activation of the unliganded estrogen receptor by prolactin in breast cancer cells. Oncogene 2009, 28, 1298–1308. [Google Scholar] [CrossRef]
- Oladimeji, P.; Skerl, R.; Rusch, C.; Diakonova, M. Synergistic activation of erα by estrogen and prolactin in breast cancer cells requires tyrosyl phosphorylation of PAK1. Cancer Res. 2016, 76, 2600–2611. [Google Scholar] [CrossRef]
- Grible, J.M.; Zot, P.; Olex, A.L.; Hedrick, S.E.; Harrell, J.C.; Woock, A.E.; Idowu, M.O.; Clevenger, C.V. The human intermediate prolactin receptor is a mammary proto-oncogene. NPJ Breast Cancer 2021, 26, 37. [Google Scholar] [CrossRef] [PubMed]
- Halim, C.E.; Deng, S.; Ong, M.S.; Yap, C.T. Involvement of STAT5 in Oncogenesis. Biomedicines 2020, 8, 316. [Google Scholar] [CrossRef] [PubMed]
- Barash, I. Stat5 in breast cancer: Potential oncogenic activity coincides with positive prognosis for the disease. Carcinogenesis 2012, 33, 2320–2325. [Google Scholar] [CrossRef]
- Igelmann, S.; Neubauer, H.A.; Ferbeyre, G. STAT3 and STAT5 activation in solid cancers. Cancers 2019, 11, 1428. [Google Scholar] [CrossRef]
- Nevalainen, M.T.; Xie, J.; Torhorst, J.; Bubendorf, L.; Haas, P.; Kononen, J.; Sauter, G.; Rui, H. Signal transducer and activator of transcription-5 activation and breast cancer prognosis. J. Clin. Oncol. 2004, 22, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.J.; Peck, A.R.; Liu, C.; Tran, T.H.; Utama, F.E.; Sjolund, A.B.; Schaber, J.D.; Witkiewicz, A.K.; Rui, H. PTP1B suppresses prolactin activation of stat5 in breast cancer cells. Am. J. Pathol. 2010, 177, 2971–2983. [Google Scholar] [CrossRef]
- da Silva, P.L.; do Amaral, V.C.; Gabrielli, M.M.; Montt Guevara, P.; Mannella, E.C.; Baracat Soares, J.M., Jr.; Simoncini, T. Prolactin promotes breast cancer cell migration through actin cytoskeleton remodeling. Front. Endocrinol. 2015, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; de Maria, J.E.; Freier, D.O.; Riegel, A.M.; Clevenger, C.V. Novel association of Vav2 and Nek3 modulates signaling through the human prolactin receptor. Mol. Endocrinol. 2005, 19, 939–949. [Google Scholar] [CrossRef]
- Bratthauer, G.L.; Strauss, B.L.; Barner, R. Reversed expression of the JAK/STAT pathway related proteins prolactin receptor and STAT5a in normal and abnormal breast epithelial cells. Breast Cancer 2008, 1, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Barcus, C.E.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells. J. Biol. Chem. 2013, 288, 12722–12732. [Google Scholar] [CrossRef]
- Barcus, C.E.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Prolactin signaling through focal adhesion complexes is amplified by stiff extracellular matrices in breast cancer cells. Oncotarget 2016, 7, 48093–48106. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Yarden, Y. EGF-ERBB signaling towards the systems level. Nat. Rev. Mol. Cell. Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef]
- Harari, D.; Yarden, Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene 2000, 19, 6102–6114. [Google Scholar] [CrossRef]
- Ren, Z.; Schaefer, T.S. ErbB-2 activates Stat3 alpha in a Src- and JAK2-dependent manner. J. Biol. Chem. 2002, 277, 38486–38493. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The erbb network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Kato, Y.; Tapping, R.I.; Huang, S.; Watson, M.H.; Ulevitch, R.J.; Lee, J.D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998, 395, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Antoon, J.W.; Martin, E.C.; Lai, R.; Salvo, V.A.; Tang, Y.; Nitzchke, A.M.; Elliott, S.; Nam, S.Y.; Xiong, W.; Rhodes, L.V.; et al. MEK5/ERK5 signaling suppresses estrogen receptor expression and promotes hormone-independent tumorigenesis. PLoS ONE 2013, 8, e69291. [Google Scholar] [CrossRef]
- Montero, J.C.; Ocaña, A.; Abad, M.; Ortiz-Ruiz, M.J.; Pandiella, A.; Esparís-Ogando, A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS ONE 2009, 4, e5565. [Google Scholar] [CrossRef] [PubMed]
- Wennbo, H.; Törnell, J. The role of prolactin and growth hormone in breast cancer. Oncogene 2000, 19, 1072–1076. [Google Scholar] [CrossRef][Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013, 3, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Witton, C.J.; Reeves, J.R.; Going, J.J.; Cooke, T.G.; Bartlett, J.M. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J. Pathol. 2003, 200, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.; Montone, K.T.; Powell, C.M.; Tomaszewski, J.E.; Clevenger, C.V. Expression of prolactin and its receptor in human breast carcinoma. Endocrinology 1997, 138, 5555–5560. [Google Scholar] [CrossRef] [PubMed]
- Touraine, P.; Martini, J.F.; Zafrani, B.; Durand, J.C.; Labaille, F.; Malet, C.; Nicolas, A.; Trivin, C.; Postel-Vinay, M.C.; Kuttenn, F.; et al. Increased expression of prolactin receptor gene assessed by quantitative polymerase chain reaction in human breast tumors versus normal breast tissues. J. Clin. Endocrinol. Metab. 1998, 83, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Peston, D.; Vonderhaar, B.K.; Shousha, S. Expression of prolactin receptors in normal, benign, and malignant breast tissue: An immunohistological study. J. Clin. Pathol. 2001, 54, 956–960. [Google Scholar] [CrossRef]
- Guo, P.; Pu, T.; Chen, S.; Qiu, Y.; Zhong, X.; Zheng, H.; Chen, L.; Bu, H.; Ye, F. Breast cancers with EGFR and HER2 co-amplification favor distant metastasis and poor clinical outcome. Oncol. Lett. 2017, 14, 6562–6570. [Google Scholar] [CrossRef][Green Version]
- Huang, Y.; Li, X.; Jiang, J.; Frank, S.J. Prolactin modulates phosphorylation, signaling and trafficking of epidermal growth factor receptor in human T47D breast cancer cells. Oncogene 2006, 25, 7565–7576. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.J. Mechanistic aspects of crosstalk between GH and PRL and ErbB receptor family signaling. J. Mammary Gland Biol. Neoplasia 2008, 13, 119–129. [Google Scholar] [CrossRef]
- Kavarthapu, R.; Dufau, M.L. Role of EGF/ERBB1 in the transcriptional regulation of the prolactin receptor independent of estrogen and prolactin in breast cancer cells. Oncotarget 2016, 7, 65602–65613. [Google Scholar] [CrossRef]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. STAT-mediated EGFR signaling in cancer. J. Cell Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef]
- Maus, M.V.; Reilly, S.C.; Clevenger, C.V. Prolactin as a chemoattractant for human breast carcinoma. Endocrinology 1999, 140, 5447–5450. [Google Scholar] [CrossRef]
- Shin, I. HER2 Signaling in Breast Cancer. Adv. Exp. Med. Biol. 2021, 1187, 53–79. [Google Scholar] [CrossRef] [PubMed]
- Schlam, I.; Swain, S.M. HER2-positive breast cancer and tyrosine kinase inhibitors: The time is now. NPJ Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Borcherding, D.C.; Hugo, E.R.; Fox, S.R.; Jacobson, E.M.; Hunt, B.G.; Merino, E.J.; Ben-Jonathan, N. Suppression of breast cancer by small molecules that block the prolactin receptor. Cancers 2021, 13, 2662. [Google Scholar] [CrossRef]
- Minami, H.; Ando, Y.; Tamura, K.; Tajima, T.; Isaacs, R. Phase I study of LFA102 in patients with advanced breast cancer or castration-resistant prostate cancer. Anticancer Res. 2020, 40, 5229–5235. [Google Scholar] [CrossRef]
- Scotti, M.L.; Langenheim, J.F.; Tomblyn, S.; Springs, A.E.B.; Chen, W.Y. Additive effects of a prolactin receptor antagonist, G129R, and herceptin on inhibition of HER2-overexpressing breast cancer cells. Breast Cancer Res. Treat. 2008, 111, 241–250. [Google Scholar] [CrossRef]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kavarthapu, R.; Anbazhagan, R.; Dufau, M.L. Crosstalk between PRLR and EGFR/HER2 Signaling Pathways in Breast Cancer. Cancers 2021, 13, 4685. https://doi.org/10.3390/cancers13184685
Kavarthapu R, Anbazhagan R, Dufau ML. Crosstalk between PRLR and EGFR/HER2 Signaling Pathways in Breast Cancer. Cancers. 2021; 13(18):4685. https://doi.org/10.3390/cancers13184685
Chicago/Turabian StyleKavarthapu, Raghuveer, Rajakumar Anbazhagan, and Maria L. Dufau. 2021. "Crosstalk between PRLR and EGFR/HER2 Signaling Pathways in Breast Cancer" Cancers 13, no. 18: 4685. https://doi.org/10.3390/cancers13184685
APA StyleKavarthapu, R., Anbazhagan, R., & Dufau, M. L. (2021). Crosstalk between PRLR and EGFR/HER2 Signaling Pathways in Breast Cancer. Cancers, 13(18), 4685. https://doi.org/10.3390/cancers13184685