
Estrogen Receptor-Alpha and p53 Status as Regulators of AMPK and mTOR in Luminal Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Reagents
2.3. Plasmid Expression/siRNA Transfections
2.4. Protein Lysis and Immunoblotting
2.5. Quantitative Real-Time PCR (qRT-PCR)
2.6. Proximity Ligation Assay (PLA)
2.7. Clonogenic Survival Assay
2.8. Statistical Analysis
3. Results
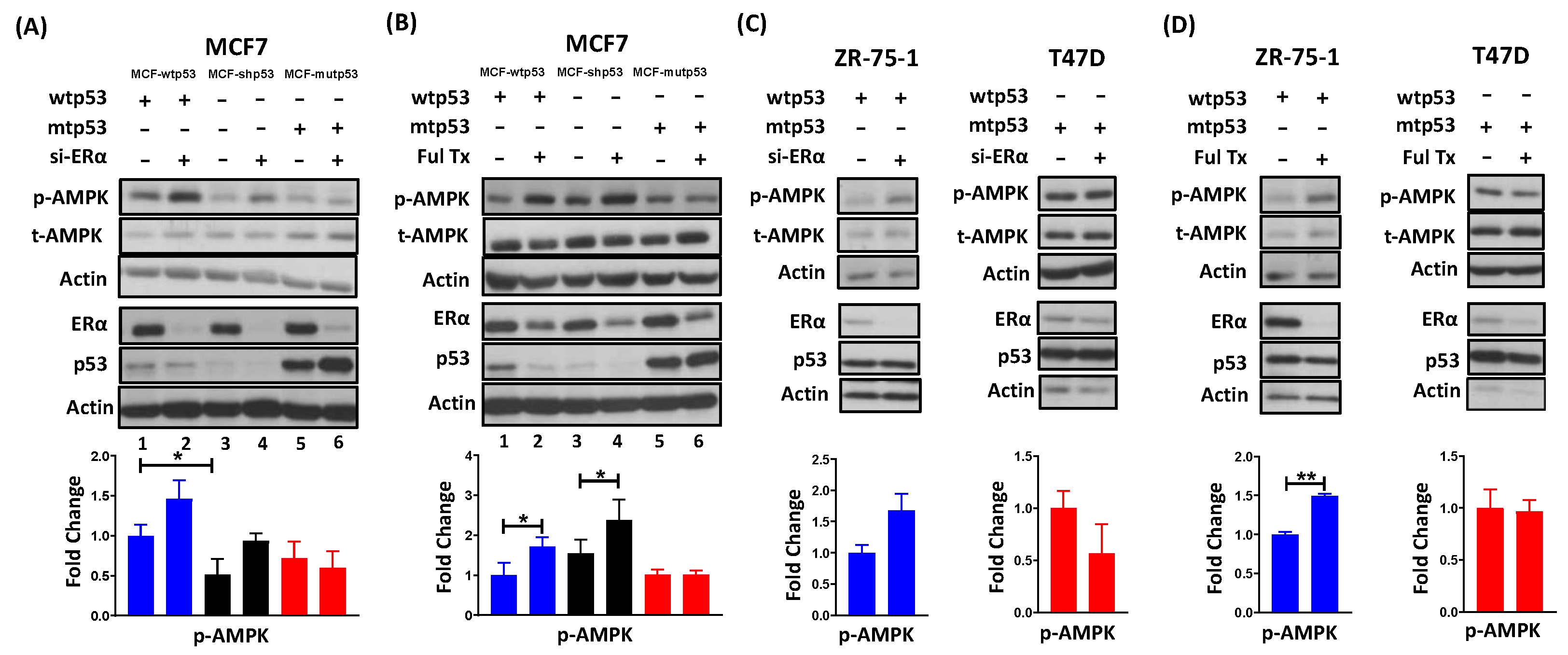
3.1. Requirement of ERα for Repressing AMPK Phosphorylation Is Dependent on p53 Mutational Status
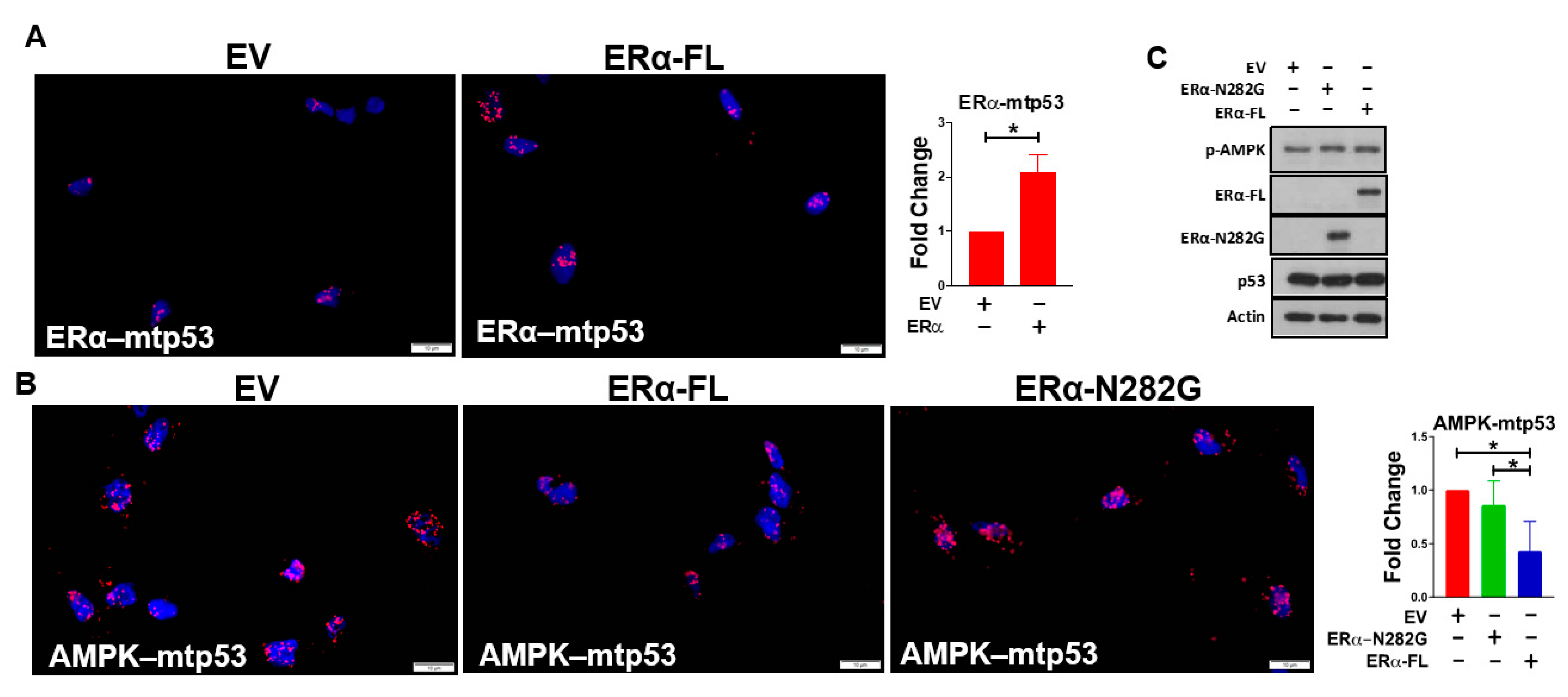
3.2. ERα Impairs Mutant p53’s Ability to Bind AMPK
3.3. ERα Regulates mTOR Signaling through AMPK-Dependent and -Independent Mechanisms
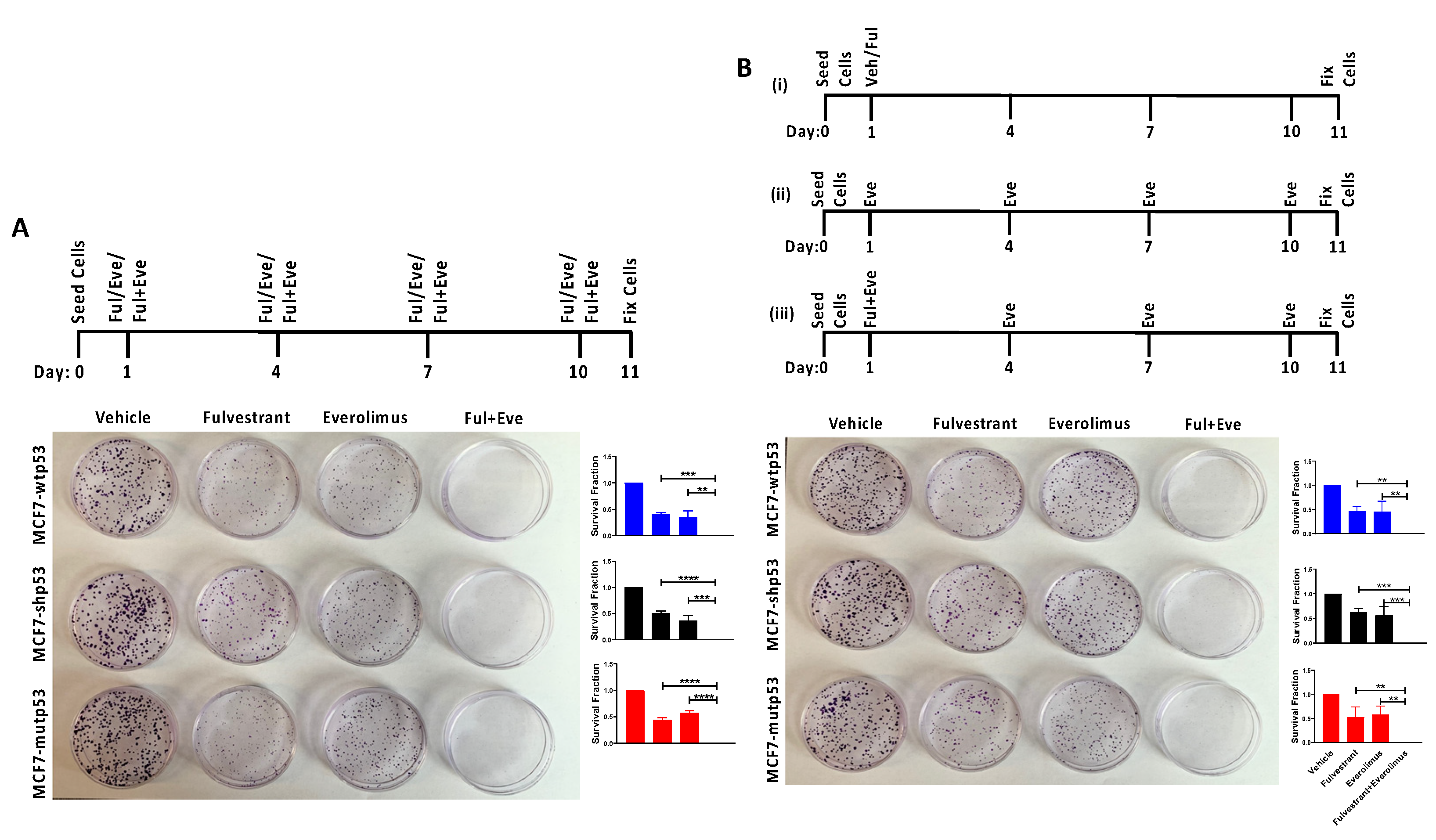
3.4. Effect of Combined mTOR Inhibition and ET on Clonogenic Survival: Drug Dosing Scheme
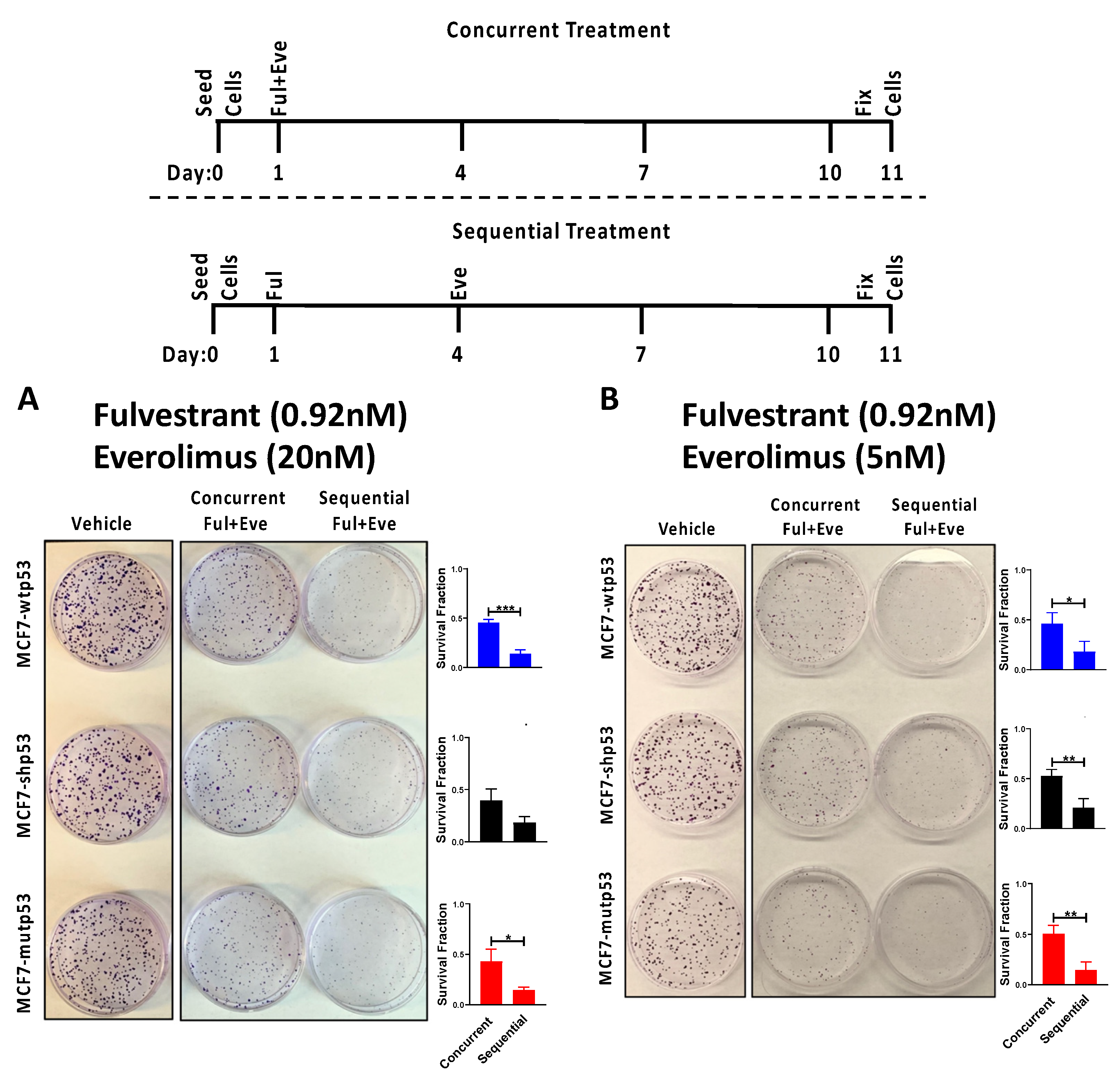
3.5. Effect of Combined mTOR Inhibition and ET on Clonogenic Survival: Drug Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Yu, D.; Hung, M.C.; Hortobagyi, G.N.; Esteva, F.J. Mechanisms of disease: Understanding resistance to HER2-targeted therapy in human breast cancer. Nat. Clin. Pract. Oncol. 2006, 3, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative, G. Aromatase inhibitors versus tamoxifen in early breast cancer: Patient-level meta-analysis of the randomised trials. Lancet 2015, 386, 1341–1352. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Ungerleider, N.A.; Rao, S.G.; Shahbandi, A.; Yee, D.; Niu, T.; Frey, W.D.; Jackson, J.G. Breast cancer survival predicted by TP53 mutation status differs markedly depending on treatment. Breast Cancer Res. 2018, 20, 115. [Google Scholar] [CrossRef]
- Berns, E.M.; Foekens, J.A.; Vossen, R.; Look, M.P.; Devilee, P.; Henzen-Logmans, S.C.; van Staveren, I.L.; van Putten, W.L.; Inganas, M.; Meijer-van Gelder, M.E.; et al. Complete sequencing of TP53 predicts poor response to systemic therapy of advanced breast cancer. Cancer Res. 2000, 60, 2155–2162. [Google Scholar]
- Konduri, S.D.; Medisetty, R.; Liu, W.; Kaipparettu, B.A.; Srivastava, P.; Brauch, H.; Fritz, P.; Swetzig, W.M.; Gardner, A.E.; Khan, S.A.; et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 15081–15086. [Google Scholar] [CrossRef]
- Yamamoto, M.; Hosoda, M.; Nakano, K.; Jia, S.; Hatanaka, K.C.; Takakuwa, E.; Hatanaka, Y.; Matsuno, Y.; Yamashita, H. p53 accumulation is a strong predictor of recurrence in estrogen receptor-positive breast cancer patients treated with aromatase inhibitors. Cancer Sci. 2014, 105, 81–88. [Google Scholar] [CrossRef]
- Yamashita, H.; Toyama, T.; Nishio, M.; Ando, Y.; Hamaguchi, M.; Zhang, Z.; Kobayashi, S.; Fujii, Y.; Iwase, H. p53 protein accumulation predicts resistance to endocrine therapy and decreased post-relapse survival in metastatic breast cancer. Breast Cancer Res. 2006, 8, R48. [Google Scholar] [CrossRef]
- Jhaveri, T.Z.; Woo, J.; Shang, X.; Park, B.H.; Gabrielson, E. AMP-activated kinase (AMPK) regulates activity of HER2 and EGFR in breast cancer. Oncotarget 2015, 6, 14754–14765. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vellon, L.; Mehmi, I.; Oza, B.P.; Ropero, S.; Colomer, R.; Lupu, R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10715–10720. [Google Scholar] [CrossRef]
- Fox, M.M.; Phoenix, K.N.; Kopsiaftis, S.G.; Claffey, K.P. AMP-Activated Protein Kinase alpha 2 Isoform Suppression in Primary Breast Cancer Alters AMPK Growth Control and Apoptotic Signaling. Genes Cancer 2013, 4, 3–14. [Google Scholar] [CrossRef]
- Zhuang, Y.; Miskimins, W.K. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J. Mol. Signal 2008, 3, 18. [Google Scholar] [CrossRef]
- Yi, Y.; Chen, D.; Ao, J.; Zhang, W.; Yi, J.; Ren, X.; Fei, J.; Li, F.; Niu, M.; Chen, H.; et al. Transcriptional suppression of AMPKalpha1 promotes breast cancer metastasis upon oncogene activation. Proc. Natl. Acad. Sci. USA 2020, 117, 8013–8021. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ross, F.A.; Gowans, G.J.; Tibarewal, P.; Leslie, N.R.; Hardie, D.G. Phosphorylation by Akt within the ST loop of AMPK-alpha1 down-regulates its activation in tumour cells. Biochem J. 2014, 459, 275–287. [Google Scholar] [CrossRef]
- Casimiro, M.C.; Di Sante, G.; Di Rocco, A.; Loro, E.; Pupo, C.; Pestell, T.G.; Bisetto, S.; Velasco-Velazquez, M.A.; Jiao, X.; Li, Z.; et al. Cyclin D1 Restrains Oncogene-Induced Autophagy by Regulating the AMPK-LKB1 Signaling Axis. Cancer Res. 2017, 77, 3391–3405. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mejia, I.C.; Lagarrigue, S.; Giralt, A.; Martinez-Carreres, L.; Zanou, N.; Denechaud, P.D.; Castillo-Armengol, J.; Chavey, C.; Orpinell, M.; Delacuisine, B.; et al. CDK4 Phosphorylates AMPKalpha2 to Inhibit Its Activity and Repress Fatty Acid Oxidation. Mol. Cell 2017, 68, 336–349 e336. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.; Jang, S.Y.; Kim, C.; Choi, Y.; Kim, A. Anticancer effect of metformin on estrogen receptor-positive and tamoxifen-resistant breast cancer cell lines. Oncol. Rep. 2016, 35, 2553–2560. [Google Scholar] [CrossRef][Green Version]
- Lipovka, Y.; Chen, H.; Vagner, J.; Price, T.J.; Tsao, T.S.; Konhilas, J.P. Oestrogen receptors interact with the alpha-catalytic subunit of AMP-activated protein kinase. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Mauro, L.; Naimo, G.D.; Gelsomino, L.; Malivindi, R.; Bruno, L.; Pellegrino, M.; Tarallo, R.; Memoli, D.; Weisz, A.; Panno, M.L.; et al. Uncoupling effects of estrogen receptor alpha on LKB1/AMPK interaction upon adiponectin exposure in breast cancer. FASEB J. 2018, 32, 4343–4355. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.; Chen, L.; Wang, J.; Zhang, M.; Yang, H.; Ma, Y.; Budanov, A.; Lee, J.H.; Karin, M.; Li, J. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J 2015, 29, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Linher-Melville, K.; Tsakiridis, T.; Singh, G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS ONE 2012, 7, e32035. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef]
- Liu, W.; Konduri, S.D.; Bansal, S.; Nayak, B.K.; Rajasekaran, S.A.; Karuppayil, S.M.; Rajasekaran, A.K.; Das, G.M. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 2006, 281, 9837–9840. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, A.; Konduri, S.D.; Liu, W.; Bansal, S.; Li, F.; Das, G.M. Estrogen receptor alpha inhibits p53-mediated transcriptional repression: Implications for the regulation of apoptosis. Cancer Res. 2007, 67, 7746–7755. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Katzenellenbogen, B.S. Estrogen Receptor-beta Modulation of the ERalpha-p53 Loop Regulating Gene Expression, Proliferation, and Apoptosis in Breast Cancer. Horm. Cancer 2017, 8, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.A.; Bhat-Nakshatri, P.; Patel, N.M.; Constantinidou, D.; Ali, S.; Nakshatri, H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: A new model for anti-estrogen resistance. J. Biol. Chem. 2001, 276, 9817–9824. [Google Scholar] [CrossRef]
- Miller, T.W.; Perez-Torres, M.; Narasanna, A.; Guix, M.; Stal, O.; Perez-Tenorio, G.; Gonzalez-Angulo, A.M.; Hennessy, B.T.; Mills, G.B.; Kennedy, J.P.; et al. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res 2009, 69, 4192–4201. [Google Scholar] [CrossRef] [PubMed]
- Beeram, M.; Tan, Q.T.; Tekmal, R.R.; Russell, D.; Middleton, A.; DeGraffenried, L.A. Akt-induced endocrine therapy resistance is reversed by inhibition of mTOR signaling. Ann. Oncol. 2007, 18, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- deGraffenried, L.A.; Friedrichs, W.E.; Russell, D.H.; Donzis, E.J.; Middleton, A.K.; Silva, J.M.; Roth, R.A.; Hidalgo, M. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin. Cancer Res. 2004, 10, 8059–8067. [Google Scholar] [CrossRef] [PubMed]
- Ghayad, S.E.; Bieche, I.; Vendrell, J.A.; Keime, C.; Lidereau, R.; Dumontet, C.; Cohen, P.A. mTOR inhibition reverses acquired endocrine therapy resistance of breast cancer cells at the cell proliferation and gene-expression levels. Cancer Sci. 2008, 99, 1992–2003. [Google Scholar] [CrossRef]
- Geter, P.A.; Ernlund, A.W.; Bakogianni, S.; Alard, A.; Arju, R.; Giashuddin, S.; Gadi, A.; Bromberg, J.; Schneider, R.J. Hyperactive mTOR and MNK1 phosphorylation of eIF4E confer tamoxifen resistance and estrogen independence through selective mRNA translation reprogramming. Genes Dev. 2017, 31, 2235–2249. [Google Scholar] [CrossRef]
- Swetzig, W.M.; Wang, J.; Das, G.M. Estrogen receptor alpha (ERalpha/ESR1) mediates the p53-independent overexpression of MDM4/MDMX and MDM2 in human breast cancer. Oncotarget 2016, 7, 16049–16069. [Google Scholar] [CrossRef]
- Mukhopadhyay, U.K.; Oturkar, C.C.; Adams, C.; Wickramasekera, N.; Bansal, S.; Medisetty, R.; Miller, A.; Swetzig, W.M.; Silwal-Pandit, L.; Borresen-Dale, A.L.; et al. TP53 Status as a Determinant of Pro- vs. Anti-Tumorigenic Effects of Estrogen Receptor-Beta in Breast Cancer. J. Natl. Cancer Inst. 2019, 111, 1202–1215. [Google Scholar] [CrossRef]
- Liu, W.; Ip, M.M.; Podgorsak, M.B.; Das, G.M. Disruption of estrogen receptor alpha-p53 interaction in breast tumors: A novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res. Treat 2009, 115, 43–50. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Levine, A.J. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010, 20, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Maruani, D.M.; Spiegel, T.N.; Harris, E.N.; Shachter, A.S.; Unger, H.A.; Herrero-Gonzalez, S.; Holz, M.K. Estrogenic regulation of S6K1 expression creates a positive regulatory loop in control of breast cancer cell proliferation. Oncogene 2012, 31, 5073–5080. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Henske, E.P. Estrogen-induced activation of mammalian target of rapamycin is mediated via tuberin and the small GTPase Ras homologue enriched in brain. Cancer Res 2006, 66, 9461–9466. [Google Scholar] [CrossRef]
- Cuesta, R.; Gritsenko, M.A.; Petyuk, V.A.; Shukla, A.K.; Tsai, C.F.; Liu, T.; McDermott, J.E.; Holz, M.K. Phosphoproteome Analysis Reveals Estrogen-ER Pathway as a Modulator of mTOR Activity Via DEPTOR. Mol. Cell Proteom. 2019, 18, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.C.; Debled, M.; Spaeth, D.; et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: A GINECO study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Kornblum, N.; Zhao, F.; Manola, J.; Klein, P.; Ramaswamy, B.; Brufsky, A.; Stella, P.J.; Burnette, B.; Telli, M.; Makower, D.F.; et al. Randomized Phase II Trial of Fulvestrant Plus Everolimus or Placebo in Postmenopausal Women With Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer Resistant to Aromatase Inhibitor Therapy: Results of PrE0102. J. Clin. Oncol. 2018, 36, 1556–1563. [Google Scholar] [CrossRef]
- Royce, M.; Bachelot, T.; Villanueva, C.; Ozguroglu, M.; Azevedo, S.J.; Cruz, F.M.; Debled, M.; Hegg, R.; Toyama, T.; Falkson, C.; et al. Everolimus Plus Endocrine Therapy for Postmenopausal Women with Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: A Clinical Trial. JAMA Oncol. 2018, 4, 977–984. [Google Scholar] [CrossRef]
- Jerusalem, G.; Mariani, G.; Ciruelos, E.M.; Martin, M.; Tjan-Heijnen, V.C.; Neven, P.; Gavila, J.G.; Michelotti, A.; Montemurro, F.; Generali, D.; et al. Safety of everolimus plus exemestane in patients with hormone-receptor-positive, HER2-negative locally advanced or metastatic breast cancer progressing on prior non-steroidal aromatase inhibitors: Primary results of a phase IIIb, open-label, single-arm, expanded-access multicenter trial (BALLET). Ann. Oncol. 2016, 27, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Steger, G.G.; Egle, D.; Bartsch, R.; Pfeiler, G.; Petru, E.; Greil, R.; Helfgott, R.; Marth, C.; Ohler, L.; Hubalek, M.; et al. Efficacy and safety of everolimus plus exemestane in patients with HR+, HER2- advanced breast cancer progressing on/after prior endocrine therapy in routine clinical practice: Primary results from the non-interventional study, STEPAUT. Breast 2020, 50, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Tesch, H.; Stoetzer, O.; Decker, T.; Kurbacher, C.M.; Marme, F.; Schneeweiss, A.; Mundhenke, C.; Distelrath, A.; Fasching, P.A.; Lux, M.P.; et al. Efficacy and safety of everolimus plus exemestane in postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative locally advanced or metastatic breast cancer: Results of the single-arm, phase IIIB 4EVER trial. Int. J. Cancer 2019, 144, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Lazar, A.A.; Bondarenko, I.; Garin, A.M.; Brincat, S.; Chow, L.; Sun, Y.; Neskovic-Konstantinovic, Z.; Guimaraes, R.C.; Fumoleau, P.; et al. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2013, 31, 195–202. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef]
- Yang, W.; Schwartz, G.N.; Marotti, J.D.; Chen, V.; Traphagen, N.A.; Gui, J.; Miller, T.W. Estrogen receptor alpha drives mTORC1 inhibitor-induced feedback activation of PI3K/AKT in ER+ breast cancer. Oncotarget 2018, 9, 8810–8822. [Google Scholar] [CrossRef]
- Olivier, M.; Langerod, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bieche, I.; et al. The clinical value of somatic TP53 gene mutations in 1794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef]
- Tsai, L.H.; Chen, P.M.; Cheng, Y.W.; Chen, C.Y.; Sheu, G.T.; Wu, T.C.; Lee, H. LKB1 loss by alteration of the NKX2-1/p53 pathway promotes tumor malignancy and predicts poor survival and relapse in lung adenocarcinomas. Oncogene 2014, 33, 3851–3860. [Google Scholar] [CrossRef][Green Version]
- Xie, B.; Nagalingam, A.; Kuppusamy, P.; Muniraj, N.; Langford, P.; Gyorffy, B.; Saxena, N.K.; Sharma, D. Benzyl Isothiocyanate potentiates p53 signaling and antitumor effects against breast cancer through activation of p53-LKB1 and p73-LKB1 axes. Sci. Rep. 2017, 7, 40070. [Google Scholar] [CrossRef]
- Alayev, A.; Salamon, R.S.; Berger, S.M.; Schwartz, N.S.; Cuesta, R.; Snyder, R.B.; Holz, M.K. mTORC1 directly phosphorylates and activates ERalpha upon estrogen stimulation. Oncogene 2016, 35, 3535–3543. [Google Scholar] [CrossRef]
- Yamnik, R.L.; Digilova, A.; Davis, D.C.; Brodt, Z.N.; Murphy, C.J.; Holz, M.K. S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J. Biol. Chem. 2009, 284, 6361–6369. [Google Scholar] [CrossRef] [PubMed]
- Yamnik, R.L.; Holz, M.K. mTOR/S6K1 and MAPK/RSK signaling pathways coordinately regulate estrogen receptor alpha serine 167 phosphorylation. FEBS Lett. 2010, 584, 124–128. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandhi, N.; Oturkar, C.C.; Das, G.M. Estrogen Receptor-Alpha and p53 Status as Regulators of AMPK and mTOR in Luminal Breast Cancer. Cancers 2021, 13, 3612. https://doi.org/10.3390/cancers13143612
Gandhi N, Oturkar CC, Das GM. Estrogen Receptor-Alpha and p53 Status as Regulators of AMPK and mTOR in Luminal Breast Cancer. Cancers. 2021; 13(14):3612. https://doi.org/10.3390/cancers13143612
Chicago/Turabian StyleGandhi, Nishant, Chetan C. Oturkar, and Gokul M. Das. 2021. "Estrogen Receptor-Alpha and p53 Status as Regulators of AMPK and mTOR in Luminal Breast Cancer" Cancers 13, no. 14: 3612. https://doi.org/10.3390/cancers13143612
APA StyleGandhi, N., Oturkar, C. C., & Das, G. M. (2021). Estrogen Receptor-Alpha and p53 Status as Regulators of AMPK and mTOR in Luminal Breast Cancer. Cancers, 13(14), 3612. https://doi.org/10.3390/cancers13143612