
Transcription Factor Homeobox D9 Drives the Malignant Phenotype of HPV18-Positive Cervical Cancer Cells via Binding to the Viral Early Promoter

 ,
,  , and
, and
Simple Summary
Abstract
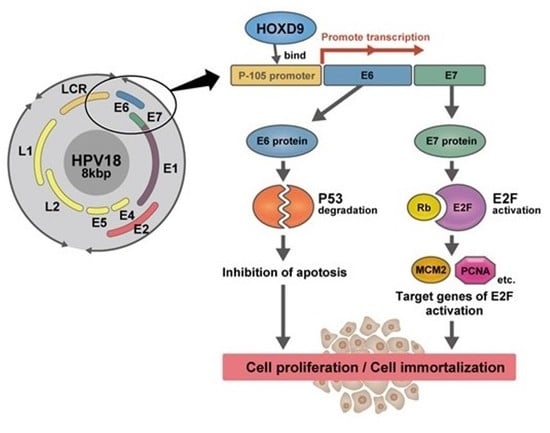
1. Introduction
2. Methods and Methods
2.1. Cell Lines
2.2. Knockdown of HOXD9
2.3. RT-PCR and qPCR
2.4. Cell Viability Assay
2.5. Cell Cycle Analysis by Flow Cytometry
2.6. Apoptosis Assay
2.7. Western Blotting
2.8. Luciferase Reporter Assay
2.9. Chromatin Immunoprecipitation
2.10. DNA Microarray and Ingenuity Pathway Analysis
2.11. Statistical Analysis
3. Results
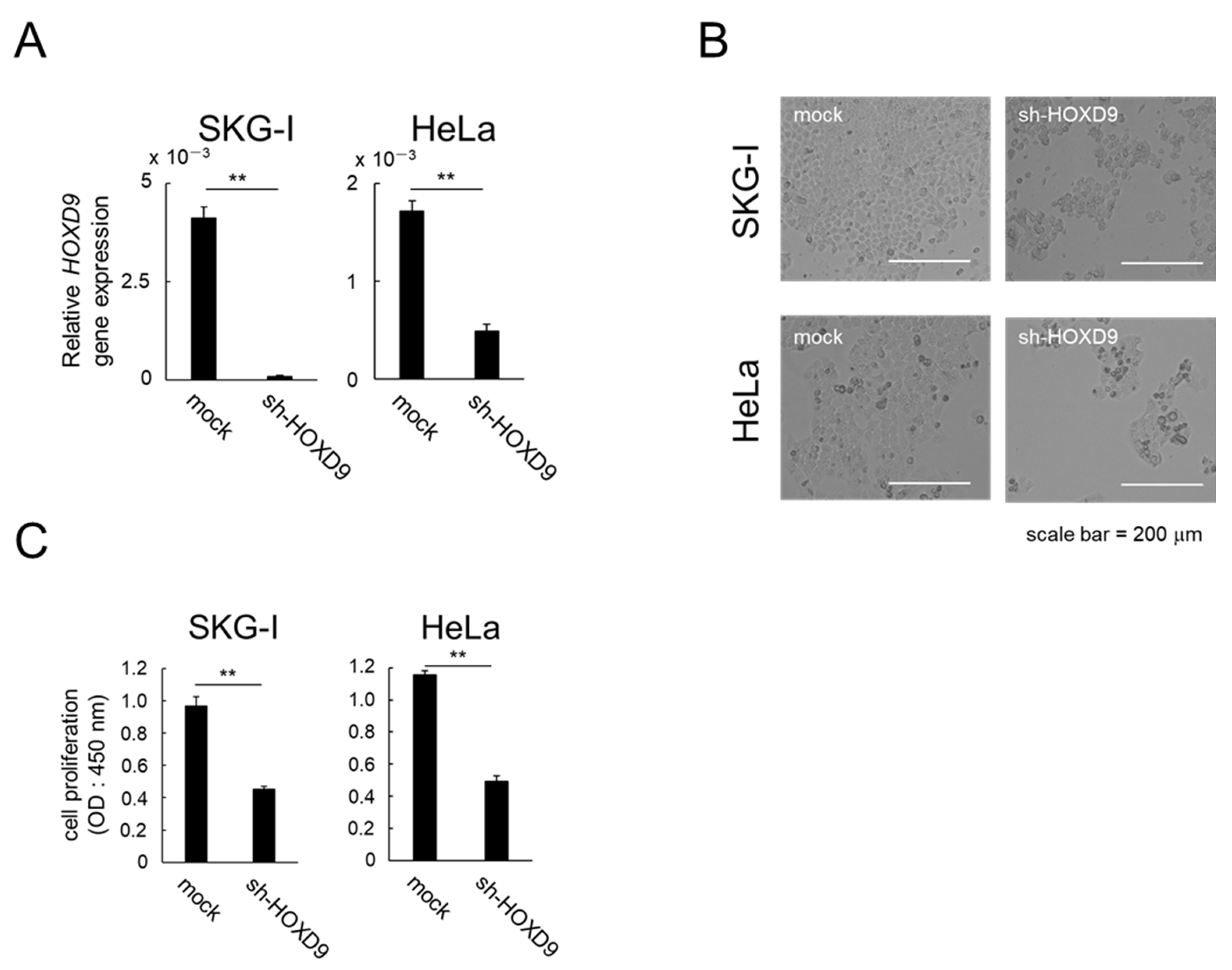
3.1. HOXD9 Is Essential for Proliferation of CC cells
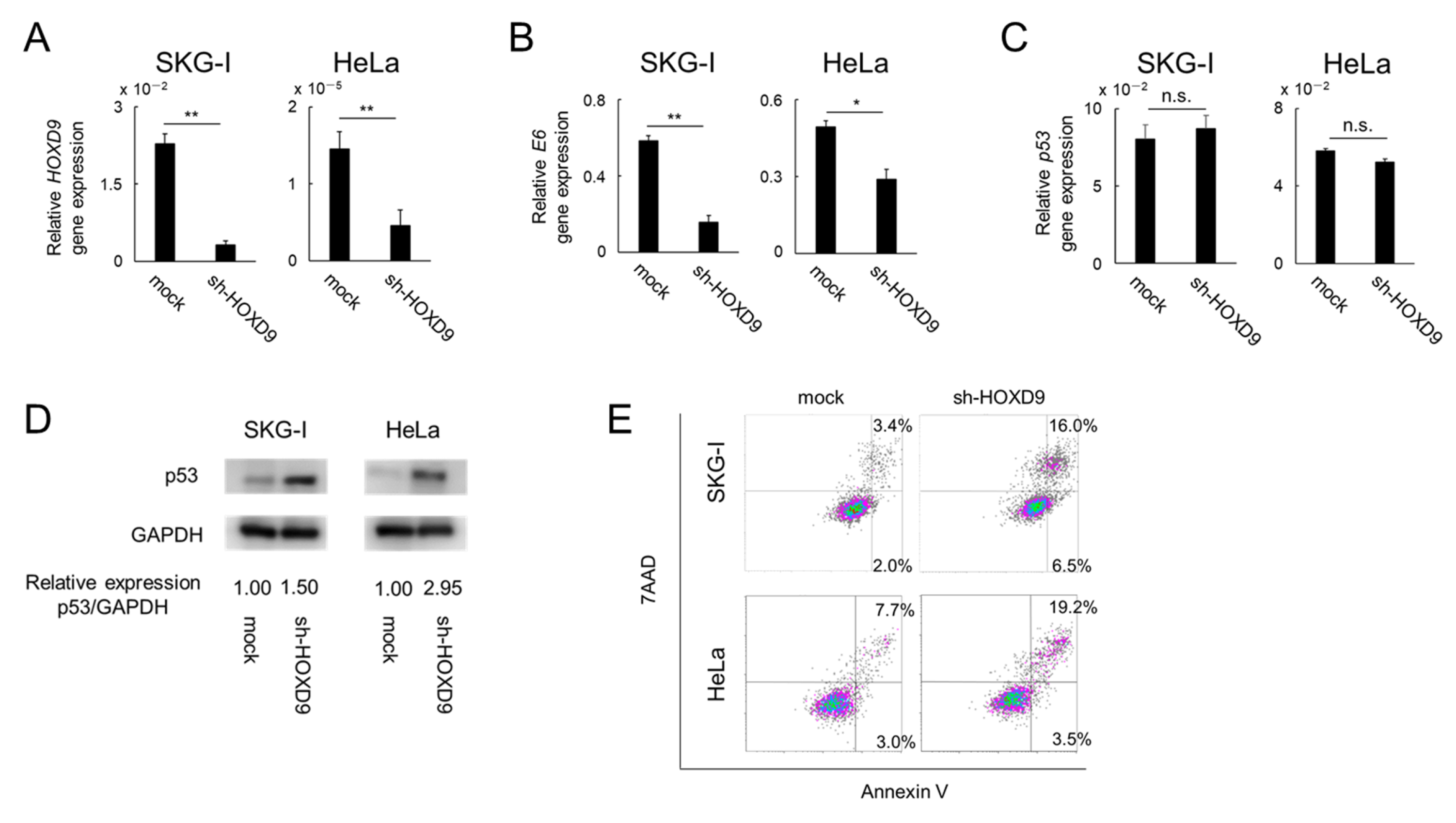
3.2. HOXD9 Represses E6 Gene Expression and Activates the p53 Pathway
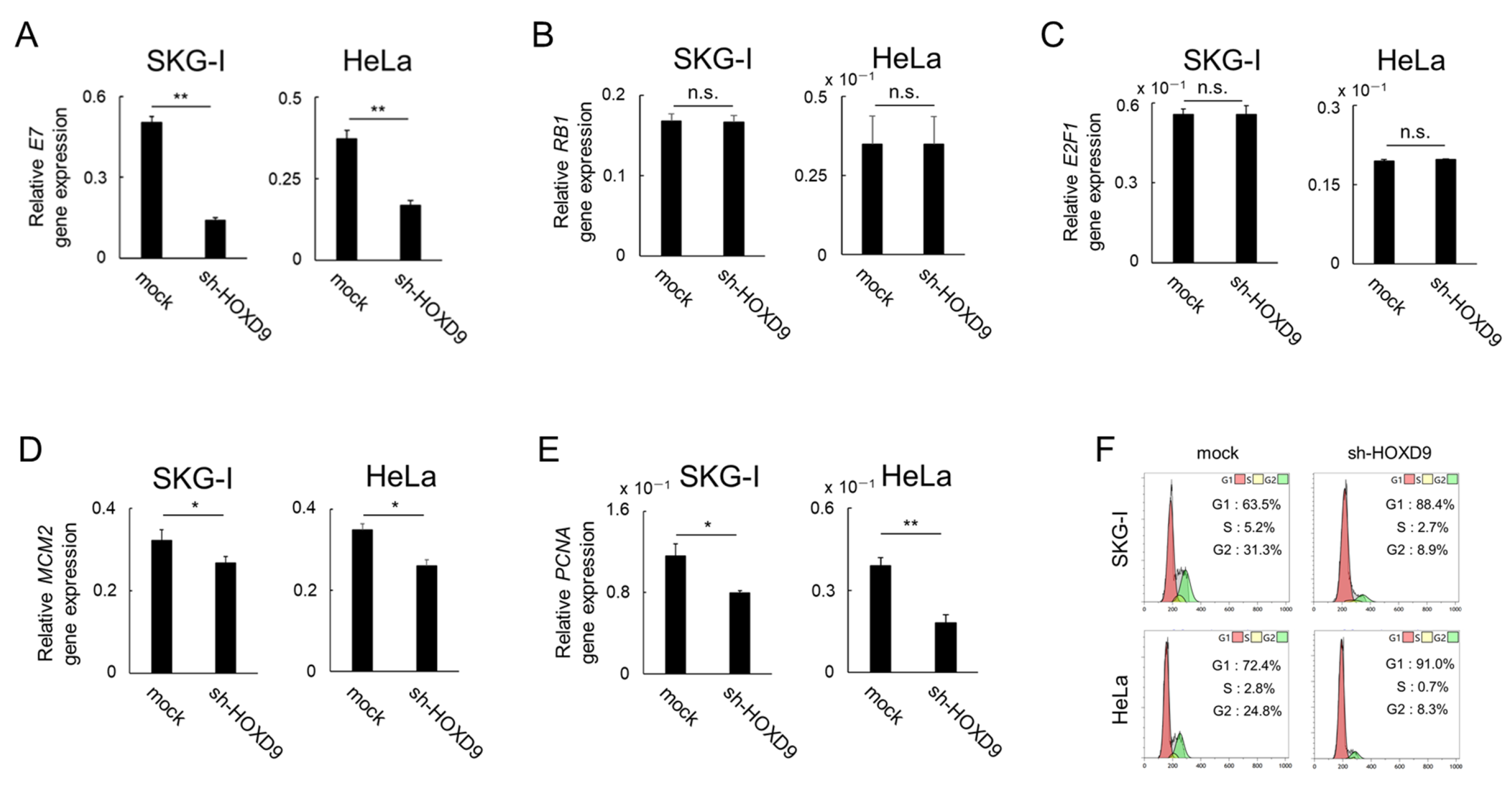
3.3. HOXD9 Represses E7 Gene Expression and Activates the E2F Pathway
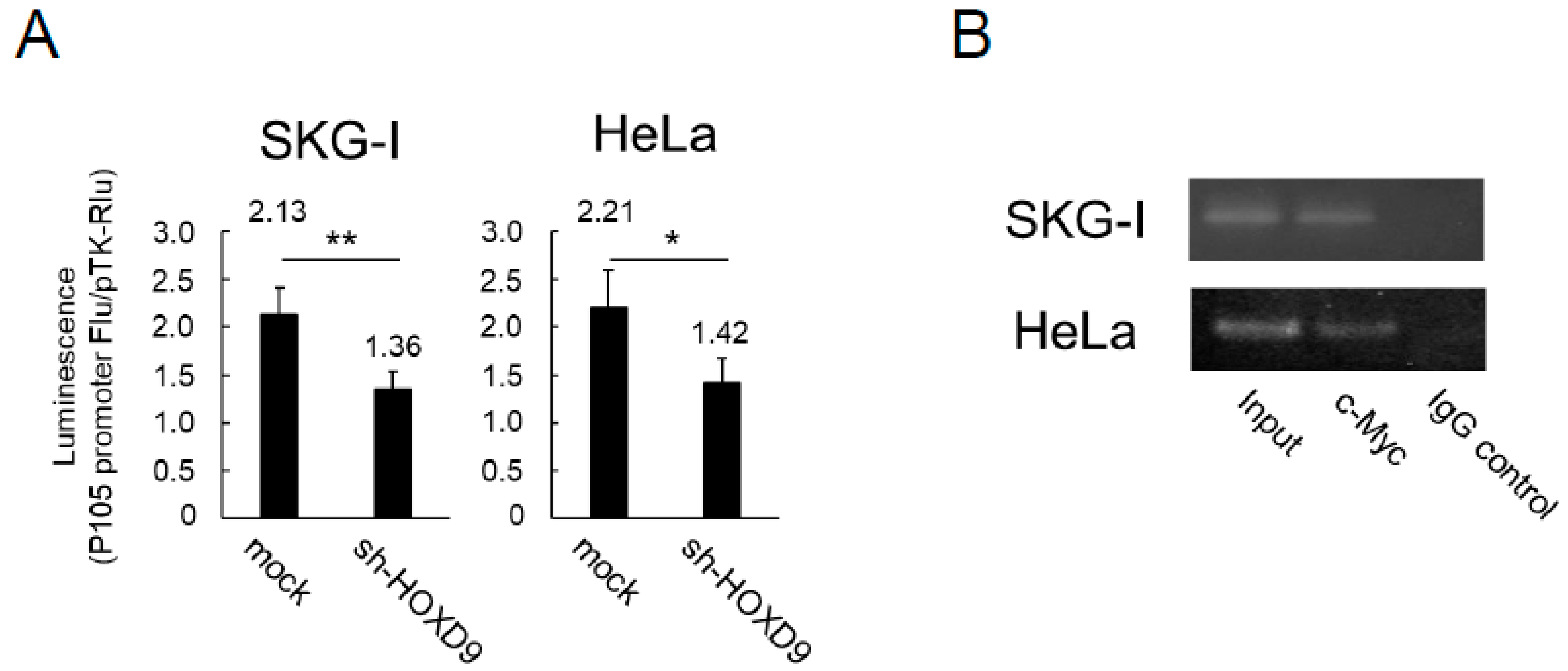
3.4. HOXD9 Regulates the P105 Promoter by Direct Binding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Human Papillomavirus and Related Diseases Report, ICO/IARC Information Centre on HPV and Cancer (HPV Information Centre). 2019. Available online: https://www.hpvcentre.net/statistics/reports/XWX.pdf (accessed on 6 September 2021).
- Arbyn, M.; Tommasino, M.; Depuydt, C.; Dillner, J. Are 20 human papillomavirus types causing cervical cancer? J. Pathol. 2014, 234, 431–435. [Google Scholar] [CrossRef]
- Munoz, N.; Bosch, F.X.; Castellsague, X.; Diaz, M.; de Sanjose, S.; Hammouda, D.; Shah, K.V.; Meijer, C.J. Against which human papillomavirus types shall we vaccinate and screen? The international perspective. Int. J. Cancer 2004, 111, 278–285. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, K.; Butz, K.; Yasui, T.; Kikutani, H.; Hoppe-Seyler, F. Regulation of human papillomavirus transcription by the differentiation-dependent epithelial factor Epoc-1/skn-1a. J. Virol. 1996, 70, 10–16. [Google Scholar] [CrossRef]
- Munger, K.; Phelps, W.C.; Bubb, V.; Howley, P.M.; Schlegel, R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 1989, 63, 4417–4421. [Google Scholar] [CrossRef]
- Sato, N.; Saga, Y.; Uchibori, R.; Tsukahara, T.; Urabe, M.; Kume, A.; Fujiwara, H.; Suzuki, M.; Ozawa, K.; Mizukami, H. Eradication of cervical cancer in vivo by an AAV vector that encodes shRNA targeting human papillomavirus type 16 E6/E7. Int. J. Oncol. 2018, 52, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; DiMaio, D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. USA 2000, 97, 12513–12518. [Google Scholar] [CrossRef]
- Shah, N.; Sukumar, S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010, 10, 361–371. [Google Scholar] [CrossRef]
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. HOX genes and their role in the development of human cancers. J. Mol. Med. 2014, 92, 811–823. [Google Scholar] [CrossRef]
- Fromental-Ramain, C.; Warot, X.; Lakkaraju, S.; Favier, B.; Haack, H.; Birling, C.; Dierich, A.; Dolle, P.; Chambon, P. Specific and redundant functions of the paralogous Hoxa-9 and Hoxd-9 genes in forelimb and axial skeleton patterning. Development 1996, 122, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Li, L.; Lv, L.; Qu, X.; Jin, S.; Li, K.; Deng, X.; Cheng, L.; He, H.; Dong, L. HOXD9 promotes epithelial-mesenchymal transition and cancer metastasis by ZEB1 regulation in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 133. [Google Scholar] [CrossRef] [PubMed]
- Tabuse, M.; Ohta, S.; Ohashi, Y.; Fukaya, R.; Misawa, A.; Yoshida, K.; Kawase, T.; Saya, H.; Thirant, C.; Chneiweiss, H.; et al. Functional analysis of HOXD9 in human gliomas and glioma cancer stem cells. Mol. Cancer 2011, 10, 60. [Google Scholar] [CrossRef]
- Hirao, N.; Iwata, T.; Tanaka, K.; Nishio, H.; Nakamura, M.; Morisada, T.; Morii, K.; Maruyama, N.; Katoh, Y.; Yaguchi, T.; et al. Transcription factor homeobox D9 is involved in the malignant phenotype of cervical cancer through direct binding to the human papillomavirus oncogene promoter. Gynecol. Oncol. 2019, 155, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Seoud, M.; Tjalma, W.A.; Ronsse, V. Cervical adenocarcinoma: Moving towards better prevention. Vaccine 2011, 29, 9148–9158. [Google Scholar] [CrossRef] [PubMed]
- Castle, P.E.; Pierz, A.; Stoler, M.H. A systematic review and meta-analysis on the attribution of human papillomavirus (HPV) in neuroendocrine cancers of the cervix. Gynecol. Oncol. 2018, 148, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chou, J.W.; Simpson, D.A.; Zhou, Y.; Mullen, T.E.; Medeiros, M.; Bushel, P.R.; Paules, R.S.; Yang, X.; Hurban, P.; et al. Profiles of global gene expression in ionizing-radiation-damaged human diploid fibroblasts reveal synchronization behind the G1 checkpoint in a G0-like state of quiescence. Environ. Health Perspect. 2006, 114, 553–559. [Google Scholar] [CrossRef][Green Version]
- Hatterschide, J.; Brantly, A.C.; Grace, M.; Munger, K.; White, E.A. A Conserved Amino Acid in the C Terminus of Human Papillomavirus E7 Mediates Binding to PTPN14 and Repression of Epithelial Differentiation. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Li, Y.Y.; Wang, L.; Lu, C.D. An E2F site in the 5’-promoter region contributes to serum-dependent up-regulation of the human proliferating cell nuclear antigen gene. FEBS Lett. 2003, 544, 112–118. [Google Scholar] [CrossRef]
- Von Knebel Doeberitz, M. Papillomaviruses in human disease: Part II. Molecular biology and immunology of papillomavirus infections and carcinogenesis. Eur. J. Med. 1992, 1, 485–491. [Google Scholar] [PubMed]
- Zur Hausen, H. Intracellular surveillance of persisting viral infections. Human genital cancer results from deficient cellular control of papillomavirus gene expression. Lancet 1986, 2, 489–491. [Google Scholar] [CrossRef]
- Kikuchi, K.; Taniguchi, A.; Yasumoto, S. Induction of the HPV16 enhancer activity by Jun-B and c-Fos through cooperation of the promoter-proximal AP-1 site and the epithelial cell type—Specific regulatory element in fibroblasts. Virus Genes 1996, 13, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Huang, L.H.; Chen, T.M. Differential effects of progestins and estrogens on long control regions of human papillomavirus types 16 and 18. Biochem. Biophys. Res. Commun. 1996, 224, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Ishiji, T.; Lace, M.J.; Parkkinen, S.; Anderson, R.D.; Haugen, T.H.; Cripe, T.P.; Xiao, J.H.; Davidson, I.; Chambon, P.; Turek, L.P. Transcriptional enhancer factor (TEF)-1 and its cell-specific co-activator activate human papillomavirus-16 E6 and E7 oncogene transcription in keratinocytes and cervical carcinoma cells. EMBO J. 1992, 11, 2271–2281. [Google Scholar] [CrossRef]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Kukimoto, I. The Transcriptional Cofactor VGLL1 Drives Transcription of Human Papillomavirus Early Genes via TEAD1. J. Virol. 2020, 94, e01945-19. [Google Scholar] [CrossRef]
- Apt, D.; Chong, T.; Liu, Y.; Bernard, H.U. Nuclear factor I and epithelial cell-specific transcription of human papillomavirus type 16. J. Virol. 1993, 67, 4455–4463. [Google Scholar] [CrossRef]
- Stunkel, W.; Huang, Z.; Tan, S.H.; O’Connor, M.J.; Bernard, H.U. Nuclear matrix attachment regions of human papillomavirus type 16 repress or activate the E6 promoter, depending on the physical state of the viral DNA. J. Virol. 2000, 74, 2489–2501. [Google Scholar] [CrossRef]
- O’Connor, M.J.; Tan, S.H.; Tan, C.H.; Bernard, H.U. YY1 represses human papillomavirus type 16 transcription by quenching AP-1 activity. J. Virol. 1996, 70, 6529–6539. [Google Scholar] [CrossRef]
- Crowe, D.L.; Nguyen, D.C.; Tsang, K.J.; Kyo, S. E2F-1 represses transcription of the human telomerase reverse transcriptase gene. Nucleic Acids Res. 2001, 29, 2789–2794. [Google Scholar] [CrossRef]
- Ohtani, K.; DeGregori, J.; Nevins, J.R. Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl. Acad. Sci. USA 1995, 92, 12146–12150. [Google Scholar] [CrossRef] [PubMed]
- Allouch, S.; Malki, A.; Allouch, A.; Gupta, I.; Vranic, S.; Al Moustafa, A.E. High-Risk HPV Oncoproteins and PD-1/PD-L1 Interplay in Human Cervical Cancer: Recent Evidence and Future Directions. Front. Oncol. 2020, 10, 914. [Google Scholar] [CrossRef] [PubMed]
- Heeren, A.M.; Punt, S.; Bleeker, M.C.; Gaarenstroom, K.N.; van der Velden, J.; Kenter, G.G.; de Gruijl, T.D.; Jordanova, E.S. Prognostic effect of different PD-L1 expression patterns in squamous cell carcinoma and adenocarcinoma of the cervix. Mod. Pathol. 2016, 29, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Dong, M.; Liu, Z.; Mi, Y.; Yang, J.; Zhang, Z.; Liu, K.; Jiang, L.; Zhang, Y.; Dong, S.; et al. Elevated PD-L1 expression predicts poor survival outcomes in patients with cervical cancer. Cancer Cell Int. 2019, 19, 146. [Google Scholar] [CrossRef] [PubMed]
- Phelan, M.L.; Sadoul, R.; Featherstone, M.S. Functional differences between HOX proteins conferred by two residues in the homeodomain N-terminal arm. Mol. Cell Biol. 1994, 14, 5066–5075. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upstream Regulator | Molecule Type | Activation z-Score | p-Value of Overlap |
|---|---|---|---|
| IL1RN | cytokine | 4.298 | 0.00197 |
| epigallocatechin-gallate | chemical drug | 4.192 | 0.00392 |
| NKX2-3 | transcription regulator | 4.135 | 0.000607 |
| Irgm1 | other | 3.978 | 0.00673 |
| TP53 | transcription regulator | 3.830 | 1.1 × 10−19 |
| CDKN1A | kinase | 3.754 | 0.00171 |
| fulvestrant | chemical drug | 3.603 | 0.000958 |
| U0126 | Chemical-kinase inhibitor | 3.494 | 0.000703 |
| PTGER4 | G-protein coupled receptor | 3.342 | 0.0123 |
| gentamicin | chemical drug | 3.219 | 0.0013 |
| PTEN | phosphatase | 3.177 | 0.000364 |
| RB1 | transcription regulator | 2.889 | 0.0024 |
| fenretinide | chemical drug | 2.866 | 0.00309 |
| MEOX2 | transcription regulator | 2.853 | 0.00603 |
| RPSA | translation regulator | 2.726 | 0.000899 |
| SP600125 | Chemical-kinase inhibitor | 2.718 | 0.00125 |
| IgG | complex | 2.705 | 0.0122 |
| ACKR2 | G-protein coupled receptor | 2.673 | 0.00926 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayashi, S.; Iwata, T.; Imagawa, R.; Sugawara, M.; Chen, G.; Tanimoto, S.; Sugawara, Y.; Tanaka, I.; Matsui, T.; Nishio, H.; et al. Transcription Factor Homeobox D9 Drives the Malignant Phenotype of HPV18-Positive Cervical Cancer Cells via Binding to the Viral Early Promoter. Cancers 2021, 13, 4613. https://doi.org/10.3390/cancers13184613
Hayashi S, Iwata T, Imagawa R, Sugawara M, Chen G, Tanimoto S, Sugawara Y, Tanaka I, Matsui T, Nishio H, et al. Transcription Factor Homeobox D9 Drives the Malignant Phenotype of HPV18-Positive Cervical Cancer Cells via Binding to the Viral Early Promoter. Cancers. 2021; 13(18):4613. https://doi.org/10.3390/cancers13184613
Chicago/Turabian StyleHayashi, Shigenori, Takashi Iwata, Ryotaro Imagawa, Masaki Sugawara, Guanliang Chen, Satoko Tanimoto, Yo Sugawara, Ikumo Tanaka, Tomoya Matsui, Hiroshi Nishio, and et al. 2021. "Transcription Factor Homeobox D9 Drives the Malignant Phenotype of HPV18-Positive Cervical Cancer Cells via Binding to the Viral Early Promoter" Cancers 13, no. 18: 4613. https://doi.org/10.3390/cancers13184613
APA StyleHayashi, S., Iwata, T., Imagawa, R., Sugawara, M., Chen, G., Tanimoto, S., Sugawara, Y., Tanaka, I., Matsui, T., Nishio, H., Nakamura, M., Katoh, Y., Mori, S., Kukimoto, I., & Aoki, D. (2021). Transcription Factor Homeobox D9 Drives the Malignant Phenotype of HPV18-Positive Cervical Cancer Cells via Binding to the Viral Early Promoter. Cancers, 13(18), 4613. https://doi.org/10.3390/cancers13184613