
Metabolic Hormones Modulate Macrophage Inflammatory Responses


Abstract
Simple Summary
Abstract
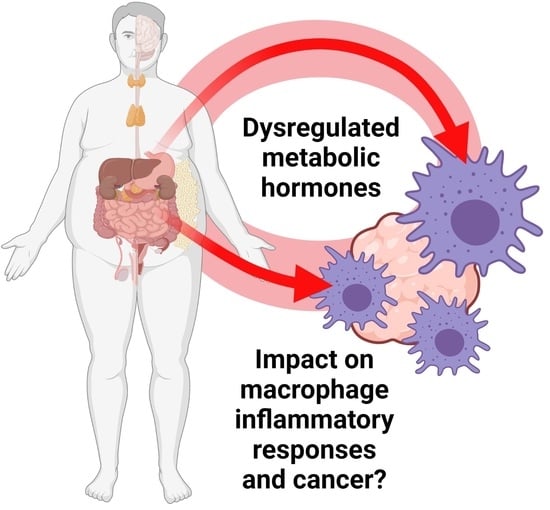
1. Metabolic Hormones Modulate Macrophage Inflammatory Responses
1.1. Introduction
1.2. The Role of Macrophages in Cancer Development
2. Metabolic Hormones Play Roles in Modulating Macrophage Inflammatory Responses
2.1. Cholecystokinin (CCK)
2.1.1. Origin and Function
2.1.2. CCK and Cancer Association
2.1.3. CCK Modulates Macrophage Inflammatory Responses
2.2. FABP4
2.2.1. Origin and Function
2.2.2. FABP4 and Cancer Association
2.2.3. FABP4 Modulates Macrophage Inflammatory Responses
2.3. Gastrin
2.3.1. Origin and Function
2.3.2. Gastrin and Cancer Association
2.3.3. Gastrin Modulates Macrophage Inflammatory Responses
2.4. Ghrelin
2.4.1. Origin and Function
2.4.2. Ghrelin and Cancer Association
2.4.3. Ghrelin Modulates Macrophage Inflammatory Responses
2.5. Incretins
2.5.1. Origin and Function
2.5.2. Incretins and Cancer Association
2.5.3. Incretins Modulate Macrophage Inflammatory Responses
2.6. Insulin
2.6.1. Origin and Function
2.6.2. Insulin and Cancer Association
2.6.3. Insulin Modulates Macrophage Inflammatory Responses
2.7. Insulin-Like Growth Factor-1
2.7.1. Origin and Function
2.7.2. IGF-1 and Cancer Association
2.7.3. IGF-1 Modulates Macrophage Inflammatory Responses
2.8. Leptin
2.8.1. Origin and Function
2.8.2. Leptin and Cancer Association
2.8.3. Leptin Modulates Macrophage Inflammatory Responses
2.9. Neuropeptide Y (NPY) and Peptide YY (PYY)
2.9.1. Origin and Function
2.9.2. NPY and PYY and Cancer Association
2.9.3. NPY and PYY Modulate Macrophage Inflammatory Responses
2.10. Estrogen
2.10.1. Origin and Function
2.10.2. Estrogen and Cancer Association
2.10.3. Estrogen Modulates Macrophage Inflammatory Responses
2.11. Testosterone
2.11.1. Origin and Function
2.11.2. Testosterone and Cancer Association
2.11.3. Testosterone Modulates Macrophage Inflammatory Responses
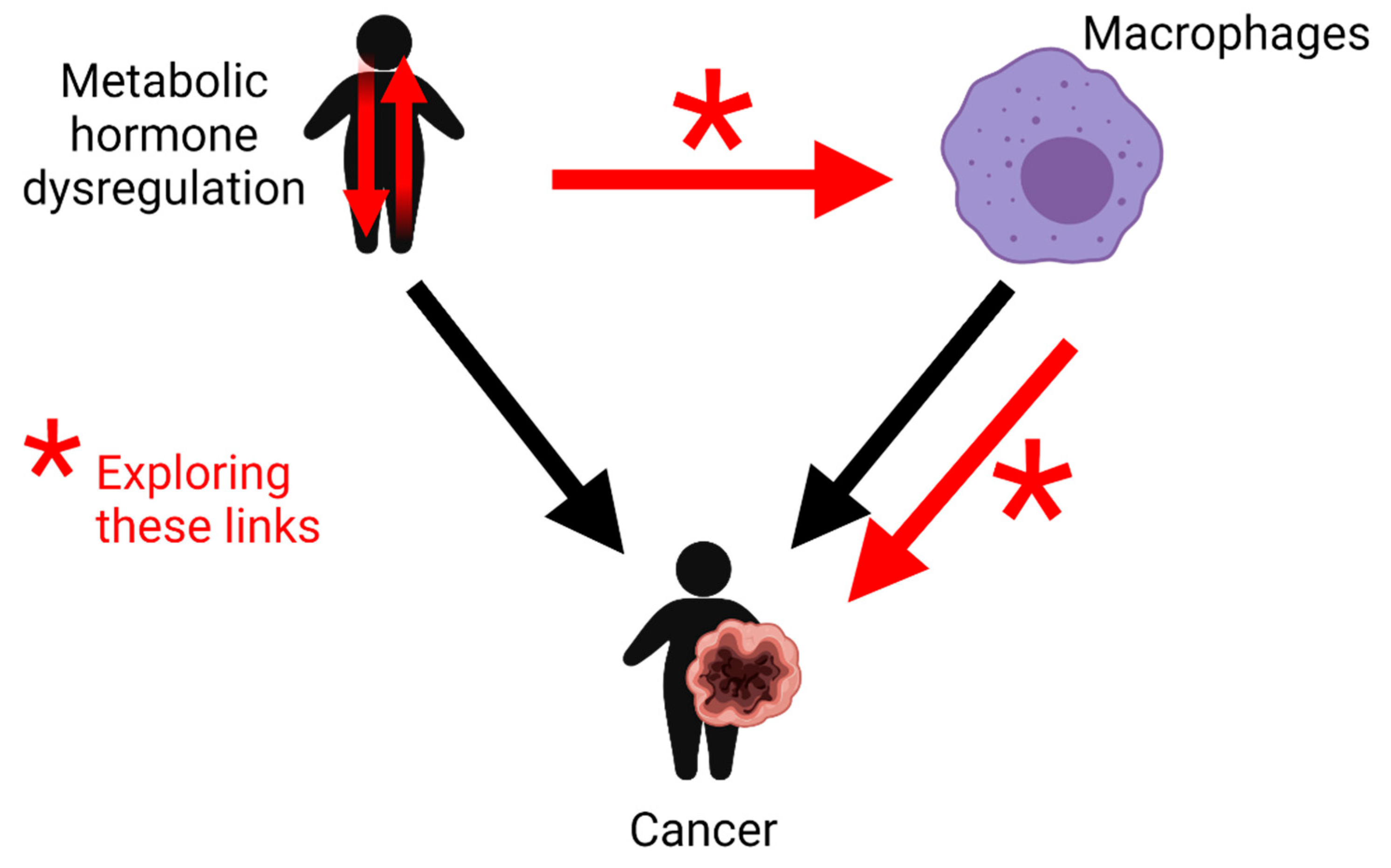
2.12. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- De Pergola, G.; Silvestris, F. Obesity as a major risk factor for cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef] [PubMed]
- Lean, M.E.J.; Malkova, D. Altered gut and adipose tissue hormones in overweight and obese individuals: Cause or consequence? Int. J. Obes. 2016, 40, 622–632. [Google Scholar] [CrossRef]
- Chawla, A.; Nguyen, K.D.; Goh, Y.P.S.S. Macrophage-Mediated Inflammation in Metabolic Disease. Nat. Rev. Immunol. 2011, 11, 738–749. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Na, Y.R.; Je, S.; Seok, S.H. Metabolic features of macrophages in inflammatory diseases and cancer. Cancer Lett. 2018, 413, 46–58. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Staels, B. Macrophage polarization in metabolic disorders: Functions and regulation. Curr. Opin. Lipidol. 2011, 22, 365–372. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; DeNardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Butcher, M.J.; Galkina, E.V. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Front. Physiol. 2012, 3, 44. [Google Scholar] [CrossRef]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Jurberg, A.D.; Cotta-de-Almeida, V.; Temerozo, J.R.; Savino, W.; Bou-Habib, D.C.; Riederer, I. Neuroendocrine Control of Macrophage Development and Function. Front. Immunol. 2018, 9, 1440. [Google Scholar] [CrossRef]
- Dufresne, M.; Seva, C.; Fourmy, D. Cholecystokinin and gastrin receptors. Physiol. Rev. 2006, 86. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.J.; Dong, M.; Harikumar, K.G.; Miller, L.J. Cholecystokinin-induced satiety, a key gut servomechanism that is affected by the membrane microenvironment of this receptor. Int. J. Obes. Suppl. 2016, 6, S22–S27. [Google Scholar] [CrossRef]
- Rehfeld, J.F. Cholecystokinin-From local gut hormone to ubiquitous messenger. Front. Endocrinol. 2017, 8, 47. [Google Scholar] [CrossRef]
- Wank, S.A.; Pisegna, J.R.; Weerth, A. De Brain and gastrointestinal cholecystokinin receptor family: Structure and functional expression. Proc. Natl. Acad. Sci. USA 1992, 89, 8691–8695. [Google Scholar] [CrossRef]
- Kopin, A.S.; Lee, Y.M.; McBride, E.W.; Miller, L.J.; Lu, M.; Lin, H.Y.; Kolakowski, L.F.; Beinborn, M. Expression cloning and characterization of the canine parietal cell gastrin receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 3605–3609. [Google Scholar] [CrossRef]
- Schjoldager, B. Role of CCK in gallbladder function. Ann. N. Y. Acad. Sci. 1994, 713, 207–218. [Google Scholar] [CrossRef]
- Nadella, S.; Burks, J.; Al-Sabban, A.; Inyang, G.; Wang, J.; Tucker, R.D.; Zamanis, M.E.; Bukowski, W.; Shivapurkar, N.; Smith, J.P. Dietary fat stimulates pancreatic cancer growth and promotes fibrosis of the tumor microenvironment through the cholecystokinin receptor. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G699–G712. [Google Scholar] [CrossRef]
- Smith, J.P.; Verderame, M.F.; McLaughlin, P.; Martenis, M.; Ballard, E.; Zagon, I.S. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Int. J. Mol. Med. 2002, 10, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Matters, G.L.; Cooper, T.K.; Mcgovern, C.O.; Gilius, E.L.; Liao, J.; Barth, B.M.; Kester, M.; Smith, J.P.; Matters, G.L.; Cooper, T.K.; et al. Cholecystokinin Mediates Progression and Metastasis of Pancreatic Cancer Associated with Dietary Fat. Dig. Dis. Sci. 2014, 59, 1180–1191. [Google Scholar] [CrossRef]
- Reubi, J.C. Targeting CCK receptors in human cancers. Curr. Top. Med. Chem. 2007, 7, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Ou, L.; Wang, W.; Guo, D.Y. Gastrin, Cholecystokinin, Signaling, and Biological Activities in Cellular Processes. Front. Endocrinol. 2020, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, J.; Guo, L.; Zou, Y.; Wang, F.; Shao, H.; Li, J.; Deng, X. Cholecystokinin protects mouse liver against ischemia and reperfusion injury. Int. Immunopharmacol. 2017, 48, 180–186. [Google Scholar] [CrossRef]
- Meng, A.H.; Ling, Y.L.; Zhang, X.P.; Zhang, J.L. Anti-inflammatory effect of cholecystokinin and its signal transduction mechanism in endotoxic shock rat. World J. Gastroenterol. 2002, 8, 712. [Google Scholar] [CrossRef]
- Bozkurt, A.; Çakir, B.; Ercan, F.; Yeǧen, B.Ç. Anti-inflammatory effects of leptin and cholecystokinin on acetic acid-induced colitis in rats: Role of capsaicin-sensitive vagal afferent fibers. Regul. Pept. 2003, 116, 109–118. [Google Scholar] [CrossRef]
- Luyer, M.D.; Greve, J.W.M.; Hadfoune, M.; Jacobs, J.A.; Dejong, C.H.; Buurman, W.A. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J. Exp. Med. 2005, 202, 1023–1029. [Google Scholar] [CrossRef]
- Saia, R.S.; Mestriner, F.L.; Bertozi, G.; Cunha, F.Q.; Cárnio, E.C. Cholecystokinin Inhibits Inducible Nitric Oxide Synthase Expression by Lipopolysaccharide-Stimulated Peritoneal Macrophages. Mediat. Inflamm. 2014, 2014, 896029. [Google Scholar] [CrossRef]
- Miyamoto, S.; Shikata, K.; Miyasaka, K.; Okada, S.; Sasaki, M.; Kodera, R.; Hirota, D.; Kajitani, N.; Takatsuka, T.; Kataoka, H.U.; et al. Cholecystokinin plays a novel protective role in diabetic kidney through anti-inflammatory actions on macrophage: Anti-inflammatory effect of cholecystokinin. Diabetes 2012, 61, 897–907. [Google Scholar] [CrossRef]
- Ling, Y.L.; Meng, A.H.; Zhao, X.Y.; Shan, B.E.; Zhang, J.L.; Zhang, X.P. Effect of cholecystokinin on cytokines during endotoxic shock in rats. World J. Gastroenterol. 2001, 7, 667. [Google Scholar] [CrossRef]
- Ye, S.; Shi, K.; Xu, J.; Wang, M.; Li, C.J. Cholecystokinin octapeptide inhibits the inflammatory response and improves neurological outcome in a porcine model of cardiopulmonary resuscitation. Exp. Ther. Med. 2018, 15, 2583–2588. [Google Scholar] [CrossRef] [PubMed]
- Zuelli, F.M.D.G.C.; Cárnio, E.C.; Saia, R.S. Cholecystokinin protects rats against sepsis induced by Staphylococcus aureus. Med. Microbiol. Immunol. 2014, 203, 165–176. [Google Scholar] [CrossRef]
- Sacerdote, P.; Wiedermann, C.J.; Wahl, L.M.; Pert, C.B.; Ruff, M.R. Visualization of cholecystokinin receptors on a subset of human monocytes and in rat spleen. Peptides 1991, 12, 167–176. [Google Scholar] [CrossRef]
- Xu, S.J.; Gao, W.J.; Cong, B.; Yao, Y.X.; Gu, Z.Y. Effect of lipopolysaccharide on expression and characterization of cholecystokinin receptors in rat pulmonary interstitial macrophages. Acta Pharmacol. Sin. 2004, 25, 1347–1353. [Google Scholar]
- Li, S.; Ni, Z.; Cong, B.; Gao, W.; Xu, S.; Wang, C.; Yao, Y.; Ma, C.; Ling, Y. CCK-8 inhibits LPS-induced IL-1β production in pulmonary interstitial macrophages by modulating PKA, p38, and NF-κB pathway. Shock 2007, 27, 678–686. [Google Scholar] [CrossRef]
- De la Fuente, M.; Campos, M.; Del Rio, M.; Hernanz, A. Inhibition of murine peritoneal macrophage functions by sulfated cholecystokinin octapeptide. Regul. Pept. 1995, 55, 47–56. [Google Scholar] [CrossRef]
- Cong, B.; Li, S.J.; Yao, Y.X.; Zhu, G.J.; Ling, Y.L. Effect of cholecystokinin octapeptide on tumor necrosis factor α transcription and nuclear factor-κB activity induced by lipopolysaccharide in rat pulmonary interstitial macrophages. World J. Gastroenterol. 2002, 8, 718. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef]
- Saye, J.A.; Lynch, K.R.; Peach, M.J. Changes in angiotensinogen messenger RNA in differentiating 3T3-F442A adipocytes. Hypertension 1990, 15, 867–871. [Google Scholar] [CrossRef]
- Hunt, C.R.; Ro, J.H.S.; Dobson, D.E.; Min, H.Y.; Spiegelman, B.M. Adipocyte P2 gene: Developmental expression and homology of 5’-flanking sequences among fat cell-specific genes. Proc. Natl. Acad. Sci. USA 1986, 83, 3786–3790. [Google Scholar] [CrossRef]
- Makowski, L.; Boord, J.B.; Maeda, K.; Babaev, V.R.; Uysal, K.T.; Morgan, M.A.; Parker, R.A.; Suttles, J.; Fazio, S.; Hotamisligil, G.S.; et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat. Med. 2001, 7, 699–705. [Google Scholar] [CrossRef]
- Pelton, P.D.; Zhou, L.; Demarest, K.T.; Burris, T.P. PPARγ activation induces the expression of the adipocyte fatty acid binding protein gene in human monocytes. Biochem. Biophys. Res. Commun. 1999, 261, 456–458. [Google Scholar] [CrossRef]
- Tontonoz, P.; Graves, R.A.; Budavari, A.I.; Erdjument-bromage, H.; Lui, M.; Hu, E.; Tempst, P.; Spiegelman, B.M. Adipocyte-specific transcription factor ARF6 is a heterodimeric complex of two nuclear hormone receptors, PPARγ and RXRa. Nucleic Acids Res. 1994, 22, 5628–5634. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Tso, A.W.K.; Cheung, B.M.Y.; Wang, Y.; Wat, N.M.S.; Fong, C.H.Y.; Yeung, D.C.Y.; Janus, E.D.; Sham, P.C.; Lam, K.S.L. Circulating adipocyte-fatty acid binding protein levels predict the development of the metabolic syndrome: A 5-year prospective study. Circulation 2007, 115, 1537–1543. [Google Scholar] [CrossRef]
- Tso, A.W.K.; Xu, A.; Sham, P.C.; Wat, N.M.S.; Wang, Y.; Fong, C.H.Y.; Cheung, B.M.Y.; Janus, E.D.; Lam, K.S.L. Serum adipocyte fatty acid-binding protein as a new biomarker predicting the development of type 2 diabetes: A 10-year prospective study in a Chinese cohort. Diabetes Care 2007, 30, 2667–2672. [Google Scholar] [CrossRef] [PubMed]
- Tuncman, G.; Erbay, E.; Hom, X.; De Vivo, I.; Campos, H.; Rimm, E.B.; Hotamisligil, G.S. A genetic variant at the fatty acid-binding protein aP2 locus reduces the risk for hypertriglyceridemia, type 2 diabetes, and cardiovascular disease. Proc. Natl. Acad. Sci. USA 2006, 103, 6970–6975. [Google Scholar] [CrossRef]
- Yeung, D.C.Y.; Xu, A.; Cheung, C.W.S.; Wat, N.M.S.; Yau, M.H.; Fong, C.H.Y.; Chau, M.T.; Lam, K.S.L. Serum adipocyte fatty acid-binding protein levels were independently associated with carotid atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1796–1802. [Google Scholar] [CrossRef]
- Hao, J.; Yan, F.; Zhang, Y.Y.; Triplett, A.; Zhang, Y.Y.; Schultz, D.A.; Sun, Y.; Zeng, J.; Silverstein, K.A.T.; Zheng, Q.; et al. Expression of adipocyte/macrophage fatty acid–binding protein in tumor-associated macrophages promotes breast cancer progression. Cancer Res. 2018, 78, 2343–2355. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.-Q.; Zhang, X.-P.; Ma, N.; Zhang, E.-B.; Li, J.-J.; Jiang, Y.-B.; Gao, Y.-Z.; Yuan, Y.-M.; Lan, S.-Q.; Xie, D.; et al. FABP4 suppresses proliferation and invasion of hepatocellular carcinoma cells and predicts a poor prognosis for hepatocellular carcinoma. Cancer Med. 2018, 7, 2629–2640. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Zhang, W.; Zhang, Y.; Zhu, T.; Hua, Y.; Li, H.; Zhang, Q.; Xia, M. FABP4 promotes invasion and metastasis of colon cancer by regulating fatty acid transport. Cancer Cell Int. 2020, 20, 512. [Google Scholar] [CrossRef]
- Gharpure, K.M.; Pradeep, S.; Sans, M.; Rupaimoole, R.; Ivan, C.; Wu, S.Y.; Bayraktar, E.; Nagaraja, A.S.; Mangala, L.S.; Zhang, X.; et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018, 9, 2923. [Google Scholar] [CrossRef]
- Harjes, U.; Bridges, E.; Gharpure, K.M.; Roxanis, I.; Sheldon, H.; Miranda, F.; Mangala, L.S.; Pradeep, S.; Lopez-Berestein, G.; Ahmed, A.; et al. Antiangiogenic and tumour inhibitory effects of downregulating tumour endothelial FABP4. Oncogene 2017, 36, 912–921. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Hardaway, A.L.; Podgorski, I. IL-1β, RAGE and FABP4: Targeting the dynamic trio in metabolic inflammation and related pathologies. Futur. Med. Chem. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Garin-Shkolnik, T.; Rudich, A.; Hotamisligil, G.S.; Rubinstein, M. FABP4 attenuates PPARγ and adipogenesis and is inversely correlated with PPARγ in adipose tissues. Diabetes 2014, 63, 900–911. [Google Scholar] [CrossRef]
- Rigamonti, E.; Chinetti-Gbaguidi, G.; Staels, B. Regulation of macrophage functions by PPAR- α, PPAR- γ, and LXRs in mice and men. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1050–1059. [Google Scholar] [CrossRef]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2007, 1771, 926–935. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L.; Brittingham, K.C.; Reynolds, J.M.; Suttles, J.; Hotamisligil, G.S. The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity: Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor γ and IκB kinase activities. J. Biol. Chem. 2005, 280, 12888–12895. [Google Scholar] [CrossRef]
- Layne, M.D.; Patel, A.; Chen, Y.-H.; Rebel, V.I.; Carvajal, I.M.; Pellacani, A.; Ith, B.; Zhao, D.; Schreiber, B.M.; Yet, S.-F.; et al. Role of macrophage-expressed adipocyte fatty acid binding protein in the development of accelerated atherosclerosis in hypercholesterolemic mice. FASEB J. 2001, 15, 1–19. [Google Scholar] [CrossRef]
- Hui, X.; Li, H.; Zhou, Z.; Lam, K.S.L.; Xiao, Y.; Wu, D.; Ding, K.; Wang, Y.; Vanhoutte, P.M.; Xu, A. Adipocyte fatty acid-binding protein modulates inflammatory responses in macrophages through a positive feedback loop involving c-Jun NH 2-terminal kinases and activator protein-1. J. Biol. Chem. 2010, 285, 10273–10280. [Google Scholar] [CrossRef]
- Boß, M.; Kemmerer, M.; Brüne, B.; Namgaladze, D. FABP4 inhibition suppresses PPARγ activity and VLDL-induced foam cell formation in IL-4-polarized human macrophages. Atherosclerosis 2015, 240, 424–430. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Nambi, P. Sirolimus upregulates aP2 expression in human monocytes and macrophages. Transplant. Proc. 2004, 36, 3229–3231. [Google Scholar] [CrossRef]
- Calatayud, S.; Alvarez, A.; Victor, V.M. Gastrin: An Acid-Releasing, Proliferative and Immunomodulatory Peptide? Mini-Rev. Med. Chem. 2010, 10, 8–19. [Google Scholar] [CrossRef]
- Hiraoka, S.; Miyazaki, Y.; Kitamura, S.; Toyota, M.; Kiyohara, T.; Shinomura, Y.; Mukaida, N.; Matsuzawa, Y. Gastrin induces CXC chemokine expression in gastric epithelial cells through activation of NF-κB. Am. J. Physiol. Liver Physiol. 2001, 281, G735–G742. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, D.; Ramalingam, S.; May, R.; Dieckgraefe, B.K.; Berg, D.E.; Pothoulakis, C.; Houchen, C.W.; Wang, T.C.; Anant, S. Gastrin-Mediated Interleukin-8 and Cyclooxygenase-2 Gene Expression: Differential Transcriptional and Posttranscriptional Mechanisms. Gastroenterology 2008, 134, 1070–1082. [Google Scholar] [CrossRef]
- Grösch, S.; Maier, T.J.; Schiffmann, S.; Geisslinger, G. Cyclooxygenase-2 (COX-2)–Independent Anticarcinogenic Effects of Selective COX-2 Inhibitors. JNCI J. Natl. Cancer Inst. 2006, 98, 736–747. [Google Scholar] [CrossRef]
- Chao, C.; Hellmich, M.R. Gastrin, inflammation, and carcinogenesis. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Bedke, J.; Hemmerlein, B.; Perske, C.; Gross, A.; Heuser, M. Tumor-associated macrophages in clear cell renal cell carcinoma express both gastrin-releasing peptide and its receptor: A possible modulatory role of immune effectors cells. World J. Urol. 2010, 28, 335–341. [Google Scholar] [CrossRef][Green Version]
- Levine, L.; Lucci, J.A.; Pazdrak, B.; Cheng, J.Z.; Guo, Y.S.; Townsend, C.M.; Hellmich, M.R. Bombesin stimulates nuclear factor κB activation and expression of proangiogenic factors in prostate cancer cells. Cancer Res. 2003, 63, 3495–3502. [Google Scholar] [PubMed]
- Mezey, É.; Palkovits, M. Localization of targets for anti-ulcer drugs in cells of the immune system. Science 1992, 258, 1662–1665. [Google Scholar] [CrossRef]
- Schmitz, F.; Schrader, H.; Otte, J.M.; Schmitz, H.; Stüber, E.; Herzig, K.H.; Schmidt, W.E. Identification of CCK-B/gastrin receptor splice variants in human peripheral blood mononuclear cells. Regul. Pept. 2001, 101, 25–33. [Google Scholar] [CrossRef]
- Iwata, N.; Murayama, T.; Matsumori, Y.; Ito, M.; Nagata, A.; Taniguchi, T.; Chihara, K.; Matsuo, Y.; Minowada, J.; Matsui, T. Autocrine loop through cholecystokinin-B/gastrin receptors involved in growth of human leukemia cells. Blood 1996, 88, 2683–2689. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.; Otte, J.M.; Stechele, H.U.; Reimann, B.; Banasiewicz, T.; Fölsch, U.R.; Schmidt, W.E.; Herzig, K.H. CCK-B/gastrin receptors in human colorectal cancer. Eur. J. Clin. Investig. 2001, 31, 812–820. [Google Scholar] [CrossRef]
- Okahata, H.; Nishi, Y.; Muraki, K.; Sumii, K.; Miyachi, Y.; Usui, T. Gastrin/cholecystokinin-like immunoreactivity in human blood cells. Life Sci. 1985, 36, 369–373. [Google Scholar] [CrossRef]
- De La Fuente, M.; Drummond, J.; Del Rio, M.; Carrasco, M.; Hernanz, A. Modulation of murine peritoneal macrophage functions by gastrin. Peptides 1996, 17, 219–224. [Google Scholar] [CrossRef]
- De La Fuente, M.; Carrasco, M.; Hernanz, A. Modulation of human neutrophil function in vitro by gastrin. J. Endocrinol. 1997, 153, 475–483. [Google Scholar] [CrossRef]
- Álvarez, Á.; Ibiza, S.; Hernández, C.; Álvarez-Barrientos, A.; Esplugues, J.V.; Calatayud, S.; Álvarez, Á.; Ibiza, S.; Hernández, C.; Álvarez-Barrientos, A.; et al. Gastrin induces leukocyte-endothelial cell interactions in vivo and contributes to the inflammation caused by Helicobacter pylori. FASEB J. 2006, 20, 2396–2398. [Google Scholar] [CrossRef]
- Kim, S.O.; Chao, C.; Townsend, C.M.; Hellmich, M.R. M1632 Interferon-γ Induces CCK2 Receptor Expression in Monocytes and Bone Marrow-Derived Cells. Gastroenterology 2009, 136, A-398. [Google Scholar] [CrossRef]
- Lefranc, F.; Mijatovic, T.; Mathieu, V.; Rorive, S.; Decaestecker, C.; Debeir, O.; Brotchi, J.; Van Ham, P.; Salmon, I.; Kiss, R. Characterization of gastrin-induced proangiogenic effects in vivo in orthotopic U373 experimental human glioblastomas and in vitro in human umbilical vein endothelial cells. Clin. Cancer Res. 2004, 10, 8250–8265. [Google Scholar] [CrossRef]
- Ibiza, S.; Álvarez, Á.; Romero, W.; Barrachina, M.D.; Esplugues, J.V.; Calatayud, S. Gastrin induces the interaction between human mononuclear leukocytes and endothelial cells through the endothelial expression of P-selectin and VCAM-1. Am. J. Physiol.-Cell Physiol. 2009, 297, C1588–C1595. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, F.; Salio, C.; Lossi, L.; Merighi, A. Ghrelin in central neurons. Curr. Neuropharmacol. 2009, 7, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Waseem, T.; Duxbury, M.; Ito, H.; Ashley, S.W.; Robinson, M.K. Exogenous ghrelin modulates release of pro-inflammatory and anti-inflammatory cytokines in LPS-stimulated macrophages through distinct signaling pathways. Surgery 2008, 143, 334–342. [Google Scholar] [CrossRef]
- Tschop, M.; Smiley, D.L.; Heiman, M.L. Ghrelin induces adiposity in rodents. Nature 2000, 407, 908–913. [Google Scholar] [CrossRef]
- Kojima, M.; Kangawa, K. Ghrelin: Structure and function. Physiol. Rev. 2005, 85, 495–522. [Google Scholar] [CrossRef]
- Garin, M.C.; Burns, C.M.; Kaul, S.; Cappola, A.R. The human experience with ghrelin administration. J. Clin. Endocrinol. Metab. 2013, 98, 1826–1837. [Google Scholar] [CrossRef] [PubMed]
- Dixit, V.D.; Palaniappan, R.; Collins, G.D.; Taub, D.D.; Sakthivel, S.K.; Lillard, J.W.; Schaffer, E.M.; Pyle, R.S.; Collins, G.D.; Sakthivel, S.K.; et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J. Clin. Investig. 2004, 114, 57–66. [Google Scholar] [CrossRef]
- Hattori, N.; Saito, T.; Yagyu, T.; Jiang, B.H.; Kitagawa, K.; Inagaki, C. GH, GH receptor, GH secretagogue receptor, and Ghrelin expression in human T cells, B cells, and neutrophils. J. Clin. Endocrinol. Metab. 2001, 86, 4284–4291. [Google Scholar] [CrossRef]
- Li, B.; Zeng, M.; He, W.; Huang, X.; Luo, L.; Zhang, H.; Deng, D.Y.B. Ghrelin protects alveolar macrophages against lipopolysaccharide-induced apoptosis through growth hormone secretagogue receptor 1a-dependent c-jun n-terminal kinase and wnt/ß-catenin signaling and suppresses lung inflammation. Endocrinology 2015, 156, 203–217. [Google Scholar] [CrossRef]
- Shiiya, T.; Nakazato, M.; Mizuta, M.; Date, Y.; Mondal, M.S.; Tanaka, M.; Nozoe, S.I.; Hosoda, H.; Kangawa, K.; Matsukura, S. Plasma ghrelin levels in lean and obese humans and the effect of glucose on ghrelin secretion. J. Clin. Endocrinol. Metab. 2002, 87, 240–244. [Google Scholar] [CrossRef]
- Naznin, F.; Toshinai, K.; Waise, T.M.Z.; Okada, T.; Sakoda, H.; Nakazato, M. Restoration of metabolic inflammation-related ghrelin resistance by weight loss. J. Mol. Endocrinol. 2018, 60, 109–118. [Google Scholar] [CrossRef]
- Soleyman-Jahi, S.; Sadeghi, F.; Pastaki Khoshbin, A.; Khani, L.; Roosta, V.; Zendehdel, K. Attribution of Ghrelin to Cancer; Attempts to Unravel an Apparent Controversy. Front. Oncol. 2019, 9, 1014. [Google Scholar] [CrossRef]
- Tian, C.; Zhang, L.; Hu, D.; Ji, J. Ghrelin induces gastric cancer cell proliferation, migration, and invasion through GHS-R/NF-κB signaling pathway. Mol. Cell. Biochem. 2013, 382, 163–172. [Google Scholar] [CrossRef]
- Murphy, G.; Kamangar, F.; Dawsey, S.M.; Stanczyk, F.Z.; Weinstein, S.J.; Taylor, P.R.; Virtamo, J.; Abnet, C.C.; Albanes, D.; Freedman, N.D. The Relationship Between Serum Ghrelin and the Risk of Gastric and Esophagogastric Junctional Adenocarcinomas. JNCI J. Natl. Cancer Inst. 2011, 103, 1123–1129. [Google Scholar] [CrossRef]
- Spiridon, I.A.; Ciobanu, D.G.A.; Giușcă, S.E.; Căruntu, I.D. Ghrelin and its role in gastrointestinal tract tumors (Review). Mol. Med. Rep. 2021, 24, 663. [Google Scholar] [CrossRef]
- Sever, S.; White, D.L.; Garcia, J.M. Is there an effect of ghrelin/ghrelin analogs on cancer? A systematic review. Endocr. Relat. Cancer 2016, 23, R393–R409. [Google Scholar] [CrossRef]
- Yuan, F.; Ma, J.; Xiang, X.; Lan, H.; Xu, Y.; Zhao, J.; Li, Y.; Zhang, W. Improvement of Adipose Macrophage Polarization in High Fat Diet-Induced Obese GHSR Knockout Mice. Biomed Res. Int. 2018, 2018, 4924325. [Google Scholar] [CrossRef]
- Lin, L.; Lee, J.H.; Buras, E.D.; Yu, K.; Wang, R.; Smith, C.W.; Wu, H.; Sheikh-Hamad, D.; Sun, Y. Ghrelin receptor regulates adipose tissue inflammation in aging. Aging 2016, 8, 178. [Google Scholar] [CrossRef]
- Ai, W.; Wu, M.; Chen, L.; Jiang, B.; Mu, M.; Liu, L.; Yuan, Z. Ghrelin ameliorates atherosclerosis by inhibiting endoplasmic reticulum stress. Fundam. Clin. Pharmacol. 2017, 31, 147–154. [Google Scholar] [CrossRef]
- Raghay, K.; Akki, R.; Bensaid, D.; Errami, M. Ghrelin as an anti-inflammatory and protective agent in ischemia/reperfusion injury. Peptides 2020, 124, 170226. [Google Scholar] [CrossRef]
- Pang, J.; Xu, Q.; Xu, X.; Yin, H.; Xu, R.; Guo, S.; Hao, W.; Wang, L.; Chen, C.; Cao, J.M. Hexarelin suppresses high lipid diet and vitamin D3-induced atherosclerosis in the rat. Peptides 2010, 31, 630–638. [Google Scholar] [CrossRef]
- Li, W.G.; Gavrila, D.; Liu, X.; Wang, L.; Gunnlaugsson, S.; Stoll, L.L.; McCormick, M.L.; Sigmund, C.D.; Tang, C.; Weintraub, N.L. Ghrelin Inhibits Proinflammatory Responses and Nuclear Factor-κB Activation in Human Endothelial Cells. Circulation 2004, 109, 2221–2226. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Q.; Ke, D.; Li, G. Ghrelin inhibits atherosclerotic plaque angiogenesis and promotes plaque stability in a rabbit atherosclerotic model. Peptides 2017, 90, 17–26. [Google Scholar] [CrossRef]
- Sun, J.; Li, X.; Jiao, K.; Zhai, Z.; Sun, D. Albiflorin inhibits the formation of THP-1-derived foam cells through the LOX-1/NF-κB pathway. Minerva Med. 2019, 110, 107–114. [Google Scholar] [CrossRef]
- Cheng, X.L.; Ding, F.; Wang, D.P.; Zhou, L.; Cao, J.M. Hexarelin attenuates atherosclerosis via inhibiting LOX-1-NF-κB signaling pathway-mediated macrophage ox-LDL uptake in ApoE-/- mice. Peptides 2019, 121, 170122. [Google Scholar] [CrossRef]
- Sun, N.; Wang, H.; Wang, L. Ghrelin inhibits oxLDL-induced inflammation in RAW264.7 mouse macrophages through down-regulation of LOX-1 expression via NF-κB signaling pathway. Cell. Mol. Biol. 2016, 62, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Liang, W.; He, W.; Huang, C.; Chen, Q.; Yi, H.; Long, L.; Deng, Y.; Zeng, M. Ghrelin attenuates sepsis-induced acute lung injury by inhibiting the NF-κB, iNOS, and Akt signaling in alveolar macrophages. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 317, L381–L391. [Google Scholar] [CrossRef]
- Nogi, Y.; Nagashima, M.; Terasaki, M.; Nohtomi, K.; Watanabe, T.; Hirano, T. Glucose-dependent insulinotropic polypeptide prevents the progression of macrophage-driven atherosclerosis in diabetic apolipoprotein E-null mice. PLoS ONE 2012, 7, e35683. [Google Scholar] [CrossRef]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Investig. 2011, 121, 2118–2125. [Google Scholar] [CrossRef]
- Eissele, R.; Göke, R.; Willemer, S.; Harthus, H.-P.; Vermeer, H.; Arnold, R.; Göke, B. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur. J. Clin. Investig. 1992, 22, 283–291. [Google Scholar] [CrossRef]
- Bell, G.I.; Santerre, R.F.; Mullenbach, G.T. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature 1983, 302, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Hirota, M.; Ohboshi, C.; Shima, K. Identification and localization of glucagon-like peptide-1 and its receptor in rat brain. Endocrinology 1987, 121, 1076–1082. [Google Scholar] [CrossRef]
- Körner, M.; Stöckli, M.; Waser, B.; Reubi, J.C. GLP-1 receptor expression in human tumors and human normal tissues: Potential for in vivo targeting. J. Nucl. Med. 2007, 48, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Vilsbøll, T.; Knop, F.K. Incretin mimetics: A novel therapeutic option for patients with type 2 diabetes—A review. Diabetes. Metab. Syndr. Obes. 2010, 3, 155–163. [Google Scholar]
- Kosowska, A.; Gallego-Colon, E.; Garczorz, W.; Kłych-Ratuszny, A.; Aghdam, M.R.F.; Woz Niak, M.; Witek, A.; Wróblewska-Czech, A.; Cygal, A.; Wojnar, J.; et al. Exenatide modulates tumor-endothelial cell interactions in human ovarian cancer cells. Endocr. Connect. 2017, 6, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Fidan-Yaylalı, G.; Dodurga, Y.; Seçme, M.; Elmas, L. Antidiabetic exendin-4 activates apoptotic pathway and inhibits growth of breast cancer cells. Tumour Biol. 2016, 37, 2647–2653. [Google Scholar] [CrossRef]
- Koehler, J.A.; Kain, T.; Drucker, D.J. Glucagon-like peptide-1 receptor activation inhibits growth and augments apoptosis in murine CT26 colon cancer cells. Endocrinology 2011, 152, 3362–3372. [Google Scholar] [CrossRef]
- He, W.; Yu, S.; Wang, L.; He, M.; Cao, X.; Li, Y.; Xiao, H. Exendin-4 inhibits growth and augments apoptosis of ovarian cancer cells. Mol. Cell. Endocrinol. 2016, 436, 240–249. [Google Scholar] [CrossRef]
- Rouette, J.; Yin, H.; Yu, O.H.Y.; Bouganim, N.; Platt, R.W.; Azoulay, L. Incretin-based drugs and risk of lung cancer among individuals with type 2 diabetes. Diabet. Med. 2020, 37, 868–875. [Google Scholar] [CrossRef]
- Chai, S.; Yu, S.; Yang, Z.; Wu, S.; Gao, L.; Wang, H.; Zhang, Y.; Zhan, S.; Ji, L.; Sun, F. Effect of incretin-based therapies on cancers of digestive system among 101 595 patients with type 2 diabetes mellitus: A systematic review and network meta-analysis combining 84 trials with a median duration of 30 weeks. BMJ Open Diabetes Res. Care 2019, 7, e000728. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.C.; Falcetta, M.R.; Rados, D.V.; Leitão, C.B.; Gross, J.L. Glucagon-like peptide-1 receptor agonists and pancreatic cancer: A meta-analysis with trial sequential analysis. Sci. Rep. 2019, 9, 2375. [Google Scholar] [CrossRef]
- Hatwal, A. Inflammation and incretins. Indian J. Endocrinol. Metab. 2012, 16, S239–S241. [Google Scholar] [CrossRef]
- Guo, C.; Huang, T.; Chen, A.; Chen, X.; Wang, L.; Shen, F.; Gu, X. Glucagon-like peptide 1 improves insulin resistance in vitro through anti-inflammation of macrophages. Braz. J. Med. Biol. Res. 2016, 49. [Google Scholar] [CrossRef]
- Lee, Y.S.; Shin, S.; Shigihara, T.; Hahm, E.; Liu, M.J.; Han, J.; Yoon, J.W.; Jun, H.S. Glucagon-like peptide-1 gene therapy in obese diabetic mice results in long-term cure of diabetes by improving insulin sensitivity and reducing hepatic gluconeogenesis. Diabetes 2007, 56, 1671–1679. [Google Scholar] [CrossRef]
- Lee, Y.S.; Park, M.S.; Choung, J.S.; Kim, S.S.; Oh, H.H.; Choi, C.S.; Ha, S.Y.; Kang, Y.; Kim, Y.; Jun, H.S. Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia 2012, 55, 2456–2468. [Google Scholar] [CrossRef]
- Lu, C.; Xie, T.; Guo, X.; Wu, D.; Li, S.; Li, X.; Lu, Y.; Wang, X. Glucagon-like peptide-1 receptor agonist exendin-4 mitigates lipopolysaccharide-induced inflammatory responses in RAW264.7 macrophages. Int. Immunopharmacol. 2019, 77, 105969. [Google Scholar] [CrossRef]
- Bułdak, Ł.; Machnik, G.; Bułdak, R.J.; Łabuzek, K.; Bołdys, A.; Belowski, D.; Basiak, M.; Okopień, B. Exenatide (a GLP-1 agonist) expresses anti-inflammatory properties in cultured human monocytes/macrophages in a protein kinase A and B/Akt manner. Pharmacol. Rep. 2016, 68, 329–337. [Google Scholar] [CrossRef]
- Bułdak, Ł.; Łabuzek, K.; Bułdak, R.J.; Machnik, G.; Bołdys, A.; Okopień, B. Exenatide (a GLP-1 agonist) improves the antioxidative potential of in vitro cultured human monocytes/macrophages. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Sun, H. Glucagon-like peptide-1 modulates RAW264.7 macrophage polarization by interfering with the JNK/STAT3 signaling pathway. Exp. Ther. Med. 2019, 17, 3573–3579. [Google Scholar] [CrossRef]
- Shiraishi, D.; Fujiwara, Y.; Komohara, Y.; Mizuta, H.; Takeya, M. Glucagon-like peptide-1 (GLP-1) induces M2 polarization of human macrophages via STAT3 activation. Biochem. Biophys. Res. Commun. 2012, 425, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Kahles, F.; Liberman, A.; Halim, C.; Rau, M.; Möllmann, J.; Mertens, R.W.; Rückbeil, M.; Diepolder, I.; Walla, B.; Diebold, S.; et al. The incretin hormone GIP is upregulated in patients with atherosclerosis and stabilizes plaques in ApoE−/− mice by blocking monocyte/macrophage activation. Mol. Metab. 2018, 14, 150–157. [Google Scholar] [CrossRef]
- Vittone, F.; Liberman, A.; Vasic, D.; Ostertag, R.; Esser, M.; Walcher, D.; Ludwig, A.; Marx, N.; Burgmaier, M. Sitagliptin reduces plaque macrophage content and stabilises arteriosclerotic lesions in Apoe−/− mice. Diabetologia 2012, 55, 2267–2275. [Google Scholar] [CrossRef]
- Ikeda, T.; Kumagai, E.; Iwata, S.; Yamakawa, A. Soluble CD26/Dipeptidyl Peptidase IV Enhances the Transcription of IL-6 and TNF-a in THP-1 Cells and Monocytes. PLoS ONE 2013, 8, e66520. [Google Scholar] [CrossRef]
- Mooradian, A.D. Dyslipidemia in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 150–159. [Google Scholar] [CrossRef]
- Lu, C.-C.; Chu, P.-Y.; Hsia, S.-M.; Wu, C.-H.; Tung, Y.-T.; Yen, G.-C. Insulin induction instigates cell proliferation and metastasis in human colorectal cancer cells. Int. J. Oncol. 2017, 50, 736–744. [Google Scholar] [CrossRef]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.C.; Tsubokawa, M.; et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985, 313, 756–761. [Google Scholar] [CrossRef]
- Ebina, Y.; Ellis, L.; Jarnagin, K.; Edery, M.; Graf, L.; Clauser, E.; Ou, J.-H.; Masiarz, F.; Kan, Y.W.; Goldfine, I.D.; et al. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signalling. Cell 1985, 40, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Gray, A.; Tam, A.W.; Yang-Feng, T.; Tsubokawa, M.; Collins, C.; Henzel, W.; Le Bon, T.; Kathuria, S.; Chen, E. Insulin-like growth factor I receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986, 5, 2503–2512. [Google Scholar] [CrossRef]
- Navé, B.T.; Haigh, R.J.; Hayward, A.C.; Siddle, K.; Shepherd, P.R. Compartment-specific regulation of phosphoinositide 3-kinase by platelet-derived growth factor and insulin in 3T3-L1 adipocytes. Biochem. J. 1996, 318, 55–60. [Google Scholar] [CrossRef]
- Haystead, C.M.M.; Gregory, P.; Shirazi, A.; Fadden, P.; Mosse, C.; Dent, P.; Haystead, T.A.J. Insulin activates a novel adipocyte mitogen-activated protein kinase kinase kinase that shows rapid phasic kinetics and is distinct from c-Raf. J. Biol. Chem. 1994, 269, 12804–12808. [Google Scholar] [CrossRef]
- Hancock, M.L.; Meyer, R.C.; Mistry, M.; Khetani, R.S.; Wagschal, A.; Shin, T.; Ho Sui, S.J.; Näär, A.M.; Flanagan, J.G. Insulin Receptor Associates with Promoters Genome-wide and Regulates Gene Expression. Cell 2019, 177, 722–736.e22. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.T.; Cutler, D.M.; Rosen, A.B. Forecasting the effects of obesity and smoking on U.S. life expectancy. N. Engl. J. Med. 2009, 361, 2252–2260. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Madarnas, Y.; Hoffman, B.; Hood, N. Fasting insulin and outcome in early-stage breast cancer: Results of a prospective cohort study. J. Clin. Oncol. 2002, 20, 42–51. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Pelicci, G.; Lanfrancone, L.; Grignani, F.; McGlade, J.; Cavallo, F.; Forni, G.; Nicoletti, I.; Grignani, F.; Pawson, T.; Pelicci, P.G. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 1992, 70, 93–104. [Google Scholar] [CrossRef]
- Cox, M.E.; Gleave, M.E.; Zakikhani, M.; Bell, R.H.; Piura, E.; Vickers, E.; Cunningham, M.; Larsson, O.; Fazli, L.; Pollak, M. Insulin receptor expression by human prostate cancers. Prostate 2009, 69, 33–40. [Google Scholar] [CrossRef]
- Kalli, K.R.; Falowo, O.I.; Bale, L.K.; Zschunke, M.A.; Roche, P.C.; Conover, C.A. Functional insulin receptors on human epithelial ovarian carcinoma cells: Implications for IGF-II mitogenic signaling. Endocrinology 2002, 143, 3259–3267. [Google Scholar] [CrossRef]
- Law, J.H.; Habibi, G.; Hu, K.; Masoudi, H.; Wang, M.Y.; Stratford, A.L.; Park, E.; Gee, J.M.W.; Finlay, P.; Jones, H.E.; et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 2008, 68, 10238–10246. [Google Scholar] [CrossRef] [PubMed]
- Sanaki, Y.; Nagata, R.; Kizawa, D.; Léopold, P.; Igaki, T. Hyperinsulinemia Drives Epithelial Tumorigenesis by Abrogating Cell Competition. Dev. Cell 2020, 53, 379–389.e5. [Google Scholar] [CrossRef] [PubMed]
- Bar, R.S.; Kahn, C.R.; Koren, H.S. Insulin inhibition of antibody-dependent cytoxicity and insulin receptors in macrophages. Nature 1977, 265, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Costa Rosa, L.F.; Safi, D.A.; Cury, Y.; Curi, R.; Rosa, L.F.B.P.C.; Safi, D.A.; Cury, Y.; Curit, R. The effect of insulin on macrophage metabolism and function. Cell Biochem. Funct. 1996, 14, 33–42. [Google Scholar] [CrossRef]
- Tada Iida, K.; Shimano, H.; Kawakami, Y.; Sone, H.; Toyoshima, H.; Suzuki, S.; Asano, T.; Okuda, Y.; Yamada, N. Insulin Up-regulates Tumor Necrosis Factor-α Production in Macrophages through an Extracellular-regulated Kinase-dependent Pathway. J. Biol. Chem. 2001, 276, 32531–32537. [Google Scholar] [CrossRef]
- Iida, K.T. Insulin Inhibits Apoptosis of Macrophage Cell Line, THP-1 Cells, via Phosphatidylinositol-3-Kinase-Dependent Pathway. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 380–386. [Google Scholar] [CrossRef]
- Senokuchi, T.; Liang, C.-P.; Seimon, T.A.; Han, S.; Matsumoto, M.; Banks, A.S.; Paik, J.-H.; DePinho, R.A.; Accili, D.; Tabas, I.; et al. Forkhead transcription factors (FoxOs) promote apoptosis of insulin-resistant macrophages during cholesterol-induced endoplasmic reticulum stress. Diabetes 2008, 57, 2967–2976. [Google Scholar] [CrossRef]
- Su, D.; Coudriet, G.M.; Hyun Kim, D.; Lu, Y.; Perdomo, G.; Qu, S.; Slusher, S.; Tse, H.M.; Piganelli, J.; Giannoukakis, N.; et al. FoxO1 links insulin resistance to proinflammatory cytokine IL-1beta production in macrophages. Diabetes 2009, 58, 2624–2633. [Google Scholar] [CrossRef]
- Yan, H.; Ma, Y.; Li, Y.; Zheng, X.; Lv, P.; Zhang, Y.; Li, J.; Ma, M.; Zhang, L.; Li, C.; et al. Insulin inhibits inflammation and promotes atherosclerotic plaque stability via PI3K-Akt pathway activation. Immunol. Lett. 2015, 170, 7–14. [Google Scholar] [CrossRef]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef]
- Park, Y.M.; Kashyap, S.R.; Major, J.A.; Silverstein, R.L. Insulin promotes macrophage foam cell formation: Potential implications in diabetes-related atherosclerosis. Lab. Investig. 2012, 92, 1171–1180. [Google Scholar] [CrossRef]
- Thibaut, R.; Gage, M.C.; Pineda-Torra, I.; Chabrier, G.; Venteclef, N.; Alzaid, F. Liver macrophages and inflammation in physiology and physiopathology of non-alcoholic fatty liver disease. FEBS J. 2021, febs.15877. [Google Scholar] [CrossRef]
- Russell-jones, D.L.; Bates, A.T.; Umpleby, A.M.; Hennessy, T.R.; Bowes, S.B.; Hopkins, K.D.; Jackson, N.; Kelly, J.; Shojaee-moradie, F.; Jones, R.H.; et al. A comparison of the effects of IGF-I and insulin on glucose metabolism, fat metabolism and the cardiovascular system in normal human volunteers. Eur. J. Clin. Investig. 1995, 25, 403–411. [Google Scholar] [CrossRef]
- Chang, H.R.; Kim, H.J.; Xu, X.; Ferrante, A.W. Macrophage and adipocyte IGF1 maintain adipose tissue homeostasis during metabolic stresses. Obesity 2016, 24, 172–183. [Google Scholar] [CrossRef]
- Spadaro, O.; Camell, C.D.; Bosurgi, L.; Nguyen, K.Y.; Youm, Y.H.; Rothlin, C.V.; Dixit, V.D. IGF1 Shapes Macrophage Activation in Response to Immunometabolic Challenge. Cell Rep. 2017, 19, 225–234. [Google Scholar] [CrossRef]
- Rom, W.N.; Pääkkö, P. Activated alveolar macrophages express the insulin-like growth factor-I receptor. Am. J. Respir. Cell Mol. Biol. 1991, 4, 432–439. [Google Scholar] [CrossRef]
- Adams, T.E.; Epa, V.C.; Garrett, T.P.J.; Ward, C.W. Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci. C 2000, 57, 1050–1093. [Google Scholar] [CrossRef]
- Lawrence, M.C.; McKern, N.M.; Ward, C.W. Insulin receptor structure and its implications for the IGF-1 receptor. Curr. Opin. Struct. Biol. 2007, 17, 699–705. [Google Scholar] [CrossRef]
- Tsuruzoe, K.; Emkey, R.; Kriauciunas, K.M.; Ueki, K.; Kahn, C.R. Insulin Receptor Substrate 3 (IRS-3) and IRS-4 Impair IRS-1- and IRS-2-Mediated Signaling. Mol. Cell. Biol. 2001, 21, 26–38. [Google Scholar] [CrossRef]
- Roith, D.L. The insulin-like growth factor system. Exp. Diabesity Res. 2003, 4, 205–212. [Google Scholar] [CrossRef]
- Murphy, N.; Carreras-Torres, R.; Song, M.; Chan, A.T.; Martin, R.M.; Papadimitriou, N.; Dimou, N.; Tsilidis, K.K.; Banbury, B.; Bradbury, K.E.; et al. Circulating Levels of Insulin-like Growth Factor 1 and Insulin-like Growth Factor Binding Protein 3 Associate With Risk of Colorectal Cancer Based on Serologic and Mendelian Randomization Analyses. Gastroenterology 2020, 158, 1300–1312.e20. [Google Scholar] [CrossRef]
- Murphy, N.; Knuppel, A.; Papadimitriou, N.; Martin, R.M.; Tsilidis, K.K.; Smith-Byrne, K.; Fensom, G.; Perez-Cornago, A.; Travis, R.C.; Key, T.J.; et al. Insulin-like growth factor-1, insulin-like growth factor-binding protein-3, and breast cancer risk: Observational and Mendelian randomization analyses with ∼430 000 women. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Párrizas, M.; Saltiel, A.R.; LeRoith, D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3’-kinase and mitogen-activated protein kinase pathways. J. Biol. Chem. 1997, 272, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Sekharam, M.; Zhao, H.; Sun, M.; Fang, Q.; Zhang, Q.; Yuan, Z.; Dan, H.C.; Boulware, D.; Cheng, J.Q.; Coppola, D. Insulin-like growth factor 1 receptor enhances invasion and induces resistance to apoptosis of colon cancer cells through the Akt/Bcl-x(L) pathway. Cancer Res. 2003, 63, 7708–7716. [Google Scholar] [PubMed]
- Kaleko, M.; Rutter, W.J.; Miller, A.D. Overexpression of the human insulinlike growth factor I receptor promotes ligand-dependent neoplastic transformation. Mol. Cell. Biol. 1990, 10, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Arkins, S.; Rebeiz, N.; Biragyn, A.; Reese, D.L.; Kelley, K.W. Murine macrophages express abundant insulin-like growth factor-I class I Ea and Eb transcripts. Endocrinology 1993, 133, 2334–2343. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef]
- Renier, G.; Clément, I.; Desfaits, A.C.; Lambert, A. Direct stimulatory effect of insulin-like growth factor-I on monocyte and macrophage tumor necrosis factor-alpha production. Endocrinology 1996, 137, 4611–4618. [Google Scholar] [CrossRef]
- Hochberg, Z.; Hertz, P.; Maor, G.; Oiknine, J.; Aviram, M. Growth hormone and insulin-like growth factor-I increase macrophage uptake and degradation of low density lipoprotein. Endocrinology 1992, 131, 430–435. [Google Scholar] [CrossRef]
- Spadaro, O.; Goldberg, E.L.; Camell, C.D.; Youm, Y.H.; Kopchick, J.J.; Nguyen, K.Y.; Bartke, A.; Sun, L.Y.; Dixit, V.D. Growth Hormone Receptor Deficiency Protects against Age-Related NLRP3 Inflammasome Activation and Immune Senescence. Cell Rep. 2016, 14, 1571–1580. [Google Scholar] [CrossRef]
- Scotece, M.; Conde, J.; López, V.; Lago, F.; Pino, J.; Gómez-Reino, J.J.; Gualillo, O. Adiponectin and leptin: New targets in inflammation. Basic Clin. Pharmacol. Toxicol. 2014, 114, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Al-Suhaimi, E.A.; Shehzad, A. Leptin, resistin and visfatin: The missing link between endocrine metabolic disorders and immunity. Eur. J. Med. Res. 2013, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Leibel, R.L. 20 years of leptin: Role of leptin in energy homeostasis in humans. J. Endocrinol. 2014, 223, T83–T96. [Google Scholar] [CrossRef]
- O’Rourke, L.; Yeaman, S.J.; Shepherd, P.R. Insulin and leptin acutely regulate cholesterol ester metabolism in macrophages by novel signaling pathways. Diabetes 2001, 50, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chen, Y.; Heiman, M.; DiMarchi, R. Leptin: Structure, Function and Biology; Academic Press: Cambridge, MA, USA, 2005; Volume 71, pp. 345–372. ISBN 0083-6729. [Google Scholar]
- Pelleymounter, M.A.; Cullen, M.J.; Baker, M.B.; Hecht, R.; Winters, D.; Boone, T.; Collins, F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995, 269, 540–543. [Google Scholar] [CrossRef]
- Lin, T.-C.; Hsiao, M. Leptin and Cancer: Updated Functional Roles in Carcinogenesis, Therapeutic Niches, and Developments. Int. J. Mol. Sci. 2021, 22, 2870. [Google Scholar] [CrossRef]
- Vuletic, M.S.; Milosevic, V.S.; Jancic, S.A.; Zujovic, J.T.; Krstic, M.S.; Vukmirovic, F.C. Clinical significance of Leptin receptor (LEPR) and Endoglin (CD105) expressions in colorectal adenocarcinoma. J. BUON 2019, 24, 2448–2457. [Google Scholar]
- Han, G.; Li, Y.; Cao, Y.; Yue, Z.; Zhang, Y.; Wang, L.; Liu, J. Overexpression of leptin receptor in human glioblastoma: Correlation with vasculogenic mimicry and poor prognosis. Oncotarget 2017, 8, 58163–58171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Newman, G.; Gonzalez-Perez, R.R. Leptin-cytokine crosstalk in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 570–582. [Google Scholar] [CrossRef]
- Chen, C.; Chang, Y.-C.; Lan, S.M.; Breslin, M. Leptin stimulates ovarian cancer cell growth and inhibits apoptosis by increasing cyclin D1 and Mcl-1 expression via the activation of the MEK/ERK1/2 and PI3K/Akt signaling pathways Corrigendum in /10.3892/ijo.2016.3564. Int. J. Oncol. 2013, 42, 1113–1119. [Google Scholar] [CrossRef]
- Babic, A.; Bao, Y.; Qian, Z.R.; Yuan, C.; Giovannucci, E.L.; Aschard, H.; Kraft, P.; Amundadottir, L.T.; Stolzenberg-Solomon, R.; Morales-Oyarvide, V.; et al. Pancreatic Cancer Risk Associated with Prediagnostic Plasma Levels of Leptin and Leptin Receptor Genetic Polymorphisms. Cancer Res. 2016, 76, 7160–7167. [Google Scholar] [CrossRef] [PubMed]
- Atoum, M.F.; Alzoughool, F.; Al-Hourani, H. Linkage Between Obesity Leptin and Breast Cancer. Breast Cancer 2020, 14, 1178223419898458. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A. The leptin receptor. J. Biol. Chem. 1997, 272, 6093–6096. [Google Scholar] [CrossRef]
- Loffreda, S.; Yang, S.Q.; Lin, H.Z.; Karp, C.L.; Brengman, M.L.; Wang, D.J.; Klein, A.S.; Bulkley, G.B.; Bao, C.; Noble, P.W.; et al. Leptin regulates proinflammatory immune responses. FASEB J. 1998, 12, 57–65. [Google Scholar] [CrossRef]
- Gainsford, T.; Willson, T.A.; Metcalf, D.; Handman, E.; McFarlane, C.; Ng, A.; Nicola, N.A.; Alexander, W.S.; Hilton, D.J. Leptin can induce proliferation, differentiation, and functional activation of hemopoietic cells. Proc. Natl. Acad. Sci. USA 1996, 93, 14564–14568. [Google Scholar] [CrossRef] [PubMed]
- Santos-Alvarez, J.; Goberna, R.; Sánchez-Margalet, V. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell. Immunol. 1999, 194, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Maeda, T.; Kobayashi, Y.; Fukazawa, Y.; Kishioka, S. Leptin enhances CC-chemokine ligand expression in cultured murine macrophage. Biochem. Biophys. Res. Commun. 2009, 384, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Tsiotra, P.C.; Boutati, E.; Dimitriadis, G.; Raptis, S.A. High insulin and leptin increase resistin and inflammatory cytokine production from human mononuclear cells. Biomed Res. Int. 2013, 2013, 487081. [Google Scholar] [CrossRef] [PubMed]
- Acedo, S.C.; Gambero, S.; Cunha, F.G.P.; Lorand-Metze, I.; Gambero, A. Participation of leptin in the determination of the macrophage phenotype: An additional role in adipocyte and macrophage crosstalk. Vitr. Cell. Dev. Biol.-Anim. 2013, 49, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef]
- Maya-Monteiro, C.M.; Almeida, P.E.; D’Ávila, H.; Martins, A.S.; Rezende, A.P.; Castro-Faria-Neto, H.; Bozza, P.T. Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase- and mammalian target of rapamycin-dependent mechanism. J. Biol. Chem. 2008, 283, 2203–2210. [Google Scholar] [CrossRef]
- Minokoshi, Y.; Kim, Y.-B.; Peroni, O.D.; Fryer, L.G.D.; Müller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef]
- Bernotiene, E.; Palmer, G.; Gabay, C. The role of leptin in innate and adaptive immune responses. Arthritis Res. Ther. 2006, 8, 217. [Google Scholar] [CrossRef][Green Version]
- Cerdá-Reverter, J.M.; Larhammar, D. Neuropeptide Y family of peptides: Structure, anatomical expression, function, and molecular evolution. Biochem. Cell Biol. 2000, 78, 371–392. [Google Scholar] [CrossRef]
- Simpson, K.A.; Martin, N.M.; Bloom, S.R. Hypothalamic regulation of food intake and clinical therapeutic applications. Arq. Bras. Endocrinol. Metabol. 2009, 53, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Taylor, I.L. Distribution and release of peptide YY in dog measured by specific radioimmunoassay. Gastroenterology 1985, 88, 731–737. [Google Scholar] [CrossRef]
- Ekblad, E.; Sundler, F. Distribution of pancreatic polypeptide and peptide YY. Peptides 2002, 23, 251–261. [Google Scholar] [CrossRef]
- Degen, L.; Oesch, S.; Casanova, M.; Graf, S.; Ketterer, S.; Drewe, J.; Beglinger, C. Effect of peptide YY3-36 on food intake in humans. Gastroenterology 2005, 129, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Mentlein, R.; Dahms, P.; Grandt, D.; Krüger, R. Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV. Regul. Pept. 1993, 49, 133–144. [Google Scholar] [CrossRef]
- Ballantyne, G.H. Peptide YY(1-36) and Peptide YY(3-36): Part I. Distribution, release and actions part II. Changes after gastointestinal and bariatric surgery will appear in the next issue (June 2006). Obes. Surg. 2006, 16, 651–658. [Google Scholar] [CrossRef]
- Bromée, T.; Sjödin, P.; Fredriksson, R.; Boswell, T.; Larsson, T.A.; Salaneck, E.; Zoorob, R.; Mohell, N.; Larhammar, D. Neuropeptide Y-family receptors Y6 and Y7 in chicken: Cloning, pharmacological characterization, tissue distribution and conserved synteny with human chromosome region. FEBS J. 2006, 273, 2048–2063. [Google Scholar] [CrossRef]
- Grandt, D.; Schimiczek, M.; Rascher, W.; Feth, F.; Shively, J.; Lee, T.D.; Davis, M.T.; Reeve, J.R.; Michel, M.C. Neuropeptide Y 3-36 is an endogenous ligand selective for Y2 receptors. Regul. Pept. 1996, 67, 33–37. [Google Scholar] [CrossRef]
- Macia, L.; Rao, P.T.; Wheway, J.; Sierro, F.; Mackay, F.; Herzog, H. Y1 signalling has a critical role in allergic airway inflammation. Immunol. Cell Biol. 2011, 89, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.M.; Klein, R.; Kruzliak, P.; Zulli, A. Role of Peptide YY in blood vessel function and atherosclerosis in a rabbit model. Clin. Exp. Pharmacol. Physiol. 2015, 42, 648–652. [Google Scholar] [CrossRef]
- Tseng, W.W.; Liu, C.D. Peptide YY and cancer: Current findings and potential clinical applications. Peptides 2002, 23, 389–395. [Google Scholar] [CrossRef]
- Li, J.; Tian, Y.; Wu, A. Neuropeptide Y receptors: A promising target for cancer imaging and therapy. Regen. Biomater. 2015, 2, 215–219. [Google Scholar] [CrossRef]
- Tilan, J.; Kitlinska, J. Neuropeptide Y (NPY) in tumor growth and progression: Lessons learned from pediatric oncology. Neuropeptides 2016, 55, 55–66. [Google Scholar] [CrossRef]
- De La Fuente, M.; Bernaez, I.; Del Rio, M.; Hernanz, A. Stimulation of murine peritoneal macrophage functions by neuropeptide Y and peptide YY. Involvement of protein kinase C. Immunology 1993, 80, 259–25965. [Google Scholar]
- Mitić, K.; Stanojević, S.; Kuštrimović, N.; Vujić, V.; Dimitrijević, M. Neuropeptide y modulates functions of inflammatory cells in the rat: Distinct role for Y1, Y2 and Y5 receptors. Peptides 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- Stanojević, S.; Vujić, V.; Kovačević-Jovanović, V.; Mitić, K.; Kosec, D.; von Hörsten, S.; Dimitrijević, M. Age-related effect of peptide YY (PYY) on paw edema in the rat: The function of Y1 receptors on inflammatory cells. Exp. Gerontol. 2006, 41, 793–799. [Google Scholar] [CrossRef]
- Dimitrijević, M.; Stanojević, S. The intriguing mission of neuropeptide y in the immune system. Amino Acids 2013, 45, 41–53. [Google Scholar] [CrossRef]
- Bedoui, S.; Kromer, A.; Gebhardt, T.; Jacobs, R.; Raber, K.; Dimitrijevic, M.; Heine, J.; von Hörsten, S. Neuropeptide Y receptor-specifically modulates human neutrophil function. J. Neuroimmunol. 2008, 195, 88–95. [Google Scholar] [CrossRef]
- Nave, H.; Bedoui, S.; Moenter, F.; Steffens, J.; Felies, M.; Gebhardt, T.; Straub, R.H.; Pabst, R.; Dimitrijevic, M.; Stanojevic, S.; et al. Reduced tissue immigration of monocytes by neuropeptide Y during endotoxemia is associated with Y2 receptor activation. J. Neuroimmunol. 2004, 155, 1–12. [Google Scholar] [CrossRef]
- Muk, T.; Stensballe, A.; Pankratova, S.; Nguyen, D.N.; Brunse, A.; Sangild, P.T.; Jiang, P.-P. Rapid Proteome Changes in Plasma and Cerebrospinal Fluid Following Bacterial Infection in Preterm Newborn Pigs. Front. Immunol. 2019, 10, 2651. [Google Scholar] [CrossRef]
- Macia, L.; Yulyaningsih, E.; Pangon, L.; Nguyen, A.D.; Lin, S.; Shi, Y.C.; Zhang, L.; Bijker, M.; Grey, S.; Mackay, F.; et al. Neuropeptide Y1 receptor in immune cells regulates inflammation and insulin resistance associated with diet-induced obesity. Diabetes 2012, 61, 3228–3238. [Google Scholar] [CrossRef] [PubMed]
- Puerto, M.; Guayerbas, N.; Álvarez, P.; De La Fuente, M. Modulation of neuropeptide Y and norepinephrine on several leucocyte functions in adult, old and very old mice. J. Neuroimmunol. 2005, 165, 33–40. [Google Scholar] [CrossRef]
- Ferreira, R.; Xapelli, S.; Santos, T.; Silva, A.P.; Cristóvão, A.; Cortes, L.; Malva, J.O. Neuropeptide Y modulation of interleukin-1β (IL-1β)-induced nitric oxide production in microglia. J. Biol. Chem. 2010, 285, 41921–41934. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.R.; Xu, Z.; Jiang, C.L. Neuropeptide Y promotes TGF-β1 production in RAW264.7 cells by activating PI3K pathway via Y1 receptor. Neurosci. Bull. 2008, 24, 155–159. [Google Scholar] [CrossRef]
- Gao, B.; Li, L.; Zhu, P.; Zhang, M.; Hou, L.; Sun, Y.; Liu, X.; Peng, X.; Gu, Y. Chronic administration of methamphetamine promotes atherosclerosis formation in ApoE-/- knockout mice fed normal diet. Atherosclerosis 2015, 243, 268–277. [Google Scholar] [CrossRef]
- De la Fuente, M.; Del Río, M.; Medina, S. Changes with aging in the modulation by neuropeptide Y of murine peritoneal macrophage functions. J. Neuroimmunol. 2001, 116, 156–167. [Google Scholar] [CrossRef]
- Hernanz, A.; Tato, E.; De la Fuente, M.; De Miguel, E.; Arnalich, F. Differential effects of gastrin-releasing peptide, neuropeptide Y, somatostatin and vasoactive intestinal peptide on interleukin-1β, interleukin-6 and tumor necrosis factor-α production by whole blood cells from healthy young and old subjects. J. Neuroimmunol. 1996, 71, 25–30. [Google Scholar] [CrossRef]
- Cheng, Y.; Tang, X.Y.; Li, Y.X.; Zhao, D.D.; Cao, Q.H.; Wu, H.X.; Yang, H.B.; Hao, K.; Yang, Y. Depression-induced neuropeptide y secretion promotes prostate cancer growth by recruiting myeloid cells. Clin. Cancer Res. 2019, 25, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Barakat, R.; Oakley, O.; Kim, H.; Jin, J.; Ko, C.M.J. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. 2016, 49, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef]
- Xu, Y.; López, M. Central regulation of energy metabolism by estrogens. Mol. Metab. 2018, 15, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J.Å. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B.; Sklar, L.A. GPR30: A G protein-coupled receptor for estrogen. Mol. Cell. Endocrinol. 2007, 265–266, 138–142. [Google Scholar] [CrossRef]
- Murphy, A.J.; Guyre, P.M.; Wira, C.R.; Pioli, P.A. Estradiol regulates expression of estrogen receptor ERα46 in human macrophages. PLoS ONE 2009, 4, e5539. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, A.; Fadini, G.P.; Tedesco, S.; Cappellari, R.; Vegeto, E.; Maggi, A.; Avogaro, A.; Bolego, C.; Cignarella, A. Alternative Activation of Human Macrophages Is Rescued by Estrogen Treatment In Vitro and Impaired by Menopausal Status. J. Clin. Endocrinol. Metab. 2015, 100, E50–E58. [Google Scholar] [CrossRef]
- Rettew, J.A.; McCall, S.H.; Marriott, I. GPR30/GPER-1 mediates rapid decreases in TLR4 expression on murine macrophages. Mol. Cell. Endocrinol. 2010, 328, 87–92. [Google Scholar] [CrossRef]
- Vegeto, E.; Bonincontro, C.; Pollio, G.; Sala, A.; Viappiani, S.; Nardi, F.; Brusadelli, A.; Viviani, B.; Ciana, P.; Maggi, A. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J. Neurosci. 2001, 21, 1809–1818. [Google Scholar] [CrossRef]
- Pelekanou, V.; Kampa, M.; Kiagiadaki, F.; Deli, A.; Theodoropoulos, P.; Agrogiannis, G.; Patsouris, E.; Tsapis, A.; Castanas, E.; Notas, G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J. Leukoc. Biol. 2016, 99, 333–347. [Google Scholar] [CrossRef]
- Leeners, B.; Geary, N.; Tobler, P.N.; Asarian, L. Ovarian hormones and obesity. Hum. Reprod. Update 2017, 23, 300–321. [Google Scholar] [CrossRef]
- Matarrese, P.; Mattia, G.; Pagano, M.T.; Pontecorvi, G.; Ortona, E.; Malorni, W.; Carè, A. The sex-related interplay between tme and cancer: On the critical role of estrogen, micrornas and autophagy. Cancers 2021, 13, 3287. [Google Scholar] [CrossRef]
- Cook, M.B.; Dawsey, S.M.; Freedman, N.D.; Inskip, P.D.; Wichner, S.M.; Quraishi, S.M.; Devesa, S.S.; McGlynn, K.A. Sex disparities in cancer incidence by period and age. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.B.; McGlynn, K.A.; Devesa, S.S.; Freedman, N.D.; Anderson, W.F. Sex disparities in cancer mortality and survival. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jin, J.; Qian, C.; Lou, J.; Lin, J.; Xu, A.; Xia, K.; Jin, L.; Liu, B.; Tao, H.; et al. Estrogen/ER in anti-tumor immunity regulation to tumor cell and tumor microenvironment. Cancer Cell Int. 2021, 21, 295. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Luo, Y.; Tai, R.; Zhang, N. Estrogen receptor β suppresses inflammation and the progression of prostate cancer. Mol. Med. Rep. 2019, 49, 3555–3563. [Google Scholar] [CrossRef]
- Yeh, C.R.; Slavin, S.; Da, J.; Hsu, I.; Luo, J.; Xiao, G.Q.; Ding, J.; Chou, F.J.; Yeh, S. Estrogen receptor α in cancer associated fibroblasts suppresses prostate cancer invasion via reducing CCL5, IL6 and macrophage infiltration in the tumor microenvironment. Mol. Cancer 2016, 15, 7. [Google Scholar] [CrossRef]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor cell–independent estrogen signaling drives disease progression through mobilization of myeloid-derived suppressor cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Pepe, G.; Braga, D.; Renzi, T.A.; Villa, A.; Bolego, C.; D’Avila, F.; Barlassina, C.; Maggi, A.; Locati, M.; Vegeto, E. Self-renewal and phenotypic conversion are the main physiological responses of macrophages to the endogenous estrogen surge. Sci. Rep. 2017, 7, 44270. [Google Scholar] [CrossRef]
- Pepe, G.; Locati, M.; Della Torre, S.; Mornata, F.; Cignarella, A.; Maggi, A.; Vegeto, E. The estrogen-macrophage interplay in the homeostasis of the female reproductive tract. Hum. Reprod. Update 2018, 24, 652–672. [Google Scholar] [CrossRef]
- Zheng, C.; Yang, Q.; Cao, J.; Xie, N.; Liu, K.; Shou, P.; Qian, F.; Wang, Y.; Shi, Y. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis. 2016, 7, e2167. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local Macrophage Proliferation, Rather than Recruitment from the Blood, Is a Signature of TH2 Inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.-L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.S.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Lhoták, Š.; Gyulay, G.; Cutz, J.; Al-Hashimi, A.; Trigatti, B.L.; Richards, C.D.; Igdoura, S.A.; Steinberg, G.R.; Bramson, J.; Ask, K.; et al. Characterization of Proliferating Lesion-Resident Cells During All Stages of Atherosclerotic Growth. J. Am. Heart Assoc. 2016, 5, e003945. [Google Scholar] [CrossRef] [PubMed]
- Gage, M.C.; Bécares, N.; Louie, R.; Waddington, K.E.; Zhang, Y.; Tittanegro, T.H.; Rodríguez-Lorenzo, S.; Jathanna, A.; Pourcet, B.; Pello, O.M.; et al. Disrupting LXRα phosphorylation promotes FoxM1 expression and modulates atherosclerosis by inducing macrophage proliferation. Proc. Natl. Acad. Sci. USA 2018, 115, E6556–E6565. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.; Emmerson, E.; Williams, H.; Saville, C.R.; Krust, A.; Chambon, P.; Mace, K.A.; Hardman, M.J. Estrogen Receptor-Alpha Promotes Alternative Macrophage Activation during Cutaneous Repair. J. Investig. Dermatol. 2014, 134, 2447–2457. [Google Scholar] [CrossRef]
- Villa, A.; Rizzi, N.; Vegeto, E.; Ciana, P.; Maggi, A. Estrogen accelerates the resolution of inflammation in macrophagic cells. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lu, Y.; Xu, Y.; Xu, L.; Zheng, W.; Wu, Y.; Li, L.; Shen, P. Estrogen represses hepatocellular carcinoma (HCC) Growth via Inhibiting Alternative Activation of Tumor-associated Macrophages (TAMs). J. Biol. Chem. 2012, 287, 40140–40149. [Google Scholar] [CrossRef]
- Kou, X.-X.; Li, C.-S.; He, D.-Q.; Wang, X.-D.; Hao, T.; Meng, Z.; Zhou, Y.-H.; Gan, Y.-H. Estradiol Promotes M1-like Macrophage Activation through Cadherin-11 To Aggravate Temporomandibular Joint Inflammation in Rats. J. Immunol. 2015, 194, 2810–2818. [Google Scholar] [CrossRef]
- Attia, D.M.A.; Ederveen, A.G.H. Opposing roles of ERα and ERβ in the genesis and progression of adenocarcinoma in the rat ventral prostate. Prostate 2012, 72, 1013–1022. [Google Scholar] [CrossRef]
- Calippe, B.; Douin-Echinard, V.; Delpy, L.; Laffargue, M.; Lélu, K.; Krust, A.; Pipy, B.; Bayard, F.; Arnal, J.-F.; Guéry, J.-C.; et al. 17β-Estradiol Promotes TLR4-Triggered Proinflammatory Mediator Production through Direct Estrogen Receptor α Signaling in Macrophages In Vivo. J. Immunol. 2010, 185, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.S.; Duffner, L.A.; Faunce, D.E.; Kovacs, E.J. Estrogen mediates the sex difference in post-burn immunosuppression. J. Endocrinol. 2000, 164, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Morishita, M.; Miyagi, M.; Iwamoto, Y. Effects of Sex Hormones on Production of Interleukin-1 by Human Peripheral Monocytes. J. Periodontol. 1999, 70, 757–760. [Google Scholar] [CrossRef]
- Ruh, M.F.; Bi, Y.; D’Alonzo, R.; Bellone, C.J. Effect of estrogens on IL-1β promoter activity. J. Steroid Biochem. Mol. Biol. 1998, 66, 203–210. [Google Scholar] [CrossRef]
- Ralston, S.H.; Russell, R.G.G.; Gowen, M. Estrogen inhibits release of tumor necrosis factor from peripheral blood mononuclear cells in postmenopausal women. J. Bone Miner. Res. 1990, 5, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Woodfork, K.A.; Schuller, K.C.; Huffman, L.J. Cytokine and nitric oxide release by J774A.1 macrophages is not regulated by estradiol. Life Sci. 2001, 69, 2287–2294. [Google Scholar] [CrossRef]
- Kassem, M.; Khosla, S.; Spelsberg, T.C.; Riggs, B.L. Cytokine production in the bone marrow microenvironment: Failure to demonstrate estrogen regulation in early postmenopausal women. J. Clin. Endocrinol. Metab. 1996, 81, 513–518. [Google Scholar] [CrossRef][Green Version]
- Pfeilschifter, J.; Köditz, R.; Pfohl, M.; Schatz, H. Changes in proinflammatory cytokine activity after menopause. Endocr. Rev. 2002, 23, 90–119. [Google Scholar] [CrossRef]
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 are novel therapeutic targets for estrogen-dependent breast cancer. Clin. Cancer Res. 2015, 21, 3794–3805. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, D.J. Androgen Physiology, Pharmacology, and Abuse. In Endocrinology: Adult and Pediatric; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 2368–2393. ISBN 9780323189071. [Google Scholar]
- Grino, P.B.; Griffin, J.E.; Wilson, J.D. Testosterone at High Concentrations Interacts with the Human Androgen Receptor Similarly to Dihydrotestosterone. Endocrinology 1990, 126, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Saltzman, A.; Yeh, S.; Young, W.; Keller, E.; Lee, H.J.; Wang, C.; Mizokami, A. Androgen receptor: An overview. Crit. Rev. Eukaryot. Gene Expr. 1995, 5, 97–125. [Google Scholar] [CrossRef]
- Lösel, R.M.; Falkenstein, E.; Feuring, M.; Schultz, A.; Tillmann, H.C.; Rossol-Haseroth, K.; Wehling, M. Nongenomic steroid action: Controversies, questions, and answers. Physiol. Rev. 2003, 83, 965–1016. [Google Scholar] [CrossRef] [PubMed]
- Fui, M.N.T.; Dupuis, P.; Grossmann, M. Lowered testosterone in male obesity: Mechanisms, morbidity and management. Asian J. Androl. 2014, 16, 223–231. [Google Scholar] [CrossRef]
- Pasquali, R. Obesity and androgens: Facts and perspectives. Fertil. Steril. 2006, 85, 1319–1340. [Google Scholar] [CrossRef]
- Zhang, L.J.; Xiong, Y.; Nilubol, N.; He, M.; Bommareddi, S.; Zhu, X.; Jia, L.; Xiao, Z.; Park, J.-W.; Xu, X.; et al. Testosterone regulates thyroid cancer progression by modifying tumor suppressor genes and tumor immunity. Carcinogenesis 2015, 36, 420–428. [Google Scholar] [CrossRef]
- Gannon, P.O.; Poisson, A.O.; Delvoye, N.; Lapointe, R.; Mes-Masson, A.M.; Saad, F. Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J. Immunol. Methods 2009, 348, 9–17. [Google Scholar] [CrossRef]
- Trigunaite, A.; Dimo, J.; Jørgensen, T.N. Suppressive effects of androgens on the immune system. Cell. Immunol. 2015, 294, 87–94. [Google Scholar] [CrossRef]
- Malkin, C.J.; Pugh, P.J.; Jones, R.D.; Jones, T.H.; Channer, K.S. Testosterone as a protective factor against atherosclerosis—Immunomodulation and influence upon plaque development and stability. J. Endocrinol. 2003, 178, 373–380. [Google Scholar] [CrossRef]
- Yao, Q.M.; Wang, B.; An, X.F.; Zhang, J.A.; Ding, L. Testosterone level and risk of type 2 diabetes in men: A systematic review and meta-analysis. Endocr. Connect. 2018, 7, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Malkin, C.J.; Pugh, P.J.; Jones, R.D.; Kapoor, D.; Channer, K.S.; Jones, T.H. The Effect of Testosterone Replacement on Endogenous Inflammatory Cytokines and Lipid Profiles in Hypogonadal Men. J. Clin. Endocrinol. Metab. 2004, 89, 3313–3318. [Google Scholar] [CrossRef] [PubMed]
- Agostino, P.; Milano, S.; Barbera, C.; Di Bella, G.; La Rosa, M.; Ferlazzo, V.; Farruggio, R.; Miceli, D.M.; Miele, M.; Castagnetta, L.; et al. Sex hormones modulate inflammatory mediators produced by macrophages a. Ann. N. Y. Acad. Sci. 1999, 876, 426–429. [Google Scholar] [CrossRef]
- Corcoran, M.P.; Meydani, M.; Lichtenstein, A.H.; Schaefer, E.J.; Dillard, A.; Lamon-Fava, S. Sex hormone modulation of proinflammatory cytokine and C-reactive protein expression in macrophages from older men and postmenopausal women. J. Endocrinol. 2010, 206, 217–224. [Google Scholar] [CrossRef]
- Fijak, M.; Schneider, E.; Klug, J.; Bhushan, S.; Hackstein, H.; Schuler, G.; Wygrecka, M.; Gromoll, J.; Meinhardt, A. Testosterone Replacement Effectively Inhibits the Development of Experimental Autoimmune Orchitis in Rats: Evidence for a Direct Role of Testosterone on Regulatory T Cell Expansion. J. Immunol. 2011, 186, 5162–5172. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.-C.; Alten, P.J.V.; Walter, R.J. Steroid Sex Hormones and Macrophage Function: Modulation of Reactive Oxygen Intermediates and Nitrite Release. Am. J. Reprod. Immunol. 1994, 32, 43–52. [Google Scholar] [CrossRef]
- Friedl, R.; Brunner, M.; Moeslinger, T.; Spieckermann, P.G. Testosterone inhibits expression of inducible nitric oxide synthase in murine macrophages. Life Sci. 2000, 68, 417–429. [Google Scholar] [CrossRef]
- Rettew, J.A.; Huet-Hudson, Y.M.; Marriott, I. Testosterone Reduces Macrophage Expression in the Mouse of Toll-Like Receptor 4, a Trigger for Inflammation and Innate Immunity. Biol. Reprod. 2008, 78, 432–437. [Google Scholar] [CrossRef]
- Quintar, A.A.; Roth, F.D.; De Paul, A.L.; Aoki, A.; Maldonado, C.A. Toll-like receptor 4 in rat prostate: Modulation by testosterone and acute bacterial infection in epithelial and stromal cells. Biol. Reprod. 2006, 75, 664–672. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.C.W.; Jian, C.Y.; Lin, P.H.; Chen, C.C.C.W.; Lieu, F.K.; Soong, C.; Hsieh, C.C.; Wan, C.Y.; Idova, G.; Hu, S.; et al. Role of testosterone in regulating induction of TNF-α in rat spleen via ERK signaling pathway. Steroids 2016, 111, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Accardo, S.; Villaggio, B.; Barone, A.; Sulli, A.; Coviello, D.A.; Carabbio, C.; Felli, L.; Miceli, D.; Farruggio, R.; et al. Androgen and estrogen receptors are present in primary cultures of human synovial macrophages. J. Clin. Endocrinol. Metab. 1996, 81, 820–827. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McCrohon, J.A.; Death, A.K.; Nakhla, S.; Jessup, W.; Handelsman, D.J.; Stanley, K.K.; Celermajer, D.S. Androgen receptor expression is greater in macrophages from male than from female donors: A sex difference with implications for atherogenesis. Circulation 2000, 101, 224–226. [Google Scholar] [CrossRef]
- Cioni, B.; Zaalberg, A.; van Beijnum, J.R.; Melis, M.H.M.; van Burgsteden, J.; Muraro, M.J.; Hooijberg, E.; Peters, D.; Hofland, I.; Lubeck, Y.; et al. Androgen receptor signalling in macrophages promotes TREM-1-mediated prostate cancer cell line migration and invasion. Nat. Commun. 2020, 11, 4498. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hormone | Trigger | Origins | Metabolic Target | Receptor | Primary Metabolic Functions |
|---|---|---|---|---|---|
| CCK | Fatty acids, proteins | Small intestine I-cells | Pancreas | CCK1R & CCK2R | Stimulates release of digestive enzymes and insulin |
| FABP4 | Lipolysis | Adipocytes Macrophages | Adipocytes Macrophages | PPARγ | Absorption of fatty acids M2 macrophage polarisation |
| Gastrin | Food intake Gastrin releasing peptide | Stomach G-cells Duodenum Pancreas | Stomach | CCK1R & CCK2R | Stomach acid regulation |
| Ghrelin | Food intake | Stomach Intestine Brain Macrophages | Brain Adipose tissue | GHSR | Regulates food intake, energy expenditure, glucose homeostasis, adiposity |
| GIP | Glucose, fatty acids | K-cells in the duodenum and jejunum | Pancreatic β-cells | GIPR | Stimulates insulin release |
| GLP-1 | Hexose, fats | L-cells of the small intestine | Pancreatic β-cells Brain | GLP1R | Stimulates insulin release Induces satiety |
| Insulin | Hyperglycaemia | Pancreatic β-cells | Muscle Liver Adipose | INSR IGF1R | Glucose uptake Inhibition of gluconeogenesis |
| IGF-1 | Growth Hormone (GH) | Liver Macrophages Adipocytes | Bones Smooth muscle Neurons | IGF1R INSR | Stimulates bone and tissue growth |
| Leptin | Food intake | Adipocytes | Brain | OBR | Regulation of food intake |
| NPY | Food intake (High levels of dietary fat and sugar) | Central nervous system | Central and peripheral nervous systems | NPY Receptors (GPCRs) | Regulation of food intake |
| PYY | Amino acids Short-chain fatty acids | L-cells of the ileum and colon | Central and peripheral nervous systems | PYY Receptors (GPCRs) | Gastric emptying Gut motility |
| Estrogen | Luteinizing hormone (LH) | Gonads, adipose tissue, bone, skin, liver & brain | Systemic | ERα Erβ GPER | Primary female sex hormone Fat distribution Metabolism |
| Testosterone | Luteinizing hormone (LH) | Leydig cells of the testis & adrenal glands | Systemic | AR | Primary male sex hormone Fat distribution Muscle mass |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batty, M.J.; Chabrier, G.; Sheridan, A.; Gage, M.C. Metabolic Hormones Modulate Macrophage Inflammatory Responses. Cancers 2021, 13, 4661. https://doi.org/10.3390/cancers13184661
Batty MJ, Chabrier G, Sheridan A, Gage MC. Metabolic Hormones Modulate Macrophage Inflammatory Responses. Cancers. 2021; 13(18):4661. https://doi.org/10.3390/cancers13184661
Chicago/Turabian StyleBatty, Matthew J., Gwladys Chabrier, Alanah Sheridan, and Matthew C. Gage. 2021. "Metabolic Hormones Modulate Macrophage Inflammatory Responses" Cancers 13, no. 18: 4661. https://doi.org/10.3390/cancers13184661
APA StyleBatty, M. J., Chabrier, G., Sheridan, A., & Gage, M. C. (2021). Metabolic Hormones Modulate Macrophage Inflammatory Responses. Cancers, 13(18), 4661. https://doi.org/10.3390/cancers13184661