
Bacitracin and Rutin Regulate Tissue Factor Production in Inflammatory Monocytes and Acute Myeloid Leukemia Blasts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Cell Lines and Cultures
2.2.2. Collection of Blood Samples
2.2.3. Isolation and Stimulation of Peripheral Blood Mononuclear Cells (PBMCs)
2.2.4. LPS Stimulation of Whole Blood
2.2.5. Cell Culture Experiments
2.2.6. Single-Stage Clotting Assay
2.2.7. Flow Cytometry
2.2.8. TF ELISA
2.2.9. Thiol Isomerase Activity Assay
2.2.10. FXa Generation Assay
2.2.11. Real-Time Quantitative PCR (qPCR)
2.2.12. Statistical Analysis
3. Results
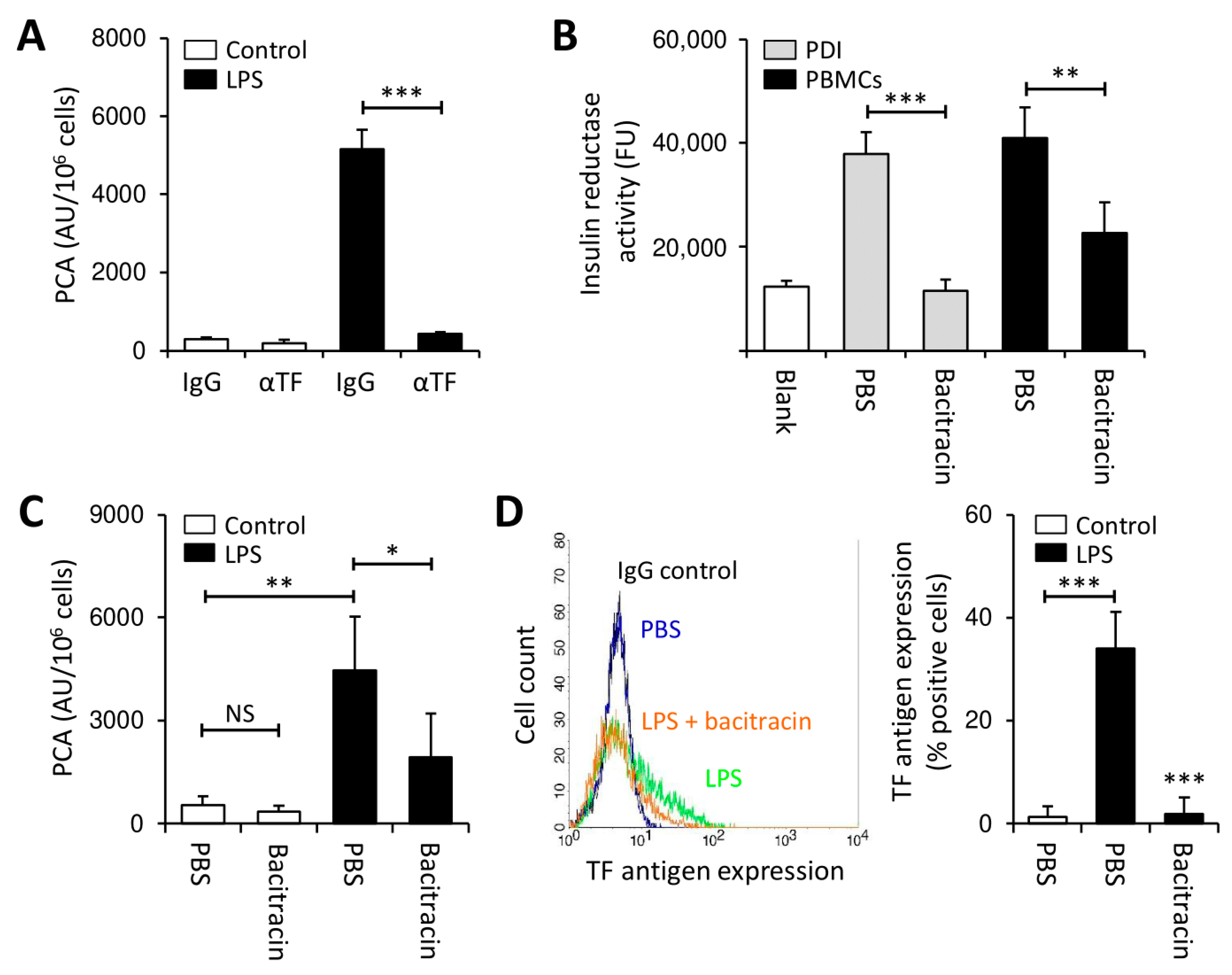
3.1. Thiol Isomerases Regulate Monocyte TF PCA under Pro-Inflammatory Conditions
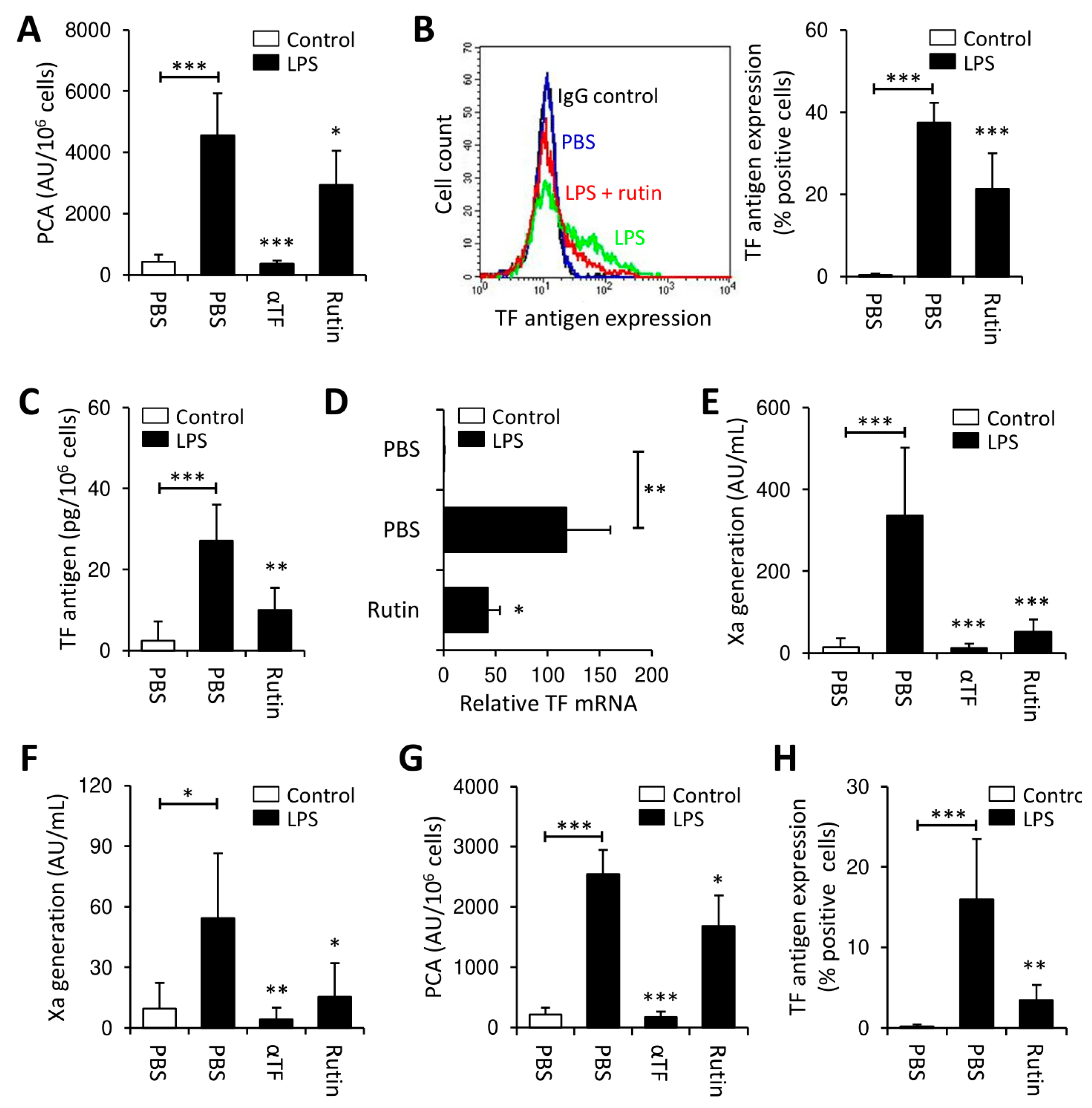
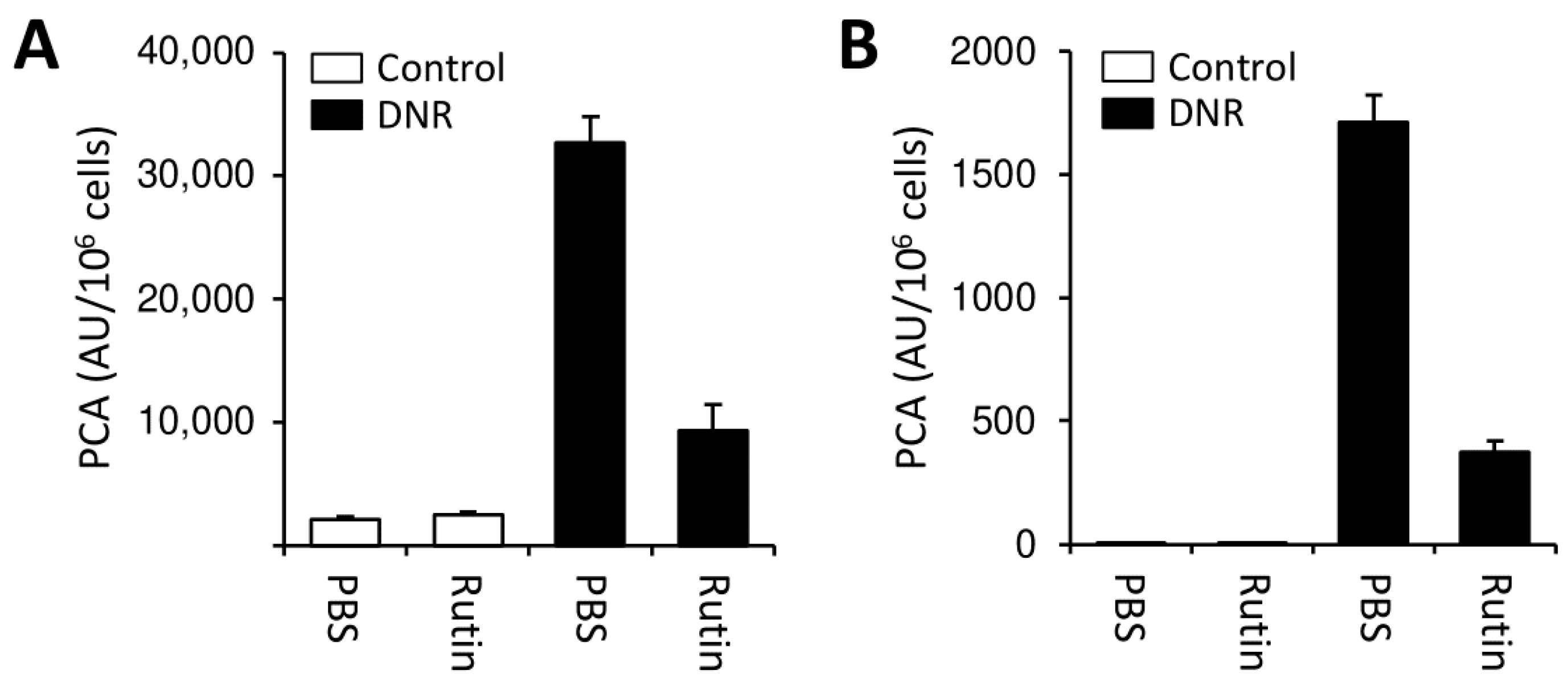
3.2. Rutin Prevents LPS-Induced TF Production by PBMCs
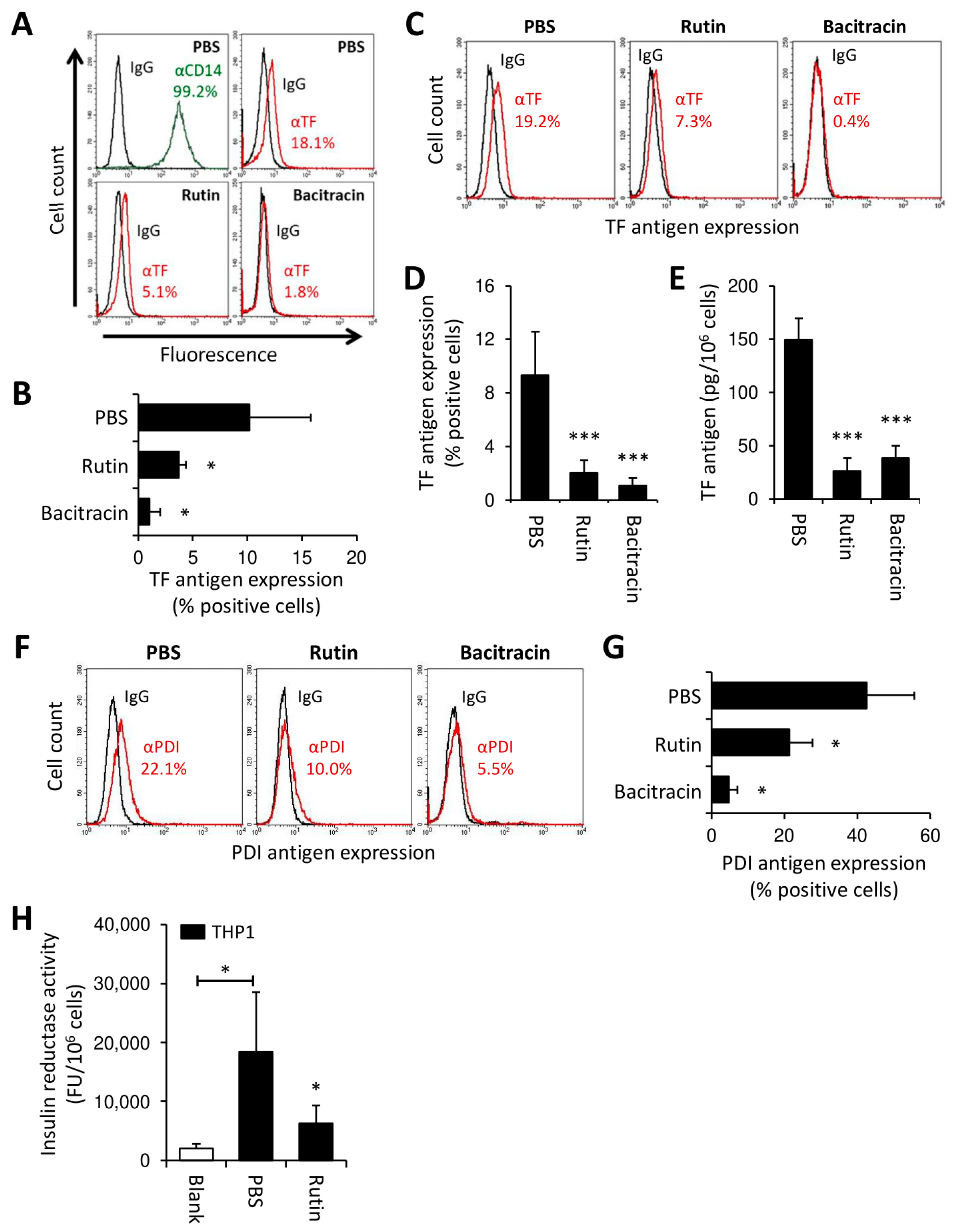
3.3. Rutin and Bacitracin Regulate Endogenous TF Expression in Monocytic Cells
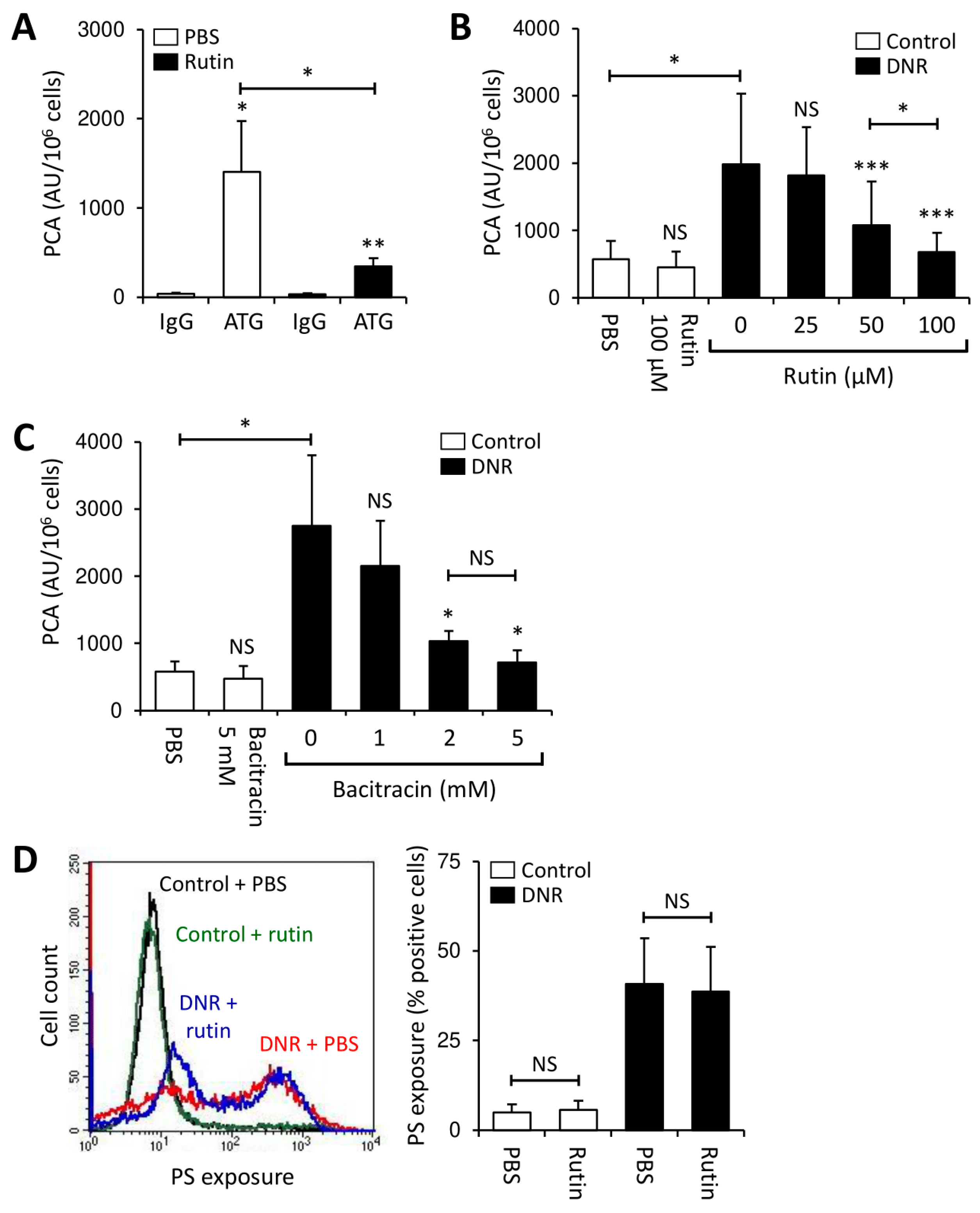
3.4. Rutin Prevents TF Activation on THP1 Cells through Downregulation of Cryptic TF Expression
3.5. Rutin Exerts Antithrombotic Activity in Myeloblasts from AML Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kwaan, H.C. Double Hazard of Thrombophilia and Bleeding in Leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2007, 2007, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Kwaan, H.C.; Cull, E.H. The coagulopathy in acute promyelocytic leukaemia—What have we learned in the past twenty years. Best Pr. Res. Clin. Haematol. 2014, 27, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Webert, K.; Cook, R.J.; Sigouin, C.S.; Rebulla, P.; Heddle, N.M. The risk of bleeding in thrombocytopenic patients with acute myeloid leukemia. Haematologica 2006, 91, 1530–1537. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Tang, L.; Wang, M.-H. Leukemia and Risk of Venous Thromboembolism: A Meta-analysis and Systematic Review of 144 Studies Comprising 162,126 Patients. Sci. Rep. 2017, 7, 1167. [Google Scholar] [CrossRef] [PubMed]
- Mohren, M.; Markmann, I.; Jentsch-Ullrich, K.; Koenigsmann, M.; Lutze, G.; Franke, A. Increased risk of venous thromboembolism in patients with acute leukaemia. Br. J. Cancer 2006, 94, 200–202. [Google Scholar] [CrossRef]
- Libourel, E.J.; Klerk, C.P.W.; Van Norden, Y.; De Maat, M.P.M.; Kruip, M.J.; Sonneveld, P.; Löwenberg, B.; Leebeek, F.W.G. Disseminated intravascular coagulation at diagnosis is a strong predictor for thrombosis in acute myeloid leukemia. Blood 2016, 128, 1854–1861. [Google Scholar] [CrossRef]
- Vu, K.; Luong, N.V.; Hubbard, J.; Zalpour, A.; Faderl, S.; Thomas, D.A.; Yang, D.; Kantarjian, H.; Kroll, M.H. A retrospective study of venous thromboembolism in acute leukemia patients treated at the University of Texas MD Anderson Cancer Center. Cancer Med. 2015, 4, 27–35. [Google Scholar] [CrossRef]
- Dicke, C.; Amirkhosravi, A.; Spath, B.; Jiménez-Alcázar, M.; Fuchs, T.; Davila, M.; Francis, J.L.; Bokemeyer, C.; Langer, F. Tissue factor-dependent and -independent pathways of systemic coagulation activation in acute myeloid leukemia: A single-center cohort study. Exp. Hematol. Oncol. 2015, 4, 22. [Google Scholar] [CrossRef]
- Breen, K.A.; Grimwade, D.; Hunt, B.J. The pathogenesis and management of the coagulopathy of acute promyelocytic leukaemia. Br. J. Haematol. 2012, 156, 24–36. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of hemostasis and Trigger of Thrombosis. Arter. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Zelaya, H.; Rothmeier, A.S.; Ruf, W. Tissue factor at the crossroad of coagulation and cell signaling. J. Thromb. Haemost. 2018, 16, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Ebert, J.; Wilgenbus, P.; Teiber, J.F.; Jurk, K.; Schwierczek, K.; Döhrmann, M.; Xia, N.; Li, H.; Spiecker, L.; Ruf, W.; et al. Paraoxonase-2 regulates coagulation activation through endothelial tissue factor. Blood 2018, 131, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Mackman, N. Tissue Factor and Cancer: Regulation, Tumor Growth, and Metastasis. Semin. Thromb. Hemost. 2019, 45, 385–395. [Google Scholar] [CrossRef]
- Han, X.; Guo, B.; Li, Y.; Zhu, B. Tissue factor in tumor microenvironment: A systematic review. J. Hematol. Oncol. 2014, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yamanishi, H. The expression of tissue factor antigen and activity on the surface of leukemic cells. Leuk. Res. 1993, 17, 103–111. [Google Scholar] [CrossRef]
- Bauer, K.A.; Conway, E.M.; Bach, R.; Konigsberg, W.H.; Griffin, J.D.; Demetri, G. Tissue factor gene expression in acute myeloblastic leukemia. Thromb. Res. 1989, 56, 425–430. [Google Scholar] [CrossRef]
- Nadir, Y.; Katz, T.; Sarig, G.; Hoffman, R.; Oliven, A.; Rowe, J.M.; Brenner, B. Hemostatic balance on the surface of leukemic cells: The role of tissue factor and urokinase plasminogen activator receptor. Haematologica 2005, 90, 1549–1556. [Google Scholar]
- Craver, B.M.; El Alaoui, K.; Scherber, R.M.; Fleischman, A.G. The Critical Role of Inflammation in the Pathogenesis and Progression of Myeloid Malignancies. Cancers 2018, 10, 104. [Google Scholar] [CrossRef]
- Langer, F.; Ruf, W. Synergies of phosphatidylserine and protein disulfide isomerase in tissue factor activation. Thromb. Haemost. 2014, 111, 590–597. [Google Scholar] [CrossRef]
- Ansari, S.A.; Pendurthi, U.R.; Rao, L.V.M. Role of Cell Surface Lipids and Thiol-Disulphide Exchange Pathways in Regulating the Encryption and Decryption of Tissue Factor. Thromb. Haemost. 2019, 119, 860–870. [Google Scholar] [CrossRef]
- Drake, T.A.; Ruf, W.; Morrissey, J.H.; Edgington, T.S. Functional tissue factor is entirely cell surface expressed on lipopolysaccharide-stimulated human blood monocytes and a constitutively tissue factor-producing neoplastic cell line. J. Cell Biol. 1989, 109, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Hatahet, F.; Ruddock, L.W. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Kennedy, D.R.; Lin, L.; Huang, M.; Merrill-Skoloff, G.; Furie, B.C.; Furie, B. Protein disulfide isomerase capture during thrombus formation in vivo depends on the presence of β3 integrins. Blood 2012, 120, 647–655. [Google Scholar] [CrossRef]
- Jasuja, R.; Furie, B.; Furie, B.C. Endothelium-derived but not platelet-derived protein disulfide isomerase is required for thrombus formation in vivo. Blood 2010, 116, 4665–4674. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, C.; Von Brühl, M.-L.; Manukyan, D.; Grahl, L.; Lorenz, M.; Altmann, B.; Dlugai, S.; Hess, S.; Konrad, I.; Orschiedt, L.; et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J. Clin. Investig. 2008, 118, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Jurk, K.; Hobohm, L.; Jäckel, S.; Saffarzadeh, M.; Schwierczek, K.; Wenzel, P.; Langer, F.; Reinhardt, C.; Ruf, W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 2017, 129, 2291–2302. [Google Scholar] [CrossRef]
- Cho, J.; Furie, B.C.; Coughlin, S.R.; Furie, B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J. Clin. Investig. 2008, 118, 1123–1131. [Google Scholar] [CrossRef]
- Jasuja, R.; Passam, F.H.; Kennedy, D.R.; Kim, S.H.; Van Hessem, L.; Lin, L.; Bowley, S.R.; Joshi, S.S.; Dilks, J.R.; Furie, B.; et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J. Clin. Investig. 2012, 122, 2104–2113. [Google Scholar] [CrossRef]
- Kim, K.; Hahm, E.; Li, J.; Holbrook, L.-M.; Sasikumar, P.; Stanley, R.G.; Ushio-Fukai, M.; Gibbins, J.M.; Cho, J. Platelet protein disulfide isomerase is required for thrombus formation but not for hemostasis in mice. Blood 2013, 122, 1052–1061. [Google Scholar] [CrossRef]
- Chen, F.; Zhao, Z.; Zhou, J.; Lu, Y.; Essex, D.W.; Wu, Y. Protein disulfide isomerase enhances tissue factor-dependent thrombin generation. Biochem. Biophys. Res. Commun. 2018, 501, 172–177. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, Y.; Chen, F.; Wang, L.; Rauova, L.; Hayes, V.M.; Poncz, M.; Li, H.; Liu, T.; Liu, J.; et al. The disulfide isomerase ERp72 supports arterial thrombosis in mice. Blood 2017, 130, 817–828. [Google Scholar] [CrossRef]
- Passam, F.H.; Lin, L.; Gopal, S.; Stopa, J.D.; Bellido-Martin, L.; Huang, M.; Furie, B.C.; Furie, B. Both platelet- and endothelial cell–derived ERp5 support thrombus formation in a laser-induced mouse model of thrombosis. Blood 2015, 125, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.; Zhou, J.; Ahmad, S.S.; Mutus, B.; Garbi, N.; Hämmerling, G.; Liu, J.; Essex, D.W. Platelet-derived ERp57 mediates platelet incorporation into a growing thrombus by regulation of the αIIbβ3 integrin. Blood 2013, 122, 3642–3650. [Google Scholar] [CrossRef]
- Stopa, J.D.; Neuberg, D.; Puligandla, M.; Furie, B.; Flaumenhaft, R.; Zwicker, J.I. Protein disulfide isomerase inhibition blocks thrombin generation in humans by interfering with platelet factor V activation. JCI Insight 2017, 2, e89373. [Google Scholar] [CrossRef]
- Zwicker, J.I.; Schlechter, B.L.; Stopa, J.D.; Liebman, H.A.; Aggarwal, A.; Puligandla, M.; Caughey, T.; Bauer, K.A.; Kuemmerle, N.; Wong, E.; et al. Targeting protein disulfide isomerase with the flavonoid isoquercetin to improve hypercoagulability in advanced cancer. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Haefliger, S.; Klebig, C.; Schaubitzer, K.; Schardt, J.; Timchenko, N.; Mueller, B.U.; Pabst, T. Protein disulfide isomerase blocks CEBPA translation and is up-regulated during the unfolded protein response in AML. Blood 2011, 117, 5931–5940. [Google Scholar] [CrossRef]
- Holstein, K.; Matysiak, A.; Witt, L.; Sievers, B.; Beckmann, L.; Haddad, M.; Renné, T.; Voigtlaender, M.; Langer, F. LPS-induced expression and release of monocyte tissue factor in patients with haemophilia. Ann. Hematol. 2020, 99, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.D.; Barcel, D.A.; Williams, J.C.; Wang, J.G.; Boles, J.C.; Manly, D.A.; Key, N.S.; Mackman, N. Pre-analytical and analytical variables affecting the measurement of plasma-derived microparticle tissue factor activity. Thromb. Res. 2012, 129, 80–85. [Google Scholar] [CrossRef]
- Basavaraj, M.G.; Olsen, J.O.; Østerud, B.; Hansen, J.-B. Differential ability of tissue factor antibody clones on detection of tissue factor in blood cells and microparticles. Thromb. Res. 2012, 130, 538–546. [Google Scholar] [CrossRef]
- Nieuwland, R.; Gardiner, C.; Dignat-George, F.; Mullier, F.; Mackman, N.; Woodhams, B.; Thaler, J. Toward standardization of assays measuring extracellular vesicle-associated tissue factor activity. J. Thromb. Haemost. 2019, 17, 1261–1264. [Google Scholar] [CrossRef]
- Beckmann, L.; Dicke, C.; Spath, B.; Lehr, C.; Sievers, B.; Klinke, A.; Baldus, S.; Rudolph, V.; Langer, F. Myeloperoxidase Is a Negative Regulator of Phospholipid-Dependent Coagulation. Thromb. Haemost. 2017, 117, 2300–2311. [Google Scholar] [CrossRef]
- Jacobsen, C.; Oechsle, K.; Hauschild, J.; Steinemann, G.; Spath, B.; Bokemeyer, C.; Ruf, W.; Honecker, F.; Langer, F. Regulation of tissue factor in NT2 germ cell tumor cells by cisplatin chemotherapy. Thromb. Res. 2015, 136, 673–681. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Langer, F.; Spath, B.; Fischer, C.; Stolz, M.; Ayuk, F.A.; Kröger, N.; Bokemeyer, C.; Ruf, W. Rapid activation of monocyte tissue factor by antithymocyte globulin is dependent on complement and protein disulfide isomerase. Blood 2013, 121, 2324–2335. [Google Scholar] [CrossRef]
- Rothmeier, A.S.; Marchese, P.; Langer, F.; Kamikubo, Y.; Schaffner, F.; Cantor, J.; Ginsberg, M.H.; Ruggeri, Z.M.; Ruf, W. Tissue Factor Prothrombotic Activity Is Regulated by Integrin-arf6 Trafficking. Arter. Thromb. Vasc. Biol. 2017, 37, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Gopal, S.; Sharda, A.; Passam, F.; Bowley, S.R.; Stopa, J.; Xue, G.; Yuan, C.; Furie, B.C.; Flaumenhaft, R.; et al. Quercetin-3-rutinoside Inhibits Protein Disulfide Isomerase by Binding to Its b’x Domain. J. Biol. Chem. 2015, 290, 23543–23552. [Google Scholar] [CrossRef] [PubMed]
- Kallakunta, V.M.; Slama-Schwok, A.; Mutus, B. Protein disulfide isomerase may facilitate the efflux of nitrite derived S-nitrosothiols from red blood cells. Redox Biol. 2013, 1, 373–380. [Google Scholar] [CrossRef]
- Voigtlaender, M.; Holstein, K.; Spath, B.; Bokemeyer, C.; Langer, F. Expression and release of platelet protein disulphide isomerase in patients with haemophilia A. Haemophilia 2016, 22, e537–e544. [Google Scholar] [CrossRef]
- Langer, F.; Amirkhosravi, A.; Loges, S.; Meyer, T.; Eifrig, B.; Hossfeld, D.K.; Fiedler, W.; Francis, J.L. An in vitro study on the mechanisms of coagulation activation in acute myelogenous leukemia (AML): Role of tissue factor regulation by cytotoxic drugs and GM-CSF. Thromb. Haemost. 2004, 92, 1136–1146. [Google Scholar] [CrossRef]
- Bekendam, R.H.; Bendapudi, P.K.; Lin, L.; Nag, P.P.; Pu, J.; Kennedy, D.R.; Feldenzer, A.; Chiu, J.; Cook, K.M.; Furie, B.; et al. A substrate-driven allosteric switch that enhances PDI catalytic activity. Nat. Commun. 2016, 7, 12579. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Ruf, W. Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity. J. Biol. Chem. 2007, 282, 25416–25424. [Google Scholar] [CrossRef] [PubMed]
- Hengel, L.G.V.D.; Osanto, S.; Reitsma, P.H.; Versteeg, H.H. Murine tissue factor coagulant activity is critically dependent on the presence of an intact allosteric disulfide. Haematologica 2013, 98, 153–158. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ahamed, J.; Versteeg, H.H.; Kerver, M.; Chen, V.M.; Mueller, B.M.; Hogg, P.J.; Ruf, W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13932–13937. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.M.; Ahamed, J.; Versteeg, H.H.; Berndt, M.C.; Ruf, W.; Hogg, P.J. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry 2006, 45, 12020–12028. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Versteeg, H.H. Tissue factor mutated at the allosteric Cys186-Cys209 disulfide bond is severely impaired in decrypted procoagulant activity. Blood 2010, 116, 500–501. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Muraoka, K.; Shimizu, K.; Sun, X.; Tani, T.; Izumi, R.; Miwa, K.; Yamamoto, K. Flavonoids exert diverse inhibitory effects on the activation of NF-kappaB. Transplant. Proc. 2002, 34, 1335–1340. [Google Scholar] [CrossRef]
- Endringer, D.C.; Pezzuto, J.M.; Braga, F.C. NF-kappaB inhibitory activity of cyclitols isolated from hancornia speciosa. Phytomedicine 2009, 16, 1064–1069. [Google Scholar] [CrossRef]
- Siriwatanametanon, N.; Heinrich, M. The Thai Medicinal Plant Gynura Pseudochina var. hispida: Chemical Composition and in vitro NF-κB Inhibitory Activity. Nat. Prod. Commun. 2011, 6, 1934578X1100600512. [Google Scholar] [CrossRef]
- Bode, M.; Mackman, N. Regulation of tissue factor gene expression in monocytes and endothelial cells: Thromboxane A2 as a new player. Vasc. Pharmacol. 2014, 62, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, A.S.; Ruf, W. Protease-activated receptor 2 signaling in inflammation. Semin. Immunopathol. 2012, 34, 133–149. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Higuchi, T.; Watanabe, Y.; Waga, I. Protein disulfide isomerase suppresses the transcriptional activity of NF-kappaB. Biochem. Biophys. Res. Commun. 2004, 318, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Sperlágh, B.; Haskó, G.; Németh, Z.; Vizi, E.S. ATP released by LPS increases nitric oxide production in raw 264.7 macrophage cell line via P2Z/P2X7 receptors. Neurochem. Int. 1998, 33, 209–215. [Google Scholar] [CrossRef]
- Furlan-Freguia, C.; Marchese, P.; Gruber, A.; Ruggeri, Z.M.; Ruf, W. P2X7 receptor signaling contributes to tissue factor-dependent thrombosis in mice. J. Clin. Investig. 2011, 121, 2932–2944. [Google Scholar] [CrossRef]
- Ansari, S.A.; Pendurthi, U.R.; Rao, L.V.M. The lipid peroxidation product 4-hydroxy-2-nonenal induces tissue factor decryption via ROS generation and the thioredoxin system. Blood Adv. 2017, 1, 2399–2413. [Google Scholar] [CrossRef]
- Rothmeier, A.S.; Marchese, P.; Petrich, B.G.; Furlan-Freguia, C.; Ginsberg, M.H.; Ruggeri, Z.M.; Ruf, W. Caspase-1-mediated pathway promotes generation of thromboinflammatory microparticles. J. Clin. Investig. 2015, 125, 1471–1484. [Google Scholar] [CrossRef]
- Xu, S.; Sankar, S.; Neamati, N. Protein disulfide isomerase: A promising target for cancer therapy. Drug Discov. Today 2014, 19, 222–240. [Google Scholar] [CrossRef] [PubMed]
- Nouri, Z.; Fakhri, S.; Nouri, K.; Wallace, C.E.; Farzaei, M.H.; Bishayee, A. Targeting Multiple Signaling Pathways in Cancer: The Rutin Therapeutic Approach. Cancers 2020, 12, 2276. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-P.; Yang, J.-S.; Lu, C.-C.; Chiang, J.-H.; Wu, C.-L.; Lin, J.-J.; Lin, H.-L.; Yang, M.-D.; Liu, K.-C.; Chiu, T.-H.; et al. Rutin inhibits the proliferation of murine leukemia WEHI-3 cells in vivo and promotes immune response in vivo. Leuk. Res. 2009, 33, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-P.; Yang, J.-S.; Lin, J.-J.; Lai, K.-C.; Lu, H.-F.; Ma, C.-Y.; Sai-Chuen Wu, R.; Wu, K.-C.; Chueh, F.-S.; Gibson Wood, W.; et al. Rutin inhibits human leukemia tumor growth in a murine xenograft model in vivo. Environ. Toxicol. 2012, 27, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Chlebowska-Tuz, J.; Sokolowska, O.; Gaj, P.; Lazniewski, M.; Firczuk, M.; Borowiec, K.; Sas-Nowosielska, H.; Bajor, M.; Malinowska, A.; Muchowicz, A.; et al. Inhibition of protein disulfide isomerase induces differentiation of acute myeloid leukemia cells. Haematologica 2018, 103, 1843–1852. [Google Scholar] [CrossRef]
- Negaard, H.F.S.; Iversen, P.O.; Ostenstad, B.; Mowinckel, M.C.; Sandset, P.M. Increased acquired activated protein C resistance in unselected patients with hematological malignancies. J. Thromb. Haemost. 2008, 6, 1482–1487. [Google Scholar] [CrossRef] [PubMed]
- Negaard, H.F.S.; Iversen, P.O.; Østenstad, B.; Iversen, N.; Holme, P.A.; Sandset, P.M. Hypercoagulability in patients with haematological neoplasia: No apparent initiation by tissue factor. Thromb. Haemost. 2008, 99, 1040–1048. [Google Scholar] [CrossRef]
- Tripodo, C.; Burocchi, A.; Piccaluga, P.P.; Chiodoni, C.; Portararo, P.; Cappetti, B.; Botti, L.; Gulino, A.; Isidori, A.; Liso, A.; et al. Persistent Immune Stimulation Exacerbates Genetically Driven Myeloproliferative Disorders via Stromal Remodeling. Cancer Res. 2017, 77, 3685–3699. [Google Scholar] [CrossRef] [PubMed]
- Karala, A.-R.; Ruddock, L.W. Bacitracin is not a specific inhibitor of protein disulfide isomerase. FEBS J. 2010, 277, 2454–2462. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.E.; Foster, P.A. Protein disulphide isomerase inhibition as a potential cancer therapeutic strategy. Cancer Med. 2021, 10, 2812–2825. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beckmann, L.; Rolling, C.C.; Voigtländer, M.; Mäder, J.; Klingler, F.; Schulenkorf, A.; Lehr, C.; Bokemeyer, C.; Ruf, W.; Langer, F. Bacitracin and Rutin Regulate Tissue Factor Production in Inflammatory Monocytes and Acute Myeloid Leukemia Blasts. Cancers 2021, 13, 3941. https://doi.org/10.3390/cancers13163941
Beckmann L, Rolling CC, Voigtländer M, Mäder J, Klingler F, Schulenkorf A, Lehr C, Bokemeyer C, Ruf W, Langer F. Bacitracin and Rutin Regulate Tissue Factor Production in Inflammatory Monocytes and Acute Myeloid Leukemia Blasts. Cancers. 2021; 13(16):3941. https://doi.org/10.3390/cancers13163941
Chicago/Turabian StyleBeckmann, Lennart, Christina Charlotte Rolling, Minna Voigtländer, Jonathan Mäder, Felix Klingler, Anita Schulenkorf, Carina Lehr, Carsten Bokemeyer, Wolfram Ruf, and Florian Langer. 2021. "Bacitracin and Rutin Regulate Tissue Factor Production in Inflammatory Monocytes and Acute Myeloid Leukemia Blasts" Cancers 13, no. 16: 3941. https://doi.org/10.3390/cancers13163941
APA StyleBeckmann, L., Rolling, C. C., Voigtländer, M., Mäder, J., Klingler, F., Schulenkorf, A., Lehr, C., Bokemeyer, C., Ruf, W., & Langer, F. (2021). Bacitracin and Rutin Regulate Tissue Factor Production in Inflammatory Monocytes and Acute Myeloid Leukemia Blasts. Cancers, 13(16), 3941. https://doi.org/10.3390/cancers13163941