
Functional Characteristics and Regulated Expression of Alternatively Spliced Tissue Factor: An Update


Simple Summary
Abstract
1. Introduction
2. asTF Is Overexpressed in Multiple Malignancies
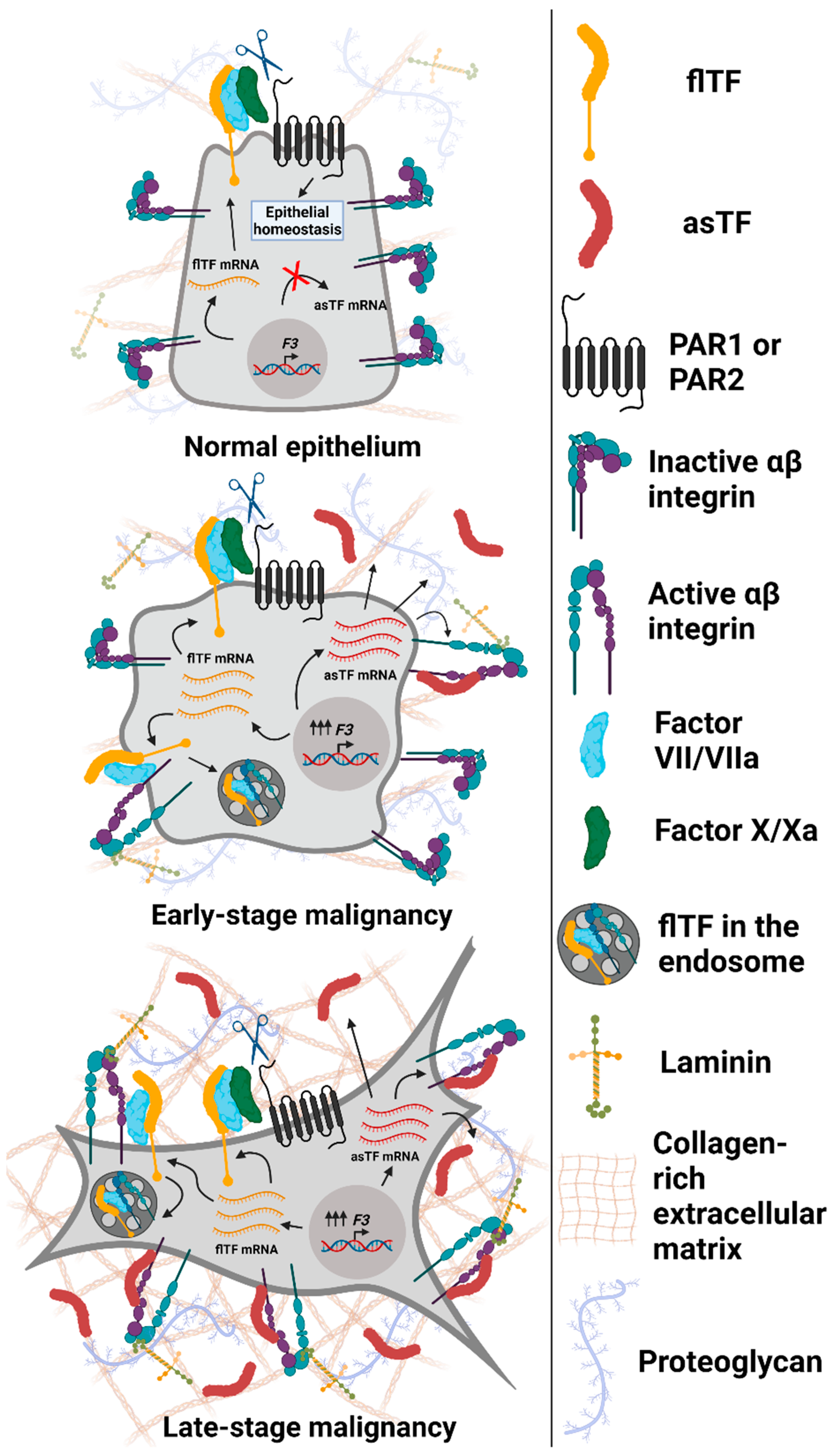
3. Consequences of asTF’s Interaction with Integrins on Surfaces of Benign and Malignant Cells
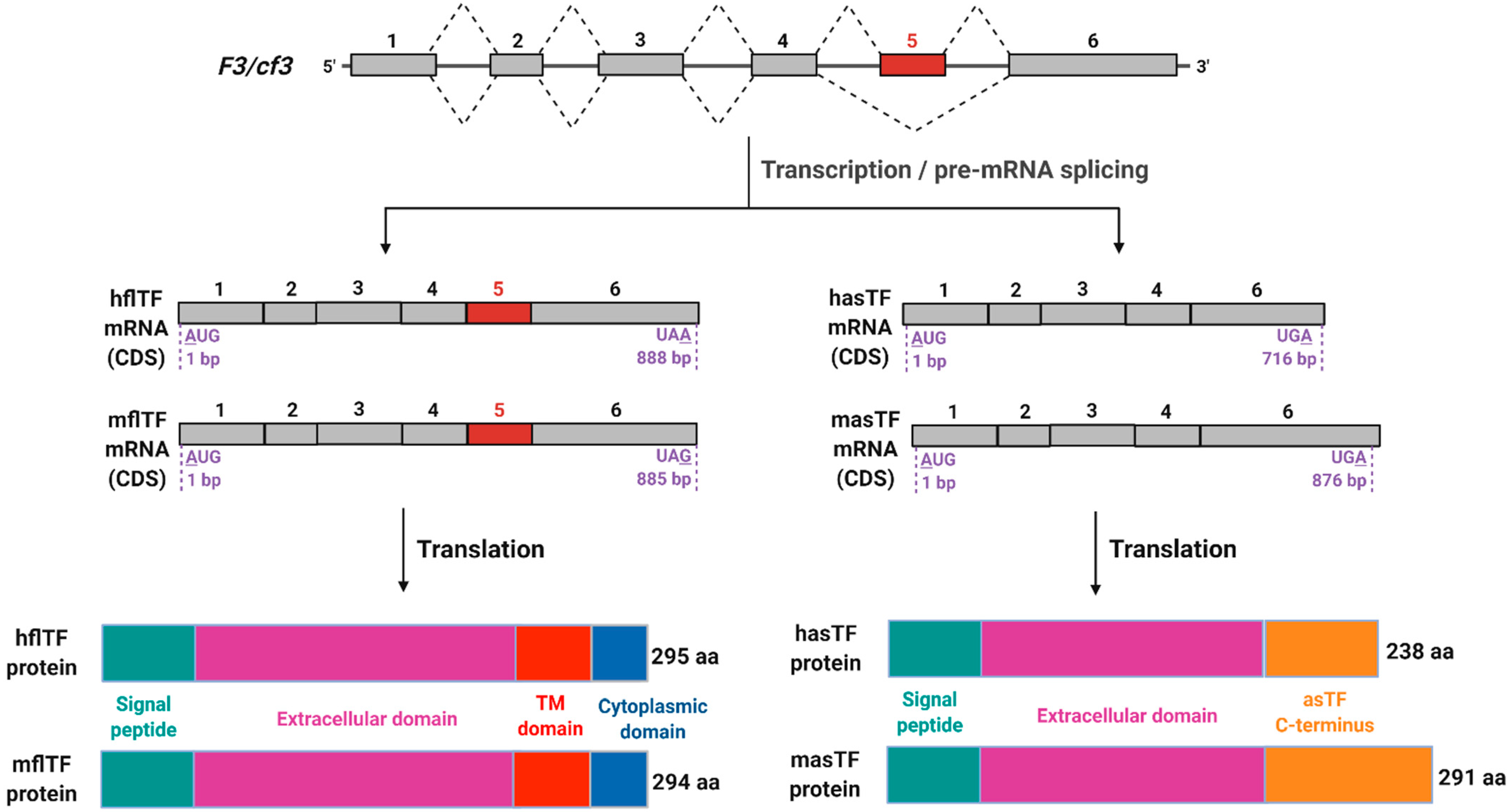
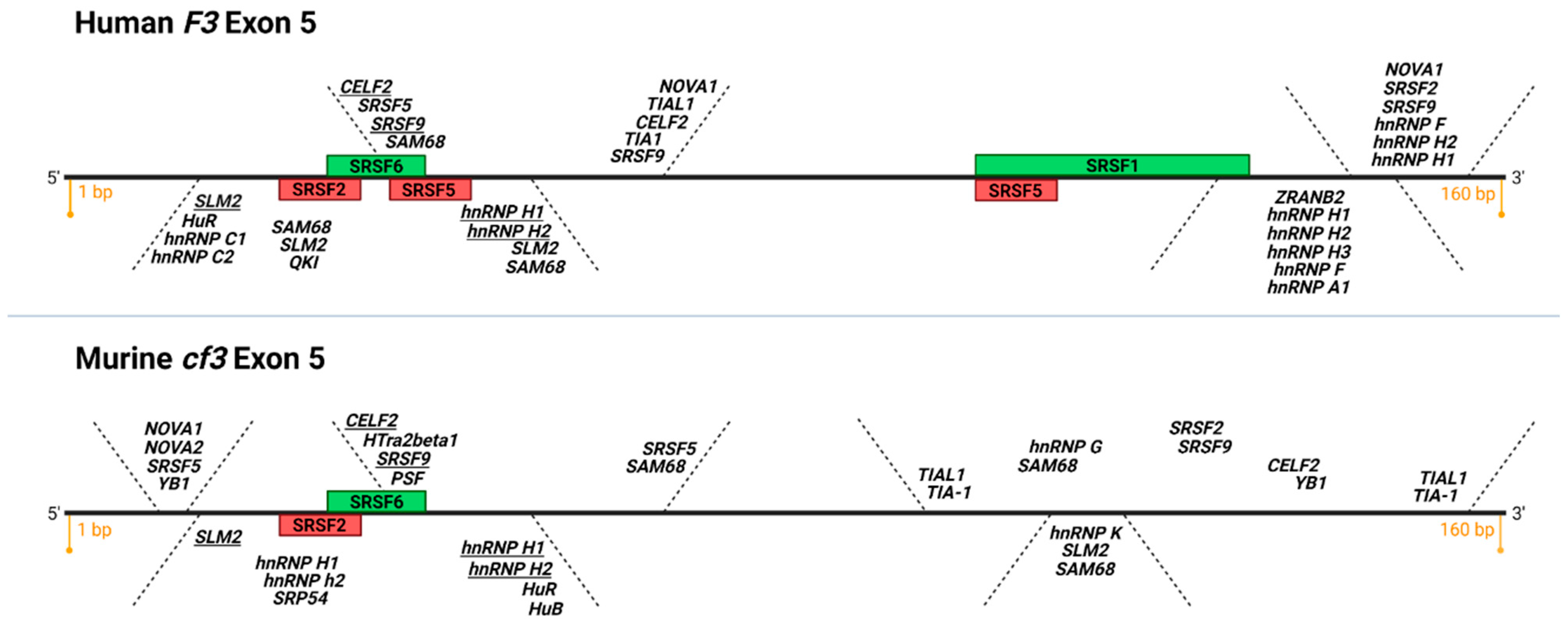
4. asTF Biosynthesis: Regulated vs. Aberrant Alternative Splicing
5. asTF and White Cell Physiology: Effect on Monocyte Recruitment and Macrophage Polarization
6. asTF’s Dispensability to Normal Hemostasis: Is This All There Is to It?
7. asTF as a Disease Biomarker
8. Therapeutic Targeting of asTF
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Spicer, E.K.; Horton, R.; Bloem, L.; Bach, R.; Williams, K.R.; Guha, A.; Kraus, J.; Lin, T.C.; Nemerson, Y.; Konigsberg, W.H. Isolation of CDNA Clones Coding for Human Tissue Factor: Primary Structure of the Protein and CDNA. Proc. Natl. Acad. Sci. USA 1987, 84, 5148–5152. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Morrissey, J.H.; Fowler, B.; Edgington, T.S. Complete Sequence of the Human Tissue Factor Gene, a Highly Regulated Cellular Receptor That Initiates the Coagulation Protease Cascade. Biochemistry 1989, 28, 1755–1762. [Google Scholar] [CrossRef]
- Witkowski, M.; Witkowski, M.; Friebel, J.; Buffa, J.A.; Li, X.S.; Wang, Z.; Sangwan, N.; Li, L.; DiDonato, J.A.; Tizian, C.; et al. Vascular Endothelial Tissue Factor Contributes to Trimethylamine N-Oxide-Enhanced Arterial Thrombosis. Cardiovasc. Res. 2021, cvab263. [Google Scholar] [CrossRef]
- Tawil, N.; Bassawon, R.; Meehan, B.; Nehme, A.; Montermini, L.; Gayden, T.; De Jay, N.; Spinelli, C.; Chennakrishnaiah, S.; Choi, D.; et al. Glioblastoma Cell Populations with Distinct Oncogenic Programs Release Podoplanin as Procoagulant Extracellular Vesicles. Blood Adv. 2021, 5, 1682–1694. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Grover, S.P.; Antoniak, S. Tissue factor expression, extracellular vesicles, and thrombosis after infection with the respiratory viruses influenza A virus and coronavirus. J. Thromb. Haemost. 2021, 1–7. [Google Scholar] [CrossRef]
- Sorensen, A.B.; Tuneew, I.; Svensson, L.A.; Persson, E.; Østergaard, H.; Overgaard, M.T.; Olsen, O.H.; Gandhi, P.S. Beating Tissue Factor at Its Own Game: Design and Properties of a Soluble Tissue Factor-Independent Coagulation Factor VIIa. J. Biol. Chem. 2020, 295, 517–528. [Google Scholar] [CrossRef]
- Vadivel, K.; Schmidt, A.E.; Cascio, D.; Padmanabhan, K.; Krishnaswamy, S.; Brandstetter, H.; Bajaj, S.P. Structure of Human Factor VIIa-Soluble Tissue Factor with Calcium, Magnesium and Rubidium. Acta Cryst. D Struct. Biol. 2021, 77, 809–819. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, W.; Wang, F.; Wang, M.; Xu, K.; Yang, A.; Wang, C.; Zhang, L.; Zhang, F.; Li, M. Tissue Factor Potentiates Adherence of Breast Cancer Cells to Human Umbilical Vein Endothelial Cells under Static and Flow Conditions. Cell Adh. Migr. 2021, 15, 74–83. [Google Scholar] [CrossRef]
- Bogdanov, V.Y.; Versteeg, H.H. “Soluble Tissue Factor” in the 21st Century: Definitions, Biochemistry, and Pathophysiological Role in Thrombus Formation. Semin. Thromb. Hemost. 2015, 41, 700–707. [Google Scholar] [CrossRef]
- Suehiro, E.; Fujiyama, Y.; Kiyohira, M.; Motoki, Y.; Nojima, J.; Suzuki, M. Probability of Soluble Tissue Factor Release Lead to the Elevation of D-Dimer as a Biomarker for Traumatic Brain Injury. Neurol. Med. Chir. 2019, 59, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, V.Y.; Kirk, R.I.; Miller, C.; Hathcock, J.J.; Vele, S.; Gazdoiu, M.; Nemerson, Y.; Taubman, M.B. Identification and Characterization of Murine Alternatively Spliced Tissue Factor. J. Thromb. Haemost. 2006, 4, 158–167. [Google Scholar] [CrossRef]
- Bogdanov, V.Y. Blood Coagulation and Alternative Pre-mRNA Splicing: An Overview. Curr. Mol. Med. 2006, 6, 859–869. [Google Scholar] [CrossRef]
- Unruh, D.; Horbinski, C. Beyond Thrombosis: The Impact of Tissue Factor Signaling in Cancer. J. Hematol. Oncol. 2020, 13, 93. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Expression of F3 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000117525-F3/pathology (accessed on 20 August 2021).
- Wang, J.; Dumartin, L.; Mafficini, A.; Ulug, P.; Sangaralingam, A.; Alamiry, N.A.; Radon, T.P.; Salvia, R.; Lawlor, R.T.; Lemoine, N.R.; et al. Splice Variants as Novel Targets in Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2017, 7, 2980. [Google Scholar] [CrossRef]
- Yang, Y.; Stang, A.; Schweickert, P.G.; Lanman, N.A.; Paul, E.N.; Monia, B.P.; Revenko, A.S.; Palumbo, J.S.; Mullins, E.S.; Elzey, B.D.; et al. Thrombin Signaling Promotes Pancreatic Adenocarcinoma through PAR-1-Dependent Immune Evasion. Cancer Res. 2019, 79, 3417–3430. [Google Scholar] [CrossRef] [PubMed]
- Nitori, N.; Ino, Y.; Nakanishi, Y.; Yamada, T.; Honda, K.; Yanagihara, K.; Kosuge, T.; Kanai, Y.; Kitajima, M.; Hirohashi, S. Prognostic Significance of Tissue Factor in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2005, 11, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Toi, M.; Koike, M.; Nakamura, S.; Tominaga, T. Tissue Factor Expression in Breast Cancer Tissues: Its Correlation with Prognosis and Plasma Concentration. Br. J. Cancer 2000, 83, 164–170. [Google Scholar] [CrossRef]
- Hron, G.; Kollars, M.; Weber, H.; Sagaster, V.; Quehenberger, P.; Eichinger, S.; Kyrle, P.A.; Weltermann, A. Tissue Factor-Positive Microparticles: Cellular Origin and Association with Coagulation Activation in Patients with Colorectal Cancer. Thromb. Haemost. 2007, 97, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Mackman, N. Tissue Factor and Cancer: Regulation, Tumor Growth, and Metastasis. Semin. Thromb. Hemost. 2019, 45, 385–395. [Google Scholar] [CrossRef]
- Haas, S.L.; Jesnowski, R.; Steiner, M.; Hummel, F.; Ringel, J.; Burstein, C.; Nizze, H.; Liebe, S.; Löhr, J.M. Expression of Tissue Factor in Pancreatic Adenocarcinoma Is Associated with Activation of Coagulation. World J. Gastroenterol. 2006, 12, 4843–4849. [Google Scholar] [CrossRef]
- Regina, S.; Rollin, J.; Bléchet, C.; Iochmann, S.; Reverdiau, P.; Gruel, Y. Tissue Factor Expression in Non-Small Cell Lung Cancer: Relationship with Vascular Endothelial Growth Factor Expression, Microvascular Density, and K-Ras Mutation. J. Thorac. Oncol. 2008, 3, 689–697. [Google Scholar] [CrossRef]
- Wu, M.; Chen, L.; Xu, T.; Xu, B.; Jiang, J.; Wu, C. Prognostic Values of Tissue Factor and Its Alternatively Splice Transcripts in Human Gastric Cancer Tissues. Oncotarget 2017, 8, 53137–53145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pan, L.; Yu, Y.; Yu, M.; Yao, S.; Mu, Q.; Luo, G.; Xu, N. Expression of FlTF and AsTF Splice Variants in Various Cell Strains and Tissues. Mol. Med. Rep. 2019, 19, 2077–2086. [Google Scholar] [CrossRef]
- Bogdanov, V.Y.; Balasubramanian, V.; Hathcock, J.; Vele, O.; Lieb, M.; Nemerson, Y. Alternatively Spliced Human Tissue Factor: A Circulating, Soluble, Thrombogenic Protein. Nat. Med. 2003, 9, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Goldin-Lang, P.; Tran, Q.-V.; Fichtner, I.; Eisenreich, A.; Antoniak, S.; Schulze, K.; Coupland, S.E.; Poller, W.; Schultheiss, H.-P.; Rauch, U. Tissue Factor Expression Pattern in Human Non-Small Cell Lung Cancer Tissues Indicate Increased Blood Thrombogenicity and Tumor Metastasis. Oncol. Rep. 2008, 20, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Kocatürk, B.; Van den Berg, Y.W.; Tieken, C.; Mieog, J.S.D.; de Kruijf, E.M.; Engels, C.C.; van der Ent, M.A.; Kuppen, P.J.; Van de Velde, C.J.; Ruf, W.; et al. Alternatively Spliced Tissue Factor Promotes Breast Cancer Growth in a Β1 Integrin-Dependent Manner. Proc. Natl. Acad. Sci. USA 2013, 110, 11517–11522. [Google Scholar] [CrossRef]
- Godby, R.C.; Van Den Berg, Y.W.; Srinivasan, R.; Sturm, R.; Hui, D.Y.; Konieczny, S.F.; Aronow, B.J.; Ozhegov, E.; Ruf, W.; Versteeg, H.H.; et al. Nonproteolytic Properties of Murine Alternatively Spliced Tissue Factor: Implications for Integrin-Mediated Signaling in Murine Models. Mol. Med. 2012, 18, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Beuneu, C.; Vosters, O.; Movahedi, B.; Remmelink, M.; Salmon, I.; Pipeleers, D.; Pradier, O.; Goldman, M.; Verhasselt, V. Human Pancreatic Duct Cells Exert Tissue Factor-Dependent Procoagulant Activity: Relevance to Islet Transplantation. Diabetes 2004, 53, 1407–1411. [Google Scholar] [CrossRef]
- Unruh, D.; Turner, K.; Srinivasan, R.; Kocatürk, B.; Qi, X.; Chu, Z.; Aronow, B.J.; Plas, D.R.; Gallo, C.A.; Kalthoff, H.; et al. Alternatively Spliced Tissue Factor Contributes to Tumor Spread and Activation of Coagulation in Pancreatic Ductal Adenocarcinoma. Int. J. Cancer 2014, 134, 9–20. [Google Scholar] [CrossRef]
- Unruh, D.; Sagin, F.; Adam, M.; Van Dreden, P.; Woodhams, B.J.; Hart, K.; Lindsell, C.J.; Ahmad, S.A.; Bogdanov, V.Y. Levels of Alternatively Spliced Tissue Factor in the Plasma of Patients with Pancreatic Cancer May Help Predict Aggressive Tumor Phenotype. Ann. Surg. Oncol. 2015, 22 (Suppl. 3), S1206–S1211. [Google Scholar] [CrossRef]
- Schlitter, A.M.; Jesinghaus, M.; Jäger, C.; Konukiewitz, B.; Muckenhuber, A.; Demir, I.E.; Bahra, M.; Denkert, C.; Friess, H.; Kloeppel, G.; et al. PT but Not PN Stage of the 8th TNM Classification Significantly Improves Prognostication in Pancreatic Ductal Adenocarcinoma. Eur. J. Cancer 2017, 84, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-J.; Hou, X.-D.; Li, Y.-M. Effect of Tissue Factor Knockdown on the Growth, Invasion, Chemoresistance and Apoptosis of Human Gastric Cancer Cells. Exp. Ther. Med. 2014, 7, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Kocatürk, B.; Versteeg, H.H. Tissue Factor-Integrin Interactions in Cancer and Thrombosis: Every Jack Has His Jill. J. Thromb. Haemost. 2013, 11 (Suppl. 1), 285–293. [Google Scholar] [CrossRef]
- Åberg, M.; Edén, D.; Siegbahn, A. Activation of Β1 Integrins and Caveolin-1 by TF/FVIIa Promotes IGF-1R Signaling and Cell Survival. Apoptosis 2020, 25, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Spoerri, P.M.; Strohmeyer, N.; Sun, Z.; Fässler, R.; Müller, D.J. Protease-Activated Receptor Signalling Initiates A5β1-Integrin-Mediated Adhesion in Non-Haematopoietic Cells. Nat. Mater. 2020, 19, 218–226. [Google Scholar] [CrossRef]
- Segal, L.; Katz, L.S.; Shapira, H.; Sandbank, J.; Geras-Raaka, E.; Gershengorn, M.C.; Oron, Y. PAR-3 Knockdown Enhances Adhesion Rate of PANC-1 Cells via Increased Expression of Integrinαv and E-Cadherin. PLoS ONE 2014, 9, e93879. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, A.S.; Marchese, P.; Langer, F.; Kamikubo, Y.; Schaffner, F.; Cantor, J.; Ginsberg, M.H.; Ruggeri, Z.M.; Ruf, W. Tissue Factor Prothrombotic Activity Is Regulated by Integrin-Arf6 Trafficking. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, Y.W.; van den Hengel, L.G.; Myers, H.R.; Ayachi, O.; Jordanova, E.; Ruf, W.; Spek, C.A.; Reitsma, P.H.; Bogdanov, V.Y.; Versteeg, H.H. Alternatively Spliced Tissue Factor Induces Angiogenesis through Integrin Ligation. Proc. Natl. Acad. Sci. USA 2009, 106, 19497–19502. [Google Scholar] [CrossRef]
- Srinivasan, R.; Ozhegov, E.; Berg, Y.W.V.D.; Aronow, B.J.; Franco, R.S.; Palascak, M.B.; Fallon, J.T.; Ruf, W.; Versteeg, H.H.; Bogdanov, V.Y. Splice Variants of Tissue Factor Promote Monocyte-Endothelial Interactions by Triggering the Expression of Cell Adhesion Molecules via Integrin-Mediated Signaling. J. Thromb. Haemost. 2011, 9, 2087–2096. [Google Scholar] [CrossRef]
- Kocatürk, B.; Tieken, C.; Vreeken, D.; Ünlü, B.; Engels, C.C.; de Kruijf, E.M.; Kuppen, P.J.; Reitsma, P.H.; Bogdanov, V.Y.; Versteeg, H.H. Alternatively Spliced Tissue Factor Synergizes with the Estrogen Receptor Pathway in Promoting Breast Cancer Progression. J. Thromb. Haemost. 2015, 13, 1683–1693. [Google Scholar] [CrossRef]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic Ductal Adenocarcinoma: Biological Hallmarks, Current Status, and Future Perspectives of Combined Modality Treatment Approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef]
- Jain, T.; Dudeja, V. The War against Pancreatic Cancer in 2020 - Advances on All Fronts. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Pourshams, A.; Sepanlou, S.G.; Ikuta, K.S.; Bisignano, C.; Safiri, S.; Roshandel, G.; Sharif, M.; Khatibian, M.; Fitzmaurice, C.; Nixon, M.R.; et al. The Global, Regional, and National Burden of Pancreatic Cancer and Its Attributable Risk Factors in 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2019, 4, 934–947. [Google Scholar] [CrossRef]
- Hobbs, J.E.; Zakarija, A.; Cundiff, D.L.; Doll, J.A.; Hymen, E.; Cornwell, M.; Crawford, S.E.; Liu, N.; Signaevsky, M.; Soff, G.A. Alternatively Spliced Human Tissue Factor Promotes Tumor Growth and Angiogenesis in a Pancreatic Cancer Tumor Model. Thromb. Res. 2007, 120 (Suppl. 2), S13–S21. [Google Scholar] [CrossRef]
- Unruh, D.; Ünlü, B.; Lewis, C.S.; Qi, X.; Chu, Z.; Sturm, R.; Keil, R.; Ahmad, S.A.; Sovershaev, T.; Adam, M.; et al. Antibody-Based Targeting of Alternatively Spliced Tissue Factor: A New Approach to Impede the Primary Growth and Spread of Pancreatic Ductal Adenocarcinoma. Oncotarget 2016, 7, 25264–25275. [Google Scholar] [CrossRef]
- Ehlen, L.; Arndt, J.; Treue, D.; Bischoff, P.; Loch, F.N.; Hahn, E.M.; Kotsch, K.; Klauschen, F.; Beyer, K.; Margonis, G.A.; et al. Novel Methods for in Vitro Modeling of Pancreatic Cancer Reveal Important Aspects for Successful Primary Cell Culture. BMC Cancer 2020, 20, 417. [Google Scholar] [CrossRef]
- Frappart, P.-O.; Hofmann, T.G. Pancreatic Ductal Adenocarcinoma (PDAC) Organoids: The Shining Light at the End of the Tunnel for Drug Response Prediction and Personalized Medicine. Cancers 2020, 12, 2750. [Google Scholar] [CrossRef]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic Cancer Stroma: An Update on Therapeutic Targeting Strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Ramchandani, D.; Unruh, D.; Lewis, C.S.; Bogdanov, V.Y.; Weber, G.F. Activation of Carbonic Anhydrase IX by Alternatively Spliced Tissue Factor under Late-Stage Tumor Conditions. Lab. Invest. 2016, 96, 1234–1245. [Google Scholar] [CrossRef]
- Ward, C.; Meehan, J.; Gray, M.; Kunkler, I.H.; Langdon, S.P.; Argyle, D.J. Carbonic Anhydrase IX (CAIX), Cancer, and Radiation Responsiveness. Metabolites 2018, 8, 13. [Google Scholar] [CrossRef]
- Moriya, H. Quantitative Nature of Overexpression Experiments. Mol. Biol. Cell. 2015, 26, 3932–3939. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Gapinske, M.; Luu, A.; Winter, J.; Woods, W.S.; Kostan, K.A.; Shiva, N.; Song, J.S.; Perez-Pinera, P. CRISPR-SKIP: Programmable Gene Splicing with Single Base Editors. Genome Biol. 2018, 19, 107. [Google Scholar] [CrossRef]
- Gooding, C.; Smith, C.W.J. Tropomyosin Exons as Models for Alternative Splicing. In Tropomyosin; Gunning, P., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2008; pp. 27–42. ISBN 978-0-387-85766-4. [Google Scholar]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and Mechanisms of Alternative Splicing in Cancer - Implications for Care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, X.; Liu, Y.; Liu, Y.; Sun, L.; Chen, F. PKM2, Function and Expression and Regulation. Cell Biosci. 2019, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Lewis, C.S.; Elnakat Thomas, H.; Orr-Asman, M.A.; Green, L.C.; Boody, R.E.; Matiash, K.; Karve, A.; Hisada, Y.M.; Davis, H.W.; Qi, X.; et al. MTOR Kinase Inhibition Reduces Tissue Factor Expression and Growth of Pancreatic Neuroendocrine Tumors. J. Thromb. Haemost. 2019, 17, 169–182. [Google Scholar] [CrossRef]
- Tardos, J.G.; Eisenreich, A.; Deikus, G.; Bechhofer, D.H.; Chandradas, S.; Zafar, U.; Rauch, U.; Bogdanov, V.Y. SR Proteins ASF/SF2 and SRp55 Participate in Tissue Factor Biosynthesis in Human Monocytic Cells. J. Thromb. Haemost. 2008, 6, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Chandradas, S.; Deikus, G.; Tardos, J.G.; Bogdanov, V.Y. Antagonistic Roles of Four SR Proteins in the Biosynthesis of Alternatively Spliced Tissue Factor Transcripts in Monocytic Cells. J. Leukoc. Biol. 2010, 87, 147–152. [Google Scholar] [CrossRef]
- Eisenreich, A.; Malz, R.; Pepke, W.; Ayral, Y.; Poller, W.; Schultheiss, H.-P.; Rauch, U. Role of the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway in Regulating Alternative Splicing of Tissue Factor MRNA in Human Endothelial Cells. Circ. J. 2009, 73, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Sanford, J.R.; Bruzik, J.P. Developmental Regulation of SR Protein Phosphorylation and Activity. Genes Dev. 1999, 13, 1513–1518. [Google Scholar] [CrossRef][Green Version]
- Cao, W.; Jamison, S.F.; Garcia-Blanco, M.A. Both Phosphorylation and Dephosphorylation of ASF/SF2 Are Required for Pre-MRNA Splicing in Vitro. RNA 1997, 3, 1456–1467. [Google Scholar] [PubMed]
- Chen, M.; Manley, J.L. Mechanisms of Alternative Splicing Regulation: Insights from Molecular and Genomics Approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol. Cells 2017, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Chabot, B. The RNA Splicing Response to DNA Damage. Biomolecules 2015, 5, 2935–2977. [Google Scholar] [CrossRef]
- Piva, F.; Giulietti, M.; Nocchi, L.; Principato, G. SpliceAid: A Database of Experimental RNA Target Motifs Bound by Splicing Proteins in Humans. Bioinformatics 2009, 25, 1211–1213. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Su, B.; Yu, P.; He, J.; Meng, L.; Xiao, Q.; Sun, J.; Zhou, K.; Xue, Y.; et al. Transcriptome-Wide Analysis and Modelling of Prognostic Alternative Splicing Signatures in Invasive Breast Cancer: A Prospective Clinical Study. Sci. Rep. 2020, 10, 16504. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, J.; Zhang, W.; Chen, D.; Wang, Y. Aberrant Alternative Splicing in Breast Cancer. J. Mol. Cell Biol. 2019, 11, 920–929. [Google Scholar] [CrossRef]
- Kawalerski, R.R.; Leach, S.D.; Escobar-Hoyos, L.F. Pancreatic Cancer Driver Mutations Are Targetable through Distant Alternative RNA Splicing Dependencies. Oncotarget 2021, 12, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Lehmann, K.-V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, Q.; Huang, K.; Wang, X.; Yu, T.; Liao, X.; Huang, J.; Zhu, G.; Gong, Y.; Han, C.; et al. Genome-Wide Profiling Reveals the Landscape of Prognostic Alternative Splicing Signatures in Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2019, 9, 511. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wei, S.; Lou, J.; Yin, S.; Zhou, L.; Zhang, W.; Zheng, S. Systematic Analysis of Alternative Splicing Landscape in Pancreatic Adenocarcinoma Reveals Regulatory Network Associated with Tumorigenesis and Immune Response. Med. Sci. Monit. 2020, 26, e925733. [Google Scholar] [CrossRef]
- Xu, L.; Pan, J.; Ding, Y.; Pan, H. Survival-Associated Alternative Splicing Events and Prognostic Signatures in Pancreatic Cancer. Front. Genet. 2020, 11, 522383. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Hoyos, L.F.; Penson, A.; Kannan, R.; Cho, H.; Pan, C.-H.; Singh, R.K.; Apken, L.H.; Hobbs, G.A.; Luo, R.; Lecomte, N.; et al. Altered RNA Splicing by Mutant P53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 2020, 38, 198–211.e8. [Google Scholar] [CrossRef]
- Frankiw, L.; Baltimore, D.; Li, G. Alternative MRNA Splicing in Cancer Immunotherapy. Nat. Rev. Immunol. 2019, 19, 675–687. [Google Scholar] [CrossRef]
- Jakubauskiene, E.; Vilys, L.; Makino, Y.; Poellinger, L.; Kanopka, A. Increased Serine-Arginine (SR) Protein Phosphorylation Changes Pre-MRNA Splicing in Hypoxia. J. Biol. Chem. 2015, 290, 18079–18089. [Google Scholar] [CrossRef]
- Bowler, E.; Porazinski, S.; Uzor, S.; Thibault, P.; Durand, M.; Lapointe, E.; Rouschop, K.M.A.; Hancock, J.; Wilson, I.; Ladomery, M. Hypoxia Leads to Significant Changes in Alternative Splicing and Elevated Expression of CLK Splice Factor Kinases in PC3 Prostate Cancer Cells. BMC Cancer 2018, 18, 355. [Google Scholar] [CrossRef]
- Vilys, L.; Peciuliene, I.; Jakubauskiene, E.; Zinkeviciute, R.; Makino, Y.; Kanopka, A. U2AF—Hypoxia-Induced Fas Alternative Splicing Regulator. Exp. Cell Res. 2021, 399, 112444. [Google Scholar] [CrossRef]
- Farina, A.R.; Cappabianca, L.; Sebastiano, M.; Zelli, V.; Guadagni, S.; Mackay, A.R. Hypoxia-Induced Alternative Splicing: The 11th Hallmark of Cancer. J. Exp. Clin. Cancer Res. 2020, 39, 110. [Google Scholar] [CrossRef]
- Zhan, W.; Shelton, C.A.; Greer, P.J.; Brand, R.E.; Whitcomb, D.C. Germline Variants and Risk for Pancreatic Cancer: A Systematic Review and Emerging Concepts. Pancreas 2018, 47, 924–936. [Google Scholar] [CrossRef]
- Chaffee, K.G.; Oberg, A.L.; McWilliams, R.R.; Majithia, N.; Allen, B.A.; Kidd, J.; Singh, N.; Hartman, A.-R.; Wenstrup, R.J.; Petersen, G.M. Prevalence of Germ-Line Mutations in Cancer Genes among Pancreatic Cancer Patients with a Positive Family History. Genet. Med. 2018, 20, 119–127. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell. Dev. Biol. 2020, 8, 607209. [Google Scholar] [CrossRef] [PubMed]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Hang, J.-J.; Han, T.; Zhuo, M.; Jiao, F.; Wang, L.-W. The M2 Phenotype of Tumor-Associated Macrophages in the Stroma Confers a Poor Prognosis in Pancreatic Cancer. Tumour Biol. 2016, 37, 8657–8664. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-Cell RNA-Seq Reveals Dynamic Change in Tumor Microenvironment during Pancreatic Ductal Adenocarcinoma Malignant Progression. EBioMedicine 2021, 66, 103315. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.-J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef]
- Srinivasan, R.; Bogdanov, V.Y. Splice Variants of Tissue Factor and Integrin-Mediated Signaling. Thromb. Res. 2012, 129 (Suppl. 2), S34–S37. [Google Scholar] [CrossRef]
- Arderiu, G.; Espinosa, S.; Peña, E.; Crespo, J.; Aledo, R.; Bogdanov, V.Y.; Badimon, L. Tissue Factor Variants Induce Monocyte Transformation and Transdifferentiation into Endothelial Cell-like Cells. J. Thromb. Haemost. 2017, 15, 1689–1703. [Google Scholar] [CrossRef]
- Harper, S.J.; Bates, D.O. VEGF-A Splicing: The Key to Anti-Angiogenic Therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Leppert, U.; Eisenreich, A. The Role of Tissue Factor Isoforms in Cancer Biology. Int. J. Cancer 2015, 137, 497–503. [Google Scholar] [CrossRef]
- Ganta, V.C.; Choi, M.; Farber, C.R.; Annex, B.H. Antiangiogenic VEGF165b Regulates Macrophage Polarization via S100A8/S100A9 in Peripheral Artery Disease. Circulation 2019, 139, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Varey, A.H.R.; Rennel, E.S.; Qiu, Y.; Bevan, H.S.; Perrin, R.M.; Raffy, S.; Dixon, A.R.; Paraskeva, C.; Zaccheo, O.; Hassan, A.B.; et al. VEGF 165 b, an Antiangiogenic VEGF-A Isoform, Binds and Inhibits Bevacizumab Treatment in Experimental Colorectal Carcinoma: Balance of pro- and Antiangiogenic VEGF-A Isoforms Has Implications for Therapy. Br. J. Cancer 2008, 98, 1366–1379. [Google Scholar] [CrossRef]
- Censarek, P.; Bobbe, A.; Grandoch, M.; Schrör, K.; Weber, A.-A. Alternatively Spliced Human Tissue Factor (AsHTF) Is Not pro-Coagulant. Thromb. Haemost. 2007, 97, 11–14. [Google Scholar] [CrossRef]
- Böing, A.N.; Hau, C.M.; Sturk, A.; Nieuwland, R. Human Alternatively Spliced Tissue Factor Is Not Secreted and Does Not Trigger Coagulation. J. Thromb. Haemost. 2009, 7, 1423–1426. [Google Scholar] [CrossRef]
- Ünlü, B.; Bogdanov, V.Y.; Versteeg, H.H. Interplay between Alternatively Spliced Tissue Factor and Full Length Tissue Factor in Modulating Coagulant Activity of Endothelial Cells. Thromb. Res. 2017, 156, 1–7. [Google Scholar] [CrossRef]
- Sluka, S.H.M.; Akhmedov, A.; Vogel, J.; Unruh, D.; Bogdanov, V.Y.; Camici, G.G.; Lüscher, T.F.; Ruf, W.; Tanner, F.C. Alternatively Spliced Tissue Factor Is Not Sufficient for Embryonic Development. PLoS ONE 2014, 9, e97793. [Google Scholar] [CrossRef]
- Szotowski, B.; Antoniak, S.; Poller, W.; Schultheiss, H.-P.; Rauch, U. Procoagulant Soluble Tissue Factor Is Released from Endothelial Cells in Response to Inflammatory Cytokines. Circ. Res. 2005, 96, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Zawaski, S.; Hammes, M.; Balasubramanian, V. Alternatively Spliced Human Tissue Factor and Thrombotic Tendencies in Hemodialysis Patients. Nephro-Urol Mon. 2010, 2, 193–199. [Google Scholar]
- Caversaccio, N.I.; Reina Caro, M.D.; Prince, R.; Müller, M.; Lewis, C.S.; Bogdanov, V.Y.; Dufour, J.-F.; Angelillo-Scherrer, A. Alternatively Spliced Tissue Factor Levels Are Elevated in the Plasma of Patients with Chronic Liver Diseases. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1470–1475. [Google Scholar] [CrossRef] [PubMed]
- Ozhegov, E.; Srinivasan, R.; Bogdanov, V. Alternatively Spliced Tissue Factor (AsTF) Is Elevated in the Plasma of Patients with Sickle Cell Disease: Pilot Studies Performed Using a Novel AsTF-Specific ELISA. Blood 2011, 118, 2240. [Google Scholar] [CrossRef]
- Pepe, M.S.; Etzioni, R.; Feng, Z.; Potter, J.D.; Thompson, M.L.; Thornquist, M.; Winget, M.; Yasui, Y. Phases of Biomarker Development for Early Detection of Cancer. J. Natl. Cancer Inst. 2001, 93, 1054–1061. [Google Scholar] [CrossRef]
- Lewis, C.S.; Karve, A.; Matiash, K.; Stone, T.; Li, J.; Wang, J.K.; Versteeg, H.H.; Aronow, B.J.; Ahmad, S.A.; Desai, P.B.; et al. A First-In-Class, Humanized Antibody Targeting Alternatively Spliced Tissue Factor: Preclinical Evaluation in an Orthotopic Model of Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 691685. [Google Scholar] [CrossRef] [PubMed]
- Bergum, P.W.; Cruikshank, A.; Maki, S.L.; Kelly, C.R.; Ruf, W.; Vlasuk, G.P. Role of Zymogen and Activated Factor X as Scaffolds for the Inhibition of the Blood Coagulation Factor VIIa-Tissue Factor Complex by Recombinant Nematode Anticoagulant Protein C2*. J. Biol. Chem. 2001, 276, 10063–10071. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Wiviott, S.D.; Stone, P.H.; Simon, D.I.; Schweiger, M.J.; Bouchard, A.; Leesar, M.A.; Goulder, M.A.; Deitcher, S.R.; McCabe, C.H.; et al. Recombinant Nematode Anticoagulant Protein C2 in Patients with Non-ST-Segment Elevation Acute Coronary Syndrome: The ANTHEM-TIMI-32 Trial. J. Am. Coll. Cardiol. 2007, 49, 2398–2407. [Google Scholar] [CrossRef]
- Hembrough, T.A.; Swartz, G.M.; Papathanassiu, A.; Vlasuk, G.P.; Rote, W.E.; Green, S.J.; Pribluda, V.S. Tissue Factor/Factor VIIa Inhibitors Block Angiogenesis and Tumor Growth through a Nonhemostatic Mechanism. Cancer Res. 2003, 63, 2997–3000. [Google Scholar]
- Zhao, J.; Aguilar, G.; Palencia, S.; Newton, E.; Abo, A. RNAPc2 Inhibits Colorectal Cancer in Mice through Tissue Factor. Clin. Cancer Res. 2009, 15, 208–216. [Google Scholar] [CrossRef]
- Jiao, J.; Kelly, A.B.; Marzec, U.M.; Nieves, E.; Acevedo, J.; Burkhardt, M.; Edwards, A.; Zhu, X.; Chavaillaz, P.-A.; Wong, A.; et al. Inhibition of Acute Vascular Thrombosis in Chimpanzees by an Anti-Human Tissue Factor Antibody Targeting the Factor X Binding Site. Thromb. Haemost. 2010, 103, 224–233. [Google Scholar] [CrossRef]
- Morris, P.E.; Steingrub, J.S.; Huang, B.Y.; Tang, S.; Liu, P.M.; Rhode, P.R.; Wong, H.C. A Phase I Study Evaluating the Pharmacokinetics, Safety and Tolerability of an Antibody-Based Tissue Factor Antagonist in Subjects with Acute Lung Injury or Acute Respiratory Distress Syndrome. BMC Pulm. Med. 2012, 12, 5. [Google Scholar] [CrossRef]
- Breij, E.C.W.; de Goeij, B.E.C.G.; Verploegen, S.; Schuurhuis, D.H.; Amirkhosravi, A.; Francis, J.; Miller, V.B.; Houtkamp, M.; Bleeker, W.K.; Satijn, D.; et al. An Antibody-Drug Conjugate That Targets Tissue Factor Exhibits Potent Therapeutic Activity against a Broad Range of Solid Tumors. Cancer Res. 2014, 74, 1214–1226. [Google Scholar] [CrossRef]
- de Bono, J.S.; Concin, N.; Hong, D.S.; Thistlethwaite, F.C.; Machiels, J.-P.; Arkenau, H.-T.; Plummer, R.; Jones, R.H.; Nielsen, D.; Windfeld, K.; et al. Tisotumab Vedotin in Patients with Advanced or Metastatic Solid Tumours (InnovaTV 201): A First-in-Human, Multicentre, Phase 1-2 Trial. Lancet Oncol. 2019, 20, 383–393. [Google Scholar] [CrossRef]
- Hong, D.S.; Concin, N.; Vergote, I.; de Bono, J.S.; Slomovitz, B.M.; Drew, Y.; Arkenau, H.-T.; Machiels, J.-P.; Spicer, J.F.; Jones, R.; et al. Tisotumab Vedotin in Previously Treated Recurrent or Metastatic Cervical Cancer. Clin. Cancer Res. 2020, 26, 1220–1228. [Google Scholar] [CrossRef]
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; González-Martín, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and Safety of Tisotumab Vedotin in Previously Treated Recurrent or Metastatic Cervical Cancer (InnovaTV 204/GOG-3023/ENGOT-Cx6): A Multicentre, Open-Label, Single-Arm, Phase 2 Study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef]
- Hong, H.; Zhang, Y.; Nayak, T.R.; Engle, J.W.; Wong, H.C.; Liu, B.; Barnhart, T.E.; Cai, W. Immuno-PET of Tissue Factor in Pancreatic Cancer. J. Nucl. Med. 2012, 53, 1748–1754. [Google Scholar] [CrossRef]
- Leung, K. 64Cu-1,4,7-Triazacyclononane-1,4,7-triacetic acid-p-isothiocyanatobenzyl-ALT-836. In Molecular Imaging and Contrast Agent Database (MICAD); National Center for Biotechnology Information: Bethesda, MD, USA, 2004. [Google Scholar]
- Shi, S.; Hong, H.; Orbay, H.; Graves, S.A.; Yang, Y.; Ohman, J.D.; Liu, B.; Nickles, R.J.; Wong, H.C.; Cai, W. ImmunoPET of Tissue Factor Expression in Triple-Negative Breast Cancer with a Radiolabeled Antibody Fab Fragment. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1295–1303. [Google Scholar] [CrossRef]
- Nayak, T.R.; Andreou, C.; Oseledchyk, A.; Marcus, W.D.; Wong, H.C.; Massagué, J.; Kircher, M.F. Tissue Factor-Specific Ultra-Bright SERRS Nanostars for Raman Detection of Pulmonary Micrometastases. Nanoscale 2017, 9, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; England, C.G.; Goel, S.; Graves, S.A.; Ai, F.; Liu, B.; Theuer, C.P.; Wong, H.C.; Nickles, R.J.; Cai, W. ImmunoPET and Near-Infrared Fluorescence Imaging of Pancreatic Cancer with a Dual-Labeled Bispecific Antibody Fragment. Mol. Pharm. 2017, 14, 1646–1655. [Google Scholar] [CrossRef]
- Hernandez, R.; England, C.G.; Yang, Y.; Valdovinos, H.F.; Liu, B.; Wong, H.C.; Barnhart, T.E.; Cai, W. ImmunoPET Imaging of Tissue Factor Expression in Pancreatic Cancer with 89Zr-Df-ALT-836. J. Control. Release 2017, 264, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Liu, Q.; Jiang, D.; Zhao, H.; Kutyreff, C.J.; Engle, J.W.; Liu, J.; Cai, W. Tissue Factor-Targeted ImmunoPET Imaging and Radioimmunotherapy of Anaplastic Thyroid Cancer. Adv. Sci. 2020, 7, 1903595. [Google Scholar] [CrossRef]
- Nielsen, C.H.; Erlandsson, M.; Jeppesen, T.E.; Jensen, M.M.; Kristensen, L.K.; Madsen, J.; Petersen, L.C.; Kjaer, A. Quantitative PET Imaging of Tissue Factor Expression Using 18F-Labeled Active Site-Inhibited Factor VII. J. Nucl. Med. 2016, 57, 89–95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsumura, R.; Sato, R.; Furuya, F.; Koga, Y.; Yamamoto, Y.; Fujiwara, Y.; Yasunaga, M.; Matsumura, Y. Feasibility Study of the Fab Fragment of a Monoclonal Antibody against Tissue Factor as a Diagnostic Tool. Int J. Oncol. 2015, 47, 2107–2114. [Google Scholar] [CrossRef]
- Takashima, H.; Tsuji, A.B.; Saga, T.; Yasunaga, M.; Koga, Y.; Kuroda, J.-I.; Yano, S.; Kuratsu, J.-I.; Matsumura, Y. Molecular Imaging Using an Anti-Human Tissue Factor Monoclonal Antibody in an Orthotopic Glioma Xenograft Model. Sci. Rep. 2017, 7, 12341. [Google Scholar] [CrossRef]
- Koga, Y.; Manabe, S.; Aihara, Y.; Sato, R.; Tsumura, R.; Iwafuji, H.; Furuya, F.; Fuchigami, H.; Fujiwara, Y.; Hisada, Y.; et al. Antitumor Effect of Antitissue Factor Antibody-MMAE Conjugate in Human Pancreatic Tumor Xenografts. Int. J. Cancer 2015, 137, 1457–1466. [Google Scholar] [CrossRef]
- Theunissen, J.-W.; Cai, A.G.; Bhatti, M.M.; Cooper, A.B.; Avery, A.D.; Dorfman, R.; Guelman, S.; Levashova, Z.; Migone, T.-S. Treating Tissue Factor-Positive Cancers with Antibody-Drug Conjugates That Do Not Affect Blood Clotting. Mol. Cancer Ther. 2018, 17, 2412–2426. [Google Scholar] [CrossRef]
- Tsumura, R.; Manabe, S.; Takashima, H.; Koga, Y.; Yasunaga, M.; Matsumura, Y. Influence of the Dissociation Rate Constant on the Intra-Tumor Distribution of Antibody-Drug Conjugate against Tissue Factor. J. Control. Release 2018, 284, 49–56. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Q.; Zhao, H.; Ma, L.; Meng, T.; Qian, J.; Jin, R.; Shen, J.; Yu, K. Pathological Expression of Tissue Factor Confers Promising Antitumor Response to a Novel Therapeutic Antibody SC1 in Triple Negative Breast Cancer and Pancreatic Adenocarcinoma. Oncotarget 2017, 8, 59086–59102. [Google Scholar] [CrossRef]
- Min, H.S.; Kim, H.J.; Ahn, J.; Naito, M.; Hayashi, K.; Toh, K.; Kim, B.S.; Matsumura, Y.; Kwon, I.C.; Miyata, K.; et al. Tuned Density of Anti-Tissue Factor Antibody Fragment onto SiRNA-Loaded Polyion Complex Micelles for Optimizing Targetability into Pancreatic Cancer Cells. Biomacromolecules 2018, 19, 2320–2329. [Google Scholar] [CrossRef]
- Sugaya, A.; Hyodo, I.; Koga, Y.; Yamamoto, Y.; Takashima, H.; Sato, R.; Tsumura, R.; Furuya, F.; Yasunaga, M.; Harada, M.; et al. Utility of Epirubicin-Incorporating Micelles Tagged with Anti-Tissue Factor Antibody Clone with No Anticoagulant Effect. Cancer Sci. 2016, 107, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Takashima, H.; Koga, Y.; Tsumura, R.; Yasunaga, M.; Tsuchiya, M.; Inoue, T.; Negishi, E.; Harada, M.; Yoshida, S.; Matsumura, Y. Reinforcement of Antitumor Effect of Micelles Containing Anticancer Drugs by Binding of an Anti-Tissue Factor Antibody without Direct Cytocidal Effects. J. Control. Release 2020, 323, 138–150. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Hyodo, I.; Koga, Y.; Tsumura, R.; Sato, R.; Obonai, T.; Fuchigami, H.; Furuya, F.; Yasunaga, M.; Harada, M.; et al. Enhanced Antitumor Effect of Anti-Tissue Factor Antibody-Conjugated Epirubicin-Incorporating Micelles in Xenograft Models. Cancer Sci. 2015, 106, 627–634. [Google Scholar] [CrossRef]
- Takashima, H.; Koga, Y.; Manabe, S.; Ohnuki, K.; Tsumura, R.; Anzai, T.; Iwata, N.; Wang, Y.; Yokokita, T.; Komori, Y.; et al. Radioimmunotherapy with an 211 At-Labeled Anti-Tissue Factor Antibody Protected by Sodium Ascorbate. Cancer Sci. 2021, 112, 1975–1986. [Google Scholar] [CrossRef]
- Teplyakov, A.; Obmolova, G.; Malia, T.J.; Raghunathan, G.; Martinez, C.; Fransson, J.; Edwards, W.; Connor, J.; Husovsky, M.; Beck, H.; et al. Structural Insights into Humanization of Anti-Tissue Factor Antibody 10H10. MAbs 2018, 10, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Magnus, N.; Meehan, B.; Garnier, D.; Hashemi, M.; Montermini, L.; Lee, T.H.; Milsom, C.; Pawlinski, R.; Ohlfest, J.; Anderson, M.; et al. The Contribution of Tumor and Host Tissue Factor Expression to Oncogene-Driven Gliomagenesis. Biochem. Biophys. Res. Commun. 2014, 454, 262–268. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | Alias | Class | Conjugate | Clinicaltrials.gov Identifier | Disease | Phase | Status | References |
|---|---|---|---|---|---|---|---|---|
| rNAPc2 | AB201 | recombinant protein | NCT04655586 | COVID-19 | Ⅱ/Ⅲ | recruiting | [110] | |
| NCT00116012 | Coronary disease | Ⅱ | completed | |||||
| Tisotumab vedotin | HUMax-TF, HuMax®-TF-ADC, TF-011-MMAE | ADC | MMAE | NCT03913741 | Solid malignancies | Ⅰ/Ⅱ | Active, not recruiting | [116] [117] [118] |
| NCT03245736 | Solid malignancies | Ⅱ | completed | |||||
| NCT03438396 | Cervical cancer | Ⅱ | Active, not recruiting | |||||
| NCT03485209 | Solid malignancies | Ⅱ | recruiting | |||||
| NCT02552121 | Solid malignancies | Ⅰ/Ⅱ | completed | |||||
| NCT03657043 | Platinum-resistant ovarian cancer | Ⅱ | Active, not recruiting | |||||
| NCT03786081 | Cervical cancer | Ⅰ/Ⅱ | Active, not recruiting | |||||
| NCT02001623 | Solid malignancies | Ⅰ/Ⅱ | completed | |||||
| NCT04697628 | Cervical cancer | Ⅲ | recruiting | |||||
| ALT-836 | TNX-832, Sunol cH36 | mAb | NCT01438853 | Acute lung injury/acute respiratory distress syndrome | Ⅰ/Ⅱ | completed | [114] | |
| NCT00879606 | ||||||||
| NCT01325558 | Solid malignancies | Ⅰ | completed | |||||
| 64Cu-NOTA-ALT-836 | Imaging reagent | p-SCN-Bn-NOTA/ Copper-64 | PDAC, triple negative breast cancer | preclinical | [119] [120] [121] | |||
| ALT-836-SERRS-NPs | Imaging reagent | SERRS-NPs | Breast cancer lung metastases | preclinical | [122] | |||
| 64Cu-NOTA-heterodimer-ZW800 | Imaging reagent | Copper-64 | PDAC | preclinical | [123] | |||
| 89Zr-DF-ALT-836 | Imaging reagent | Zirconium-89 | PDAC | preclinical | [124] | |||
| IRDye 800CW-ALT-836 | Imaging reagent | IRDye 800CW | Anaplastic thyroid cancer | preclinical | [125] | |||
| 18F-Fvllai | Imaging reagent | Fluorine-18 | PDAC | preclinical | [126] | |||
| 1849-Alexa-Fluor-647 | Imaging reagent | Alexa Fluor 647 | PDAC, glioblastoma multiforme | preclinical | [127] [128] | |||
| 1849-MMAE | ADC | MMAE | PDAC, HNSCC, ovarian cancer, GAC | preclinical | [129] [130] [131] | |||
| SC1-MMAE | ADC | MMAE | PDAC, triple negative breast cancer, HNSCC, ovarian cancer, GAC | preclinical | [132] [130] | |||
| anti-TF Fab'-installed PIC micelles | ADC | siRNA loaded polyion complex micelles | PDAC | preclinical | [133] | |||
| anti-TF1849-NC-6300 | ADC | epirubicin | PDAC | preclinical | [134] | |||
| anti-TF1859-NC-6300 | ADC | epirubicin | PDAC | preclinical | [134] [135] [136] | |||
| 1084-MMAE | ADC | MMAE | PDAC | preclinical | [131] | |||
| 1084(211At-anti-TF mAb) | radioimmunotherapeutic | astatine-211 | GAC | preclinical | [137] | |||
| 131I-ALT-836 | radioimmunotherapeutic | lodine-131 | Anaplastic thyroid cancer | preclinical | [125] | |||
| 10h10 | mAb | Glioma | preclinical | [138] | ||||
| hRabMab1 | Rb1 | mAb | PDAC, breast cancer | preclinical | [28] [47] [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matiash, K.; Lewis, C.S.; Bogdanov, V.Y. Functional Characteristics and Regulated Expression of Alternatively Spliced Tissue Factor: An Update. Cancers 2021, 13, 4652. https://doi.org/10.3390/cancers13184652
Matiash K, Lewis CS, Bogdanov VY. Functional Characteristics and Regulated Expression of Alternatively Spliced Tissue Factor: An Update. Cancers. 2021; 13(18):4652. https://doi.org/10.3390/cancers13184652
Chicago/Turabian StyleMatiash, Kateryna, Clayton S. Lewis, and Vladimir Y. Bogdanov. 2021. "Functional Characteristics and Regulated Expression of Alternatively Spliced Tissue Factor: An Update" Cancers 13, no. 18: 4652. https://doi.org/10.3390/cancers13184652
APA StyleMatiash, K., Lewis, C. S., & Bogdanov, V. Y. (2021). Functional Characteristics and Regulated Expression of Alternatively Spliced Tissue Factor: An Update. Cancers, 13(18), 4652. https://doi.org/10.3390/cancers13184652