
Development of Multi-Scale X-ray Fluorescence Tomography for Examination of Nanocomposite-Treated Biological Samples


 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Nanocomposite Preparation
2.2. Cell Culture and Nanocomposite Treatments
2.3. Flow Cytometry
2.4. Protein Isolation and Western Blots
2.5. HeLa Cell Treatment and Sample Preparation for X-ray Fluoresence Microscopy
2.6. X-ray Fluorescence Microscopy and Tomography
2.7. X-ray Fluorescence Data Analysis and Visualization
3. Results
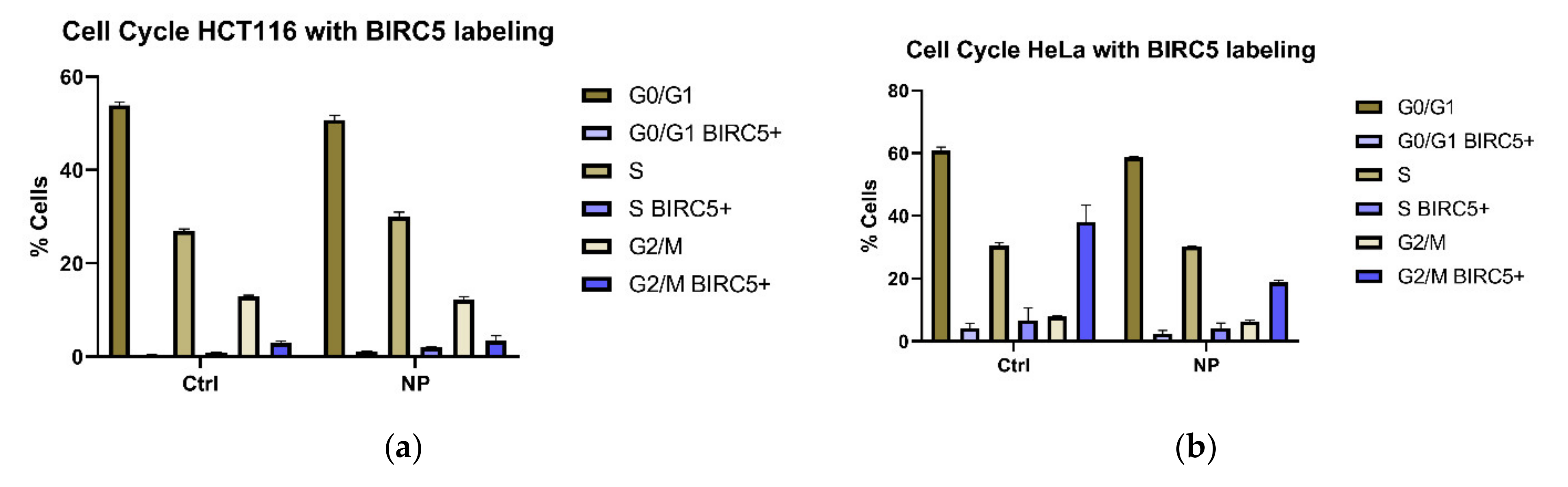
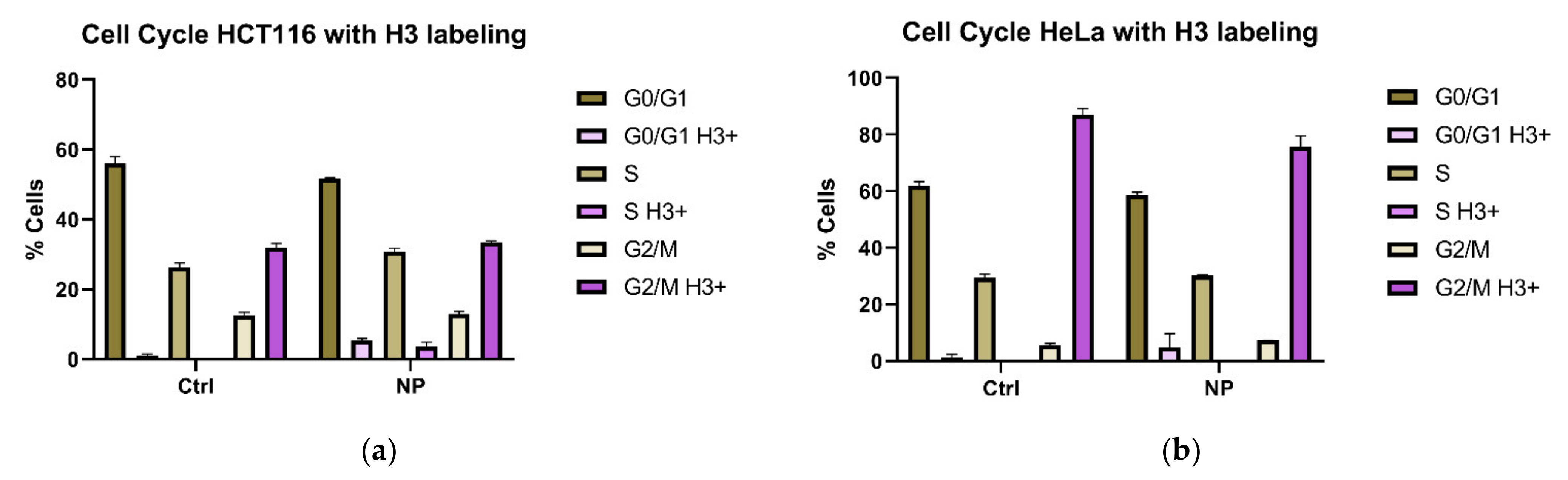
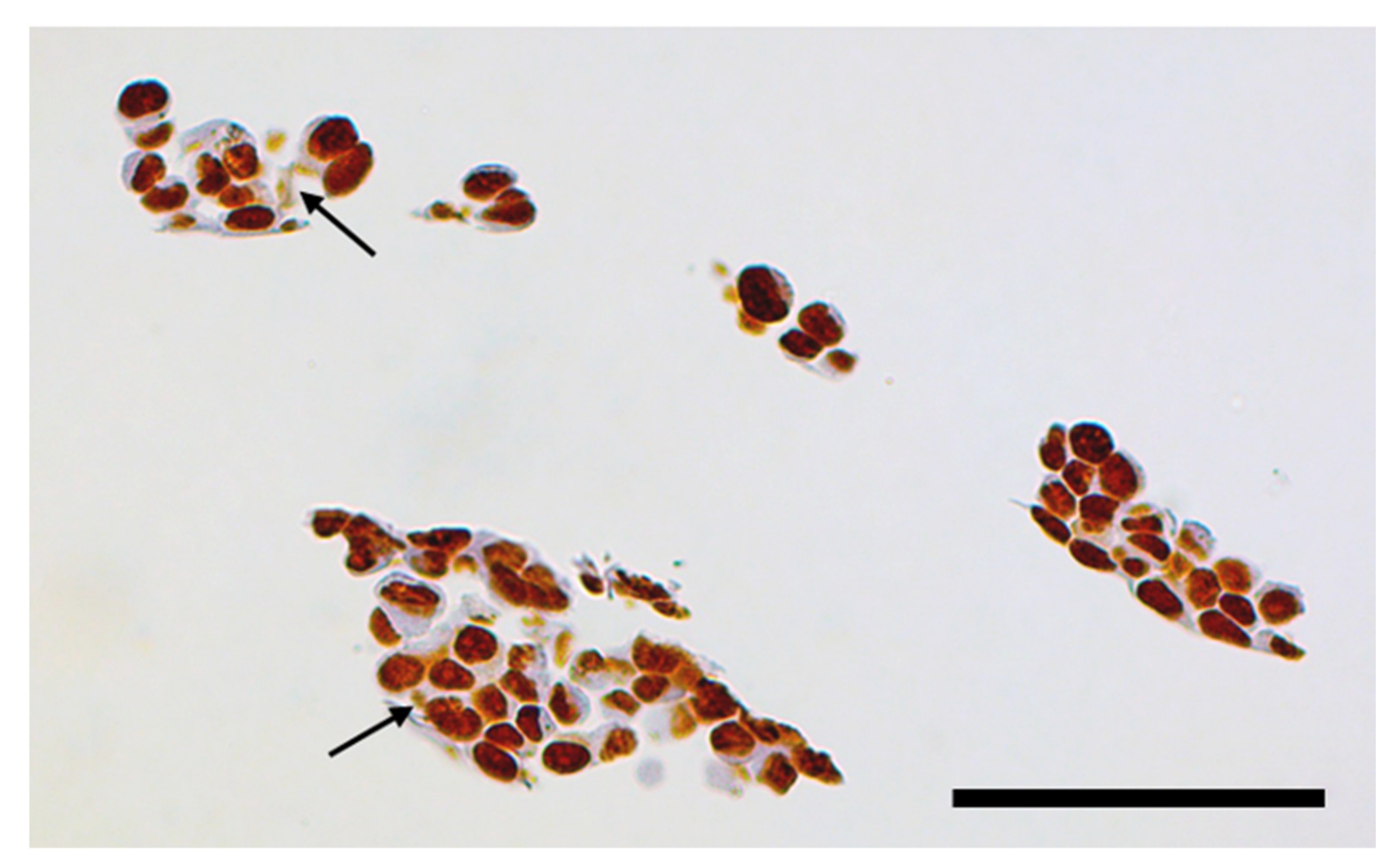
3.1. Cell Cycle Changes in Cells Exposed to Non-Targeted Nanocomposites
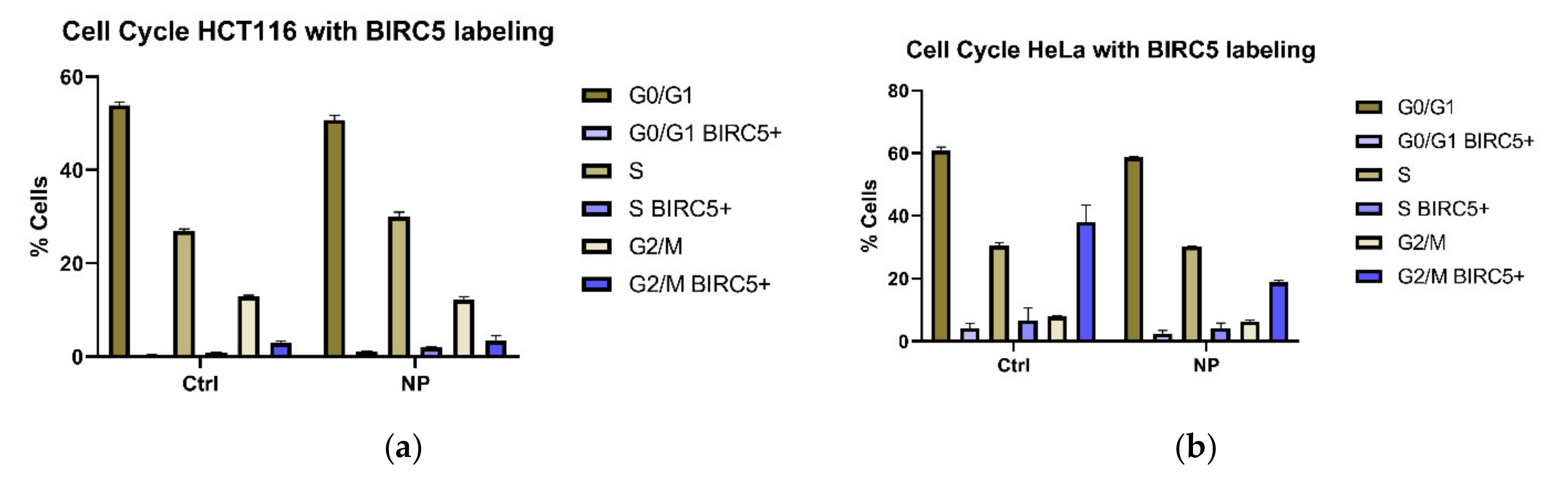
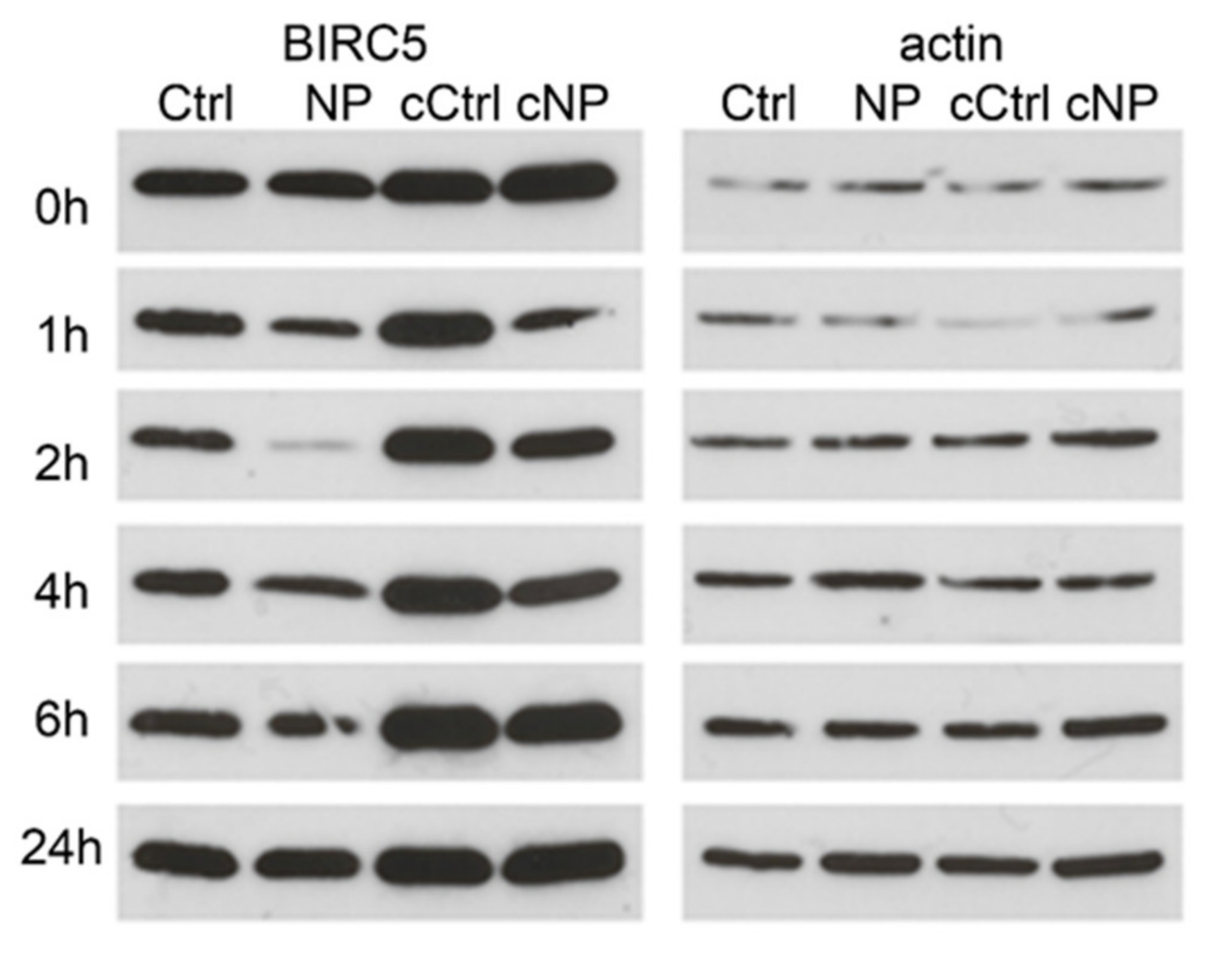
3.2. BIRC5 Participates in Nanocomposite Protein Corona
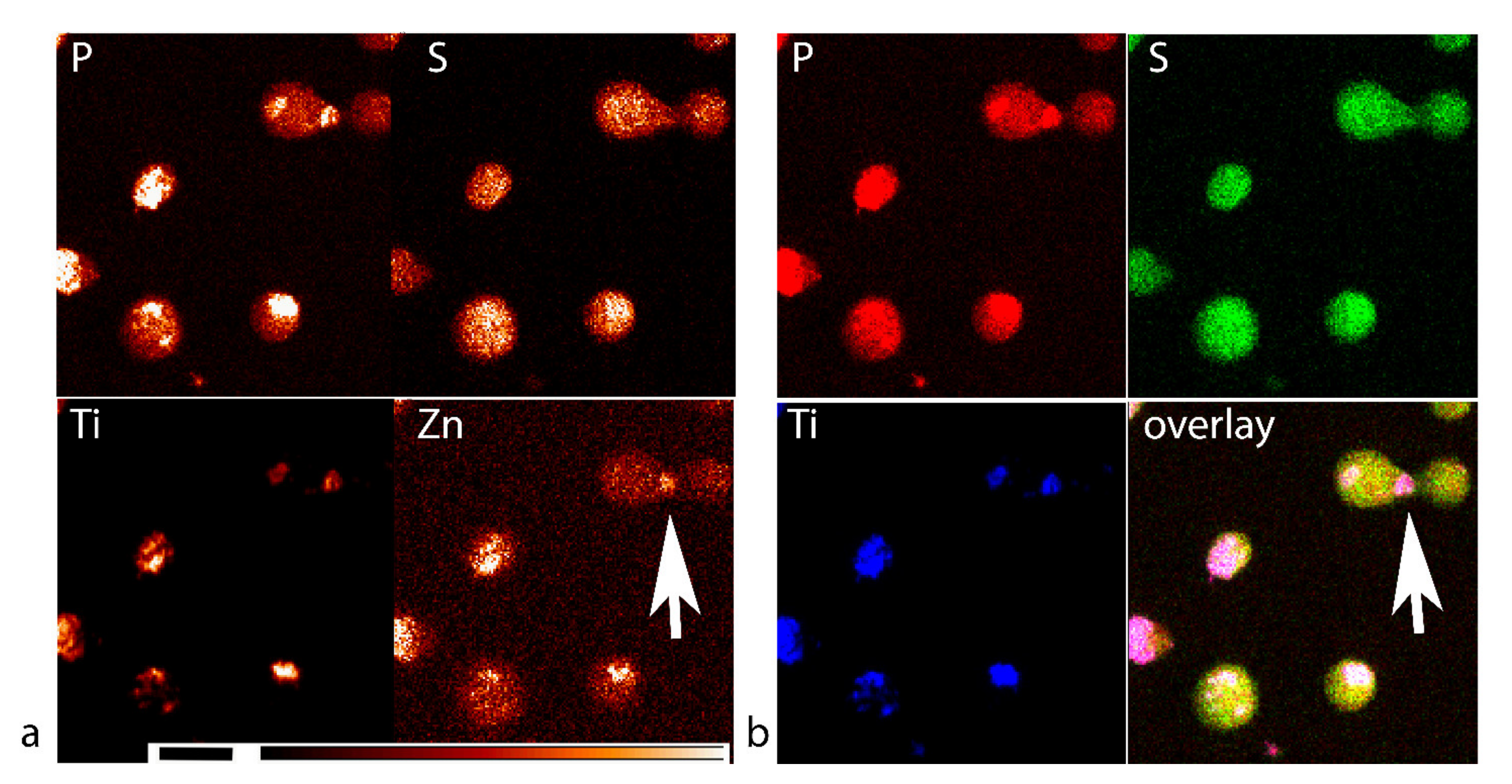
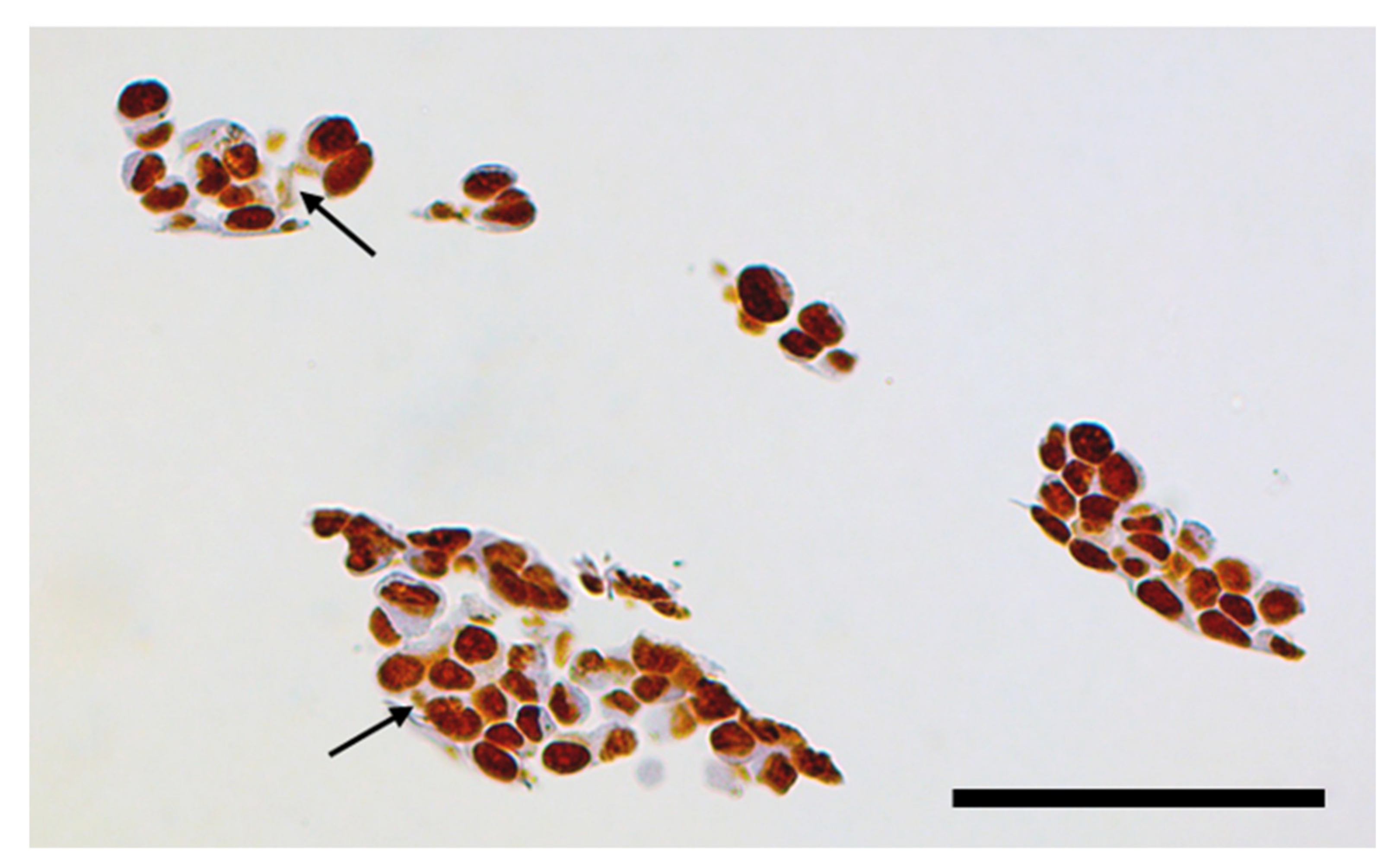
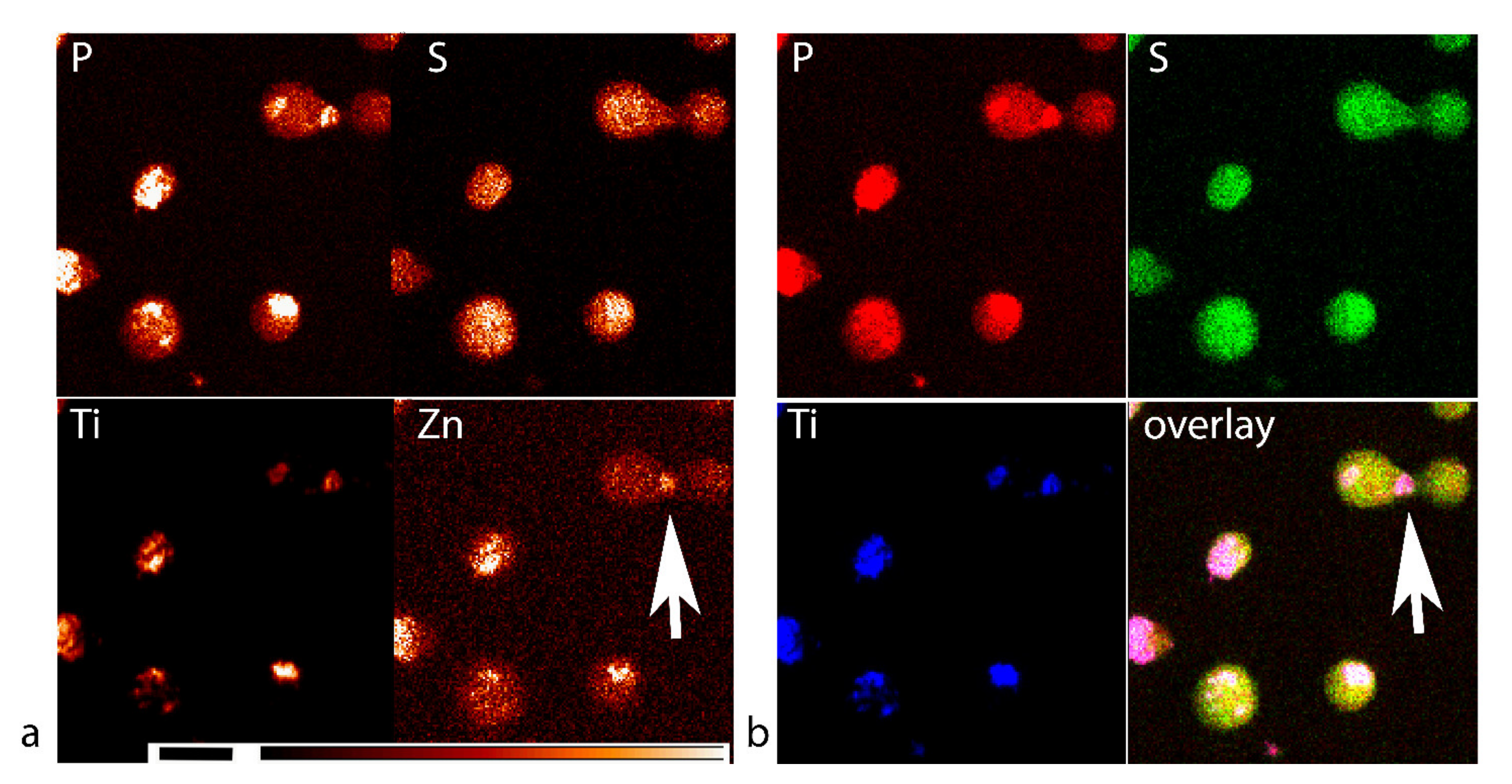
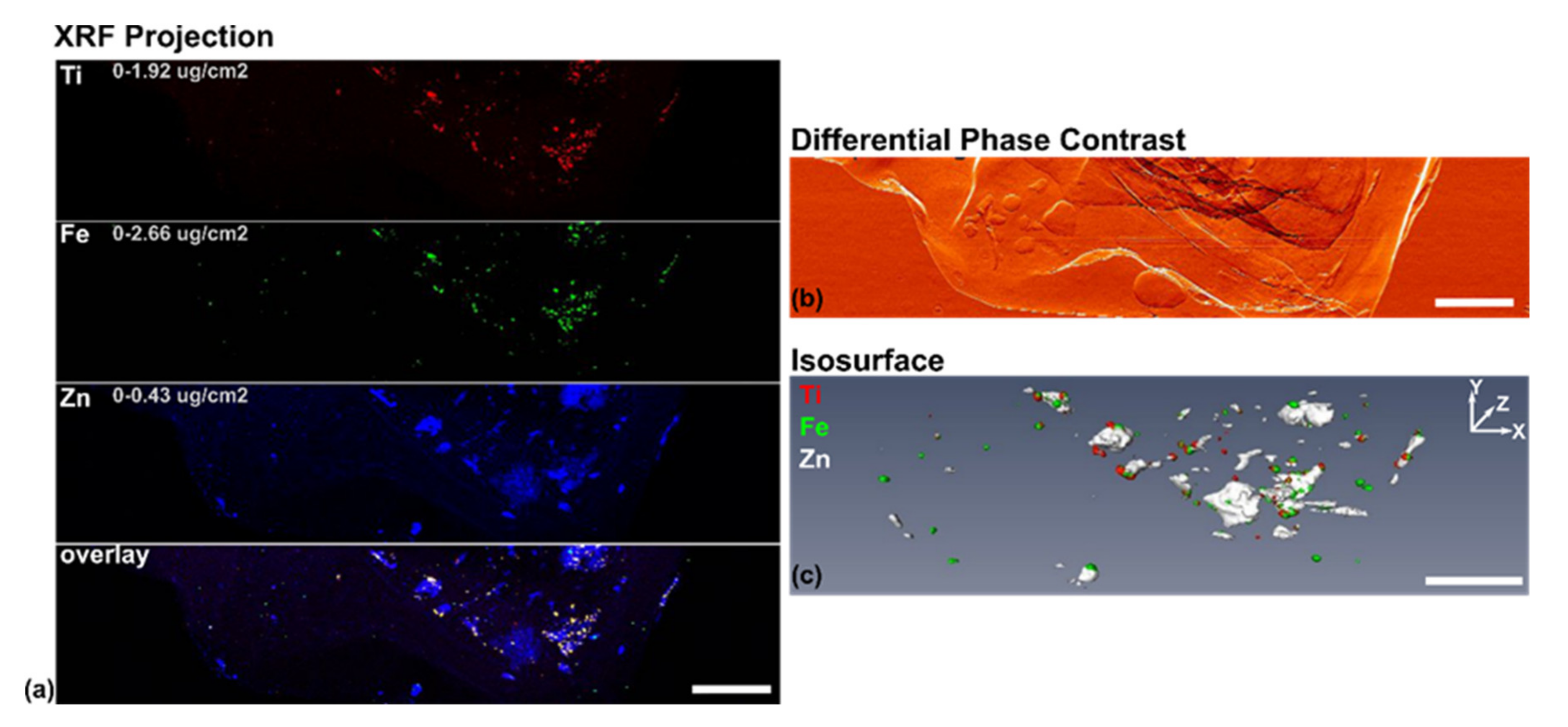
3.3. XFM Permits Imaging of Nanocomposites Inside Cells
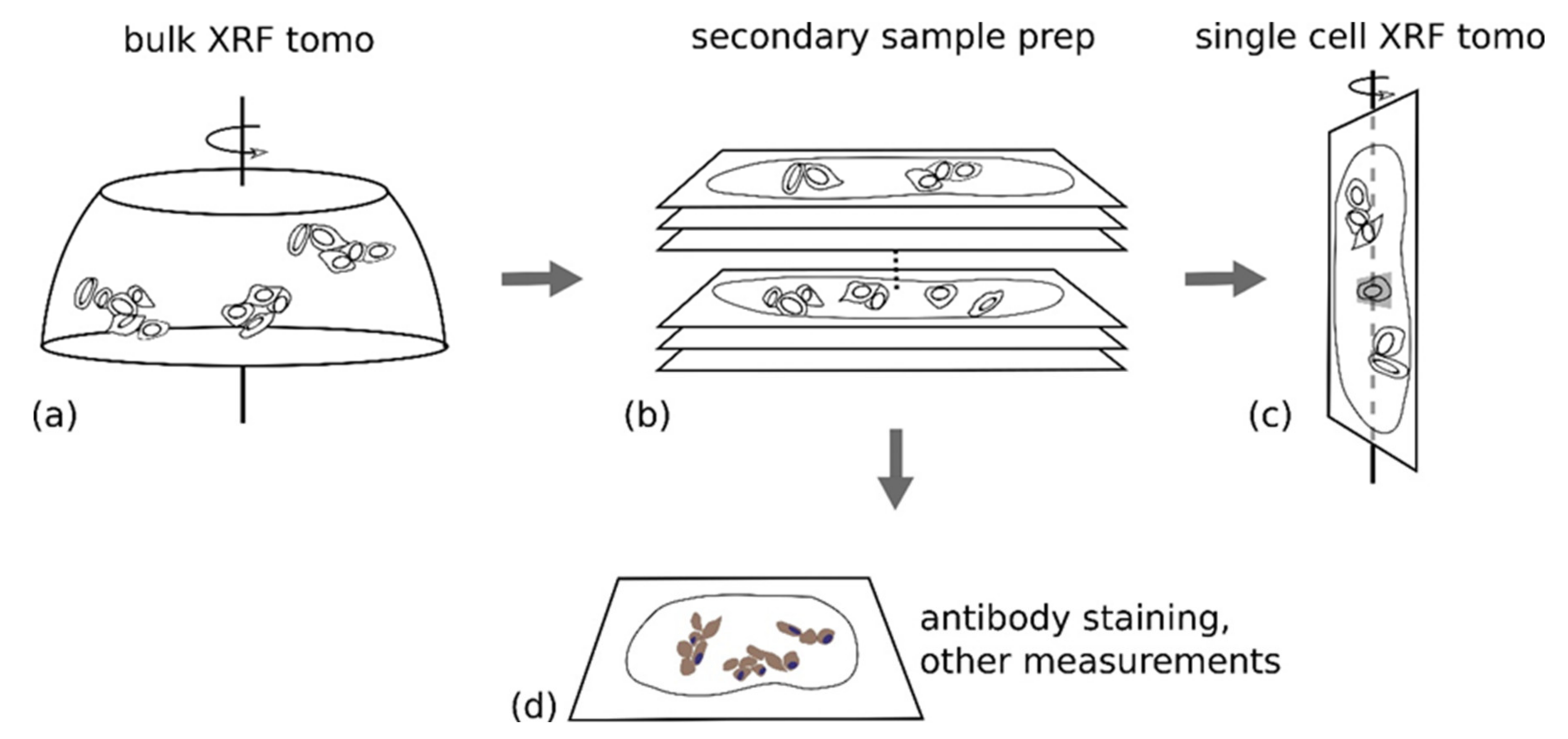
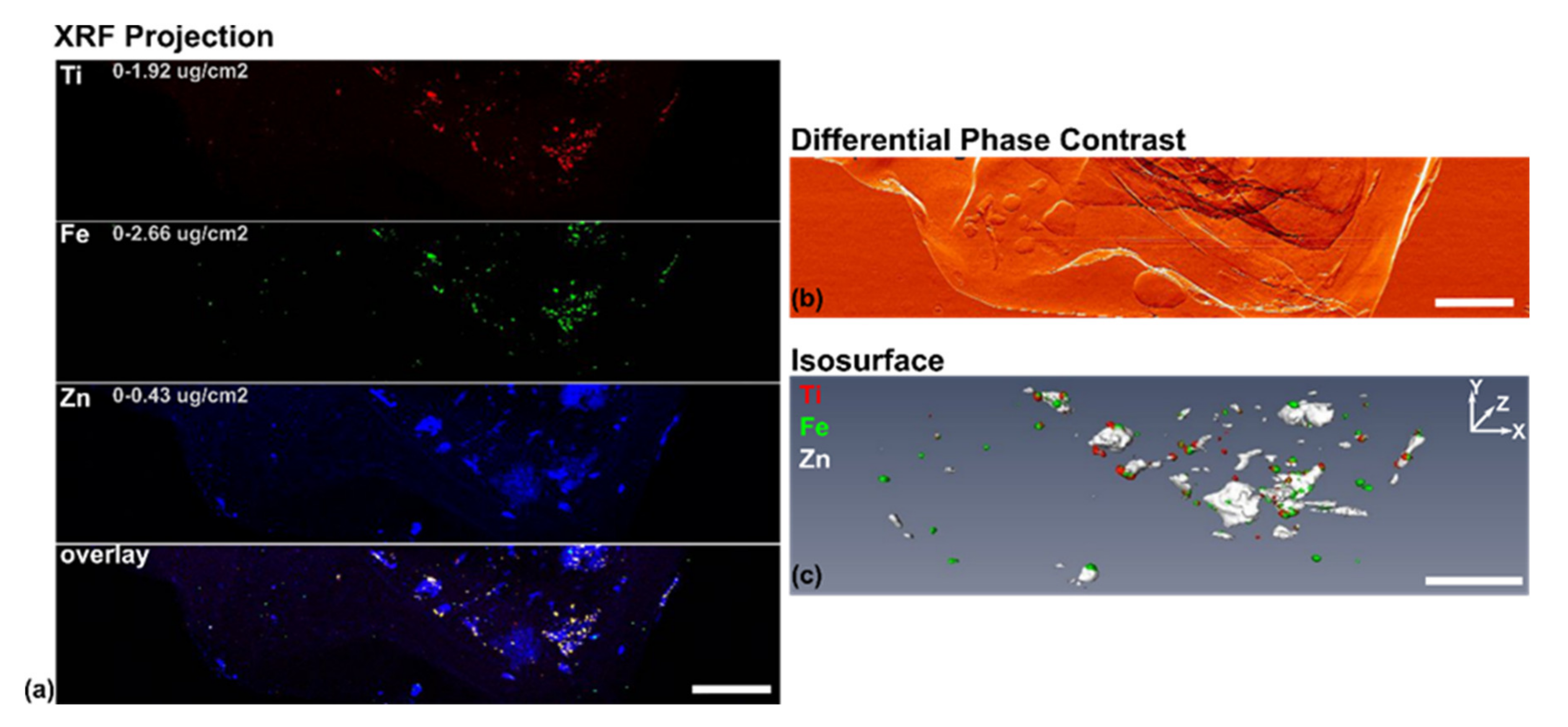
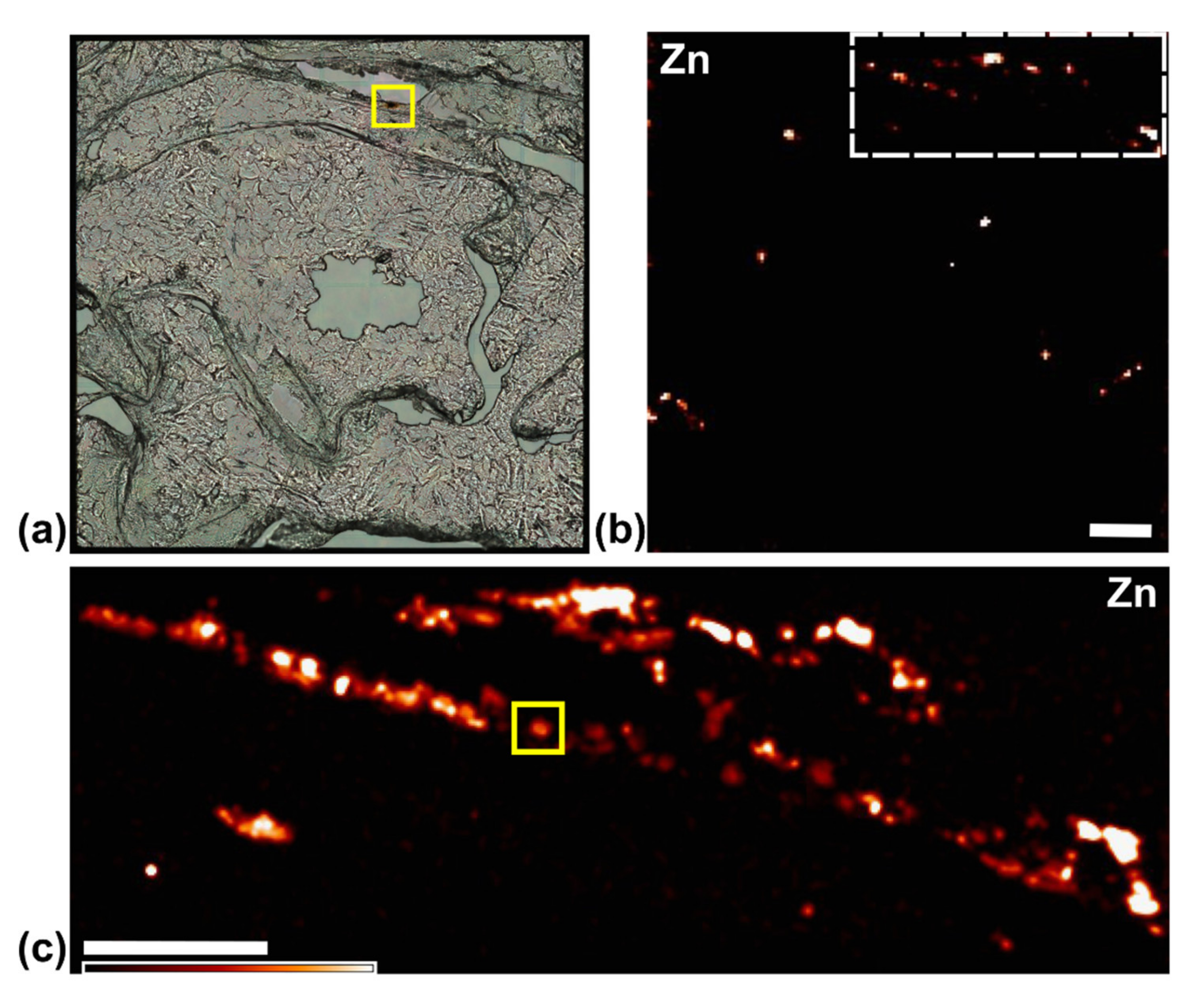
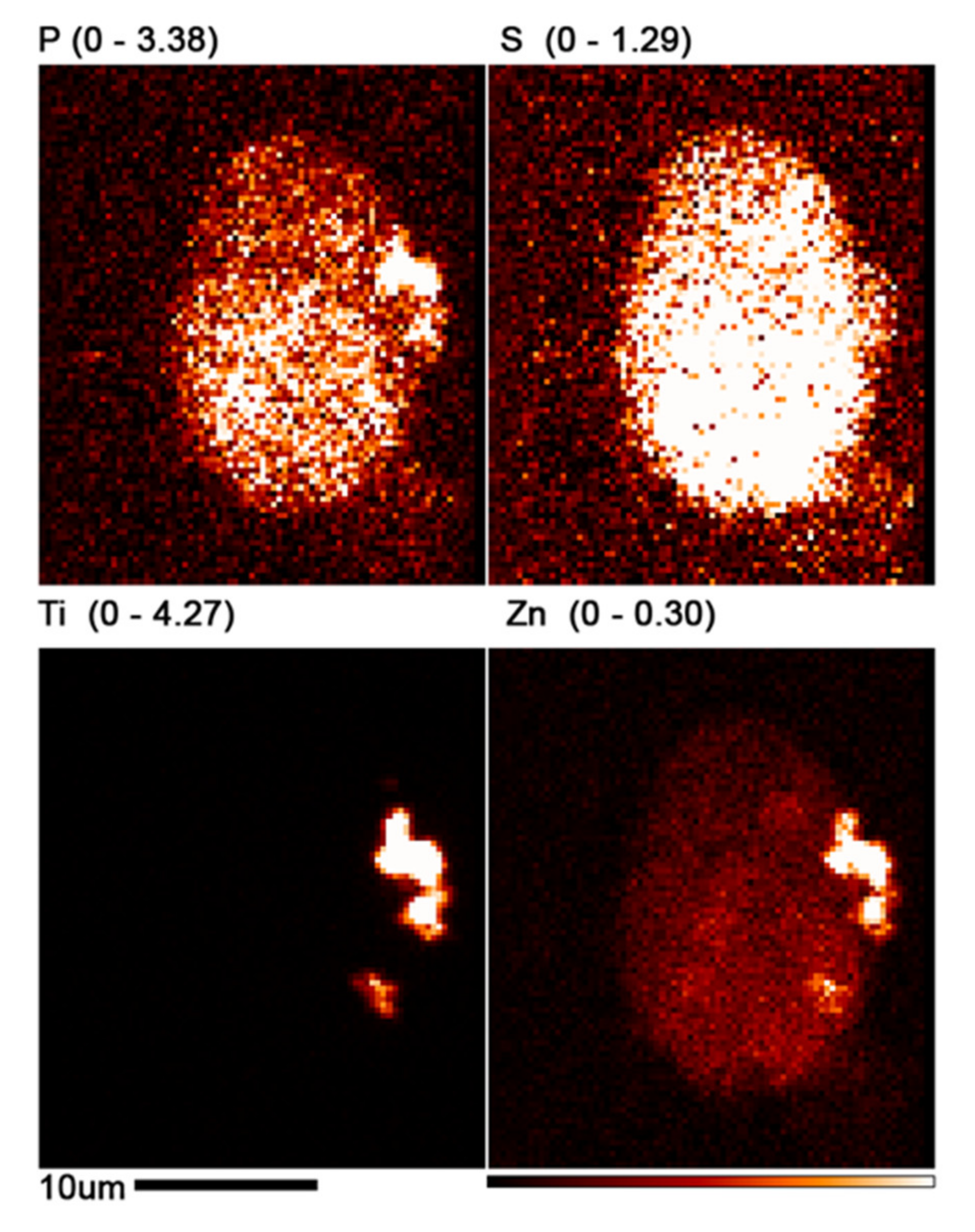
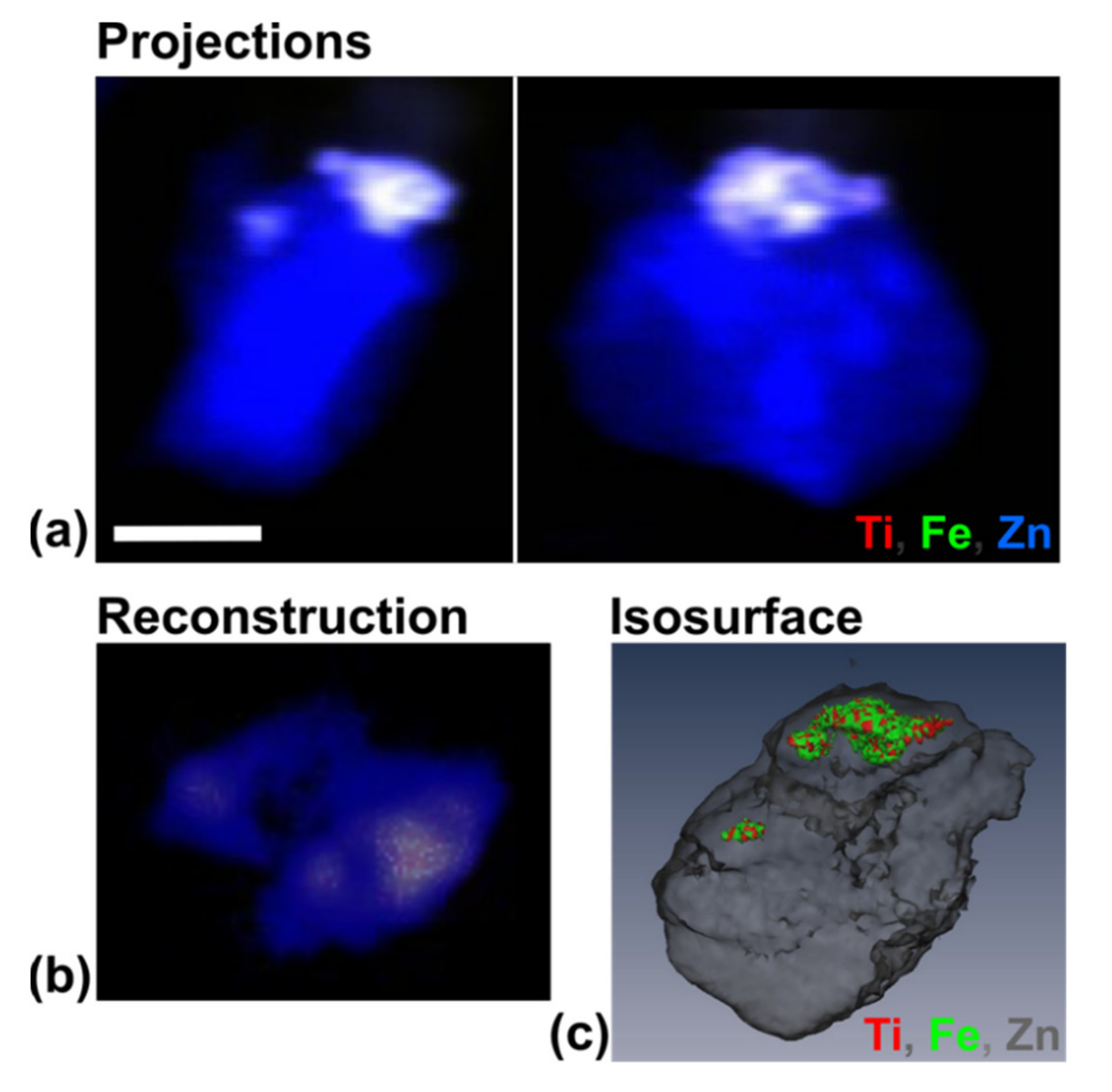
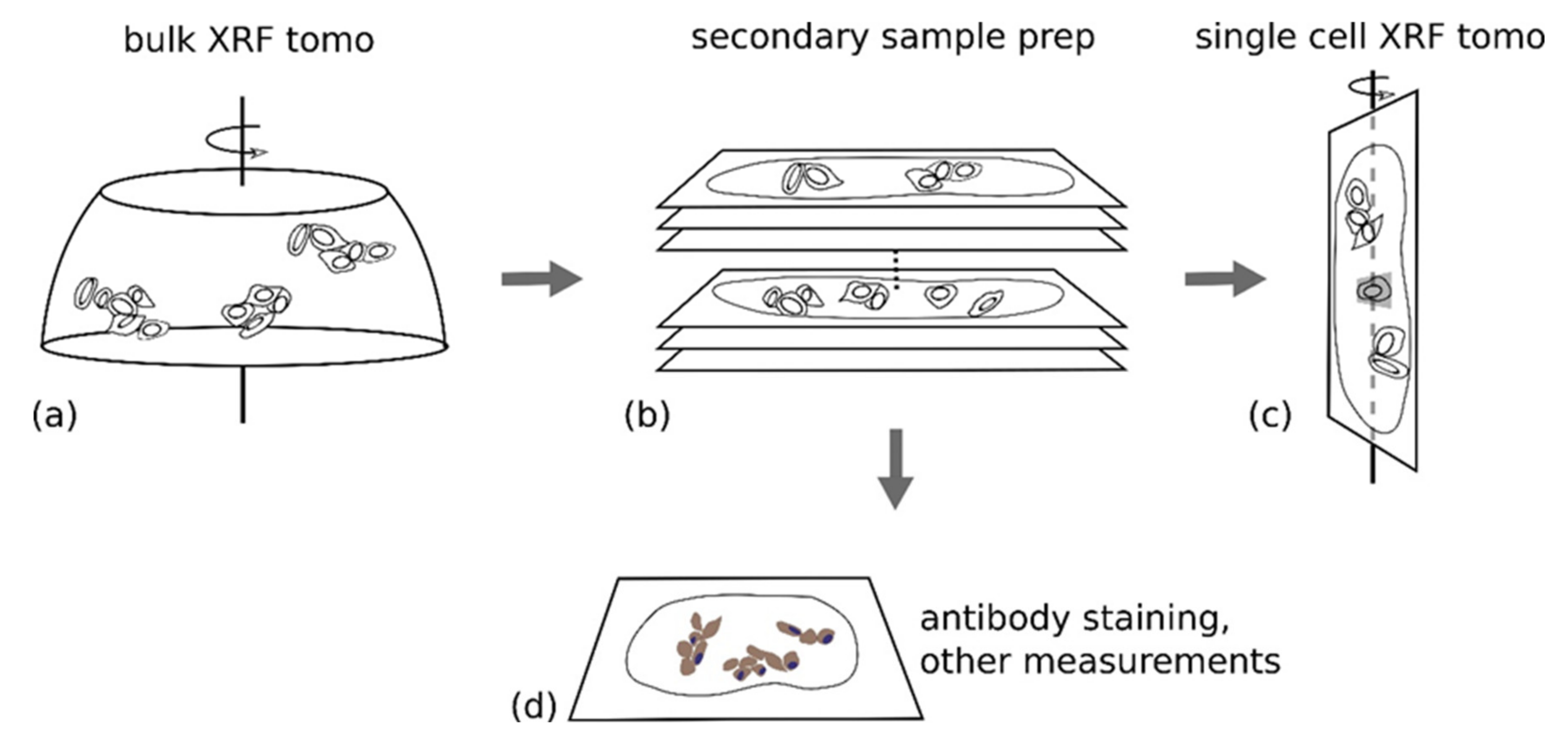
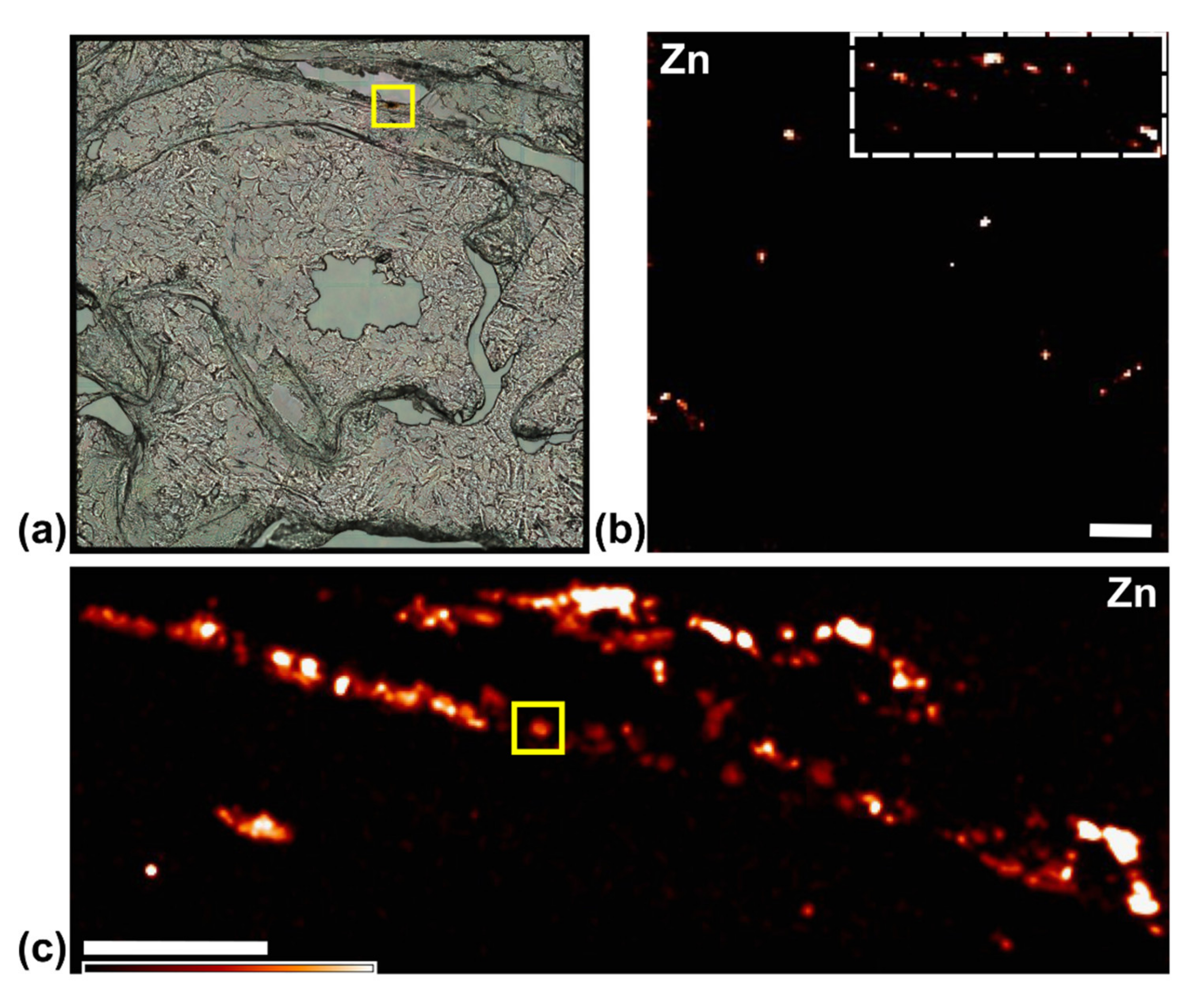
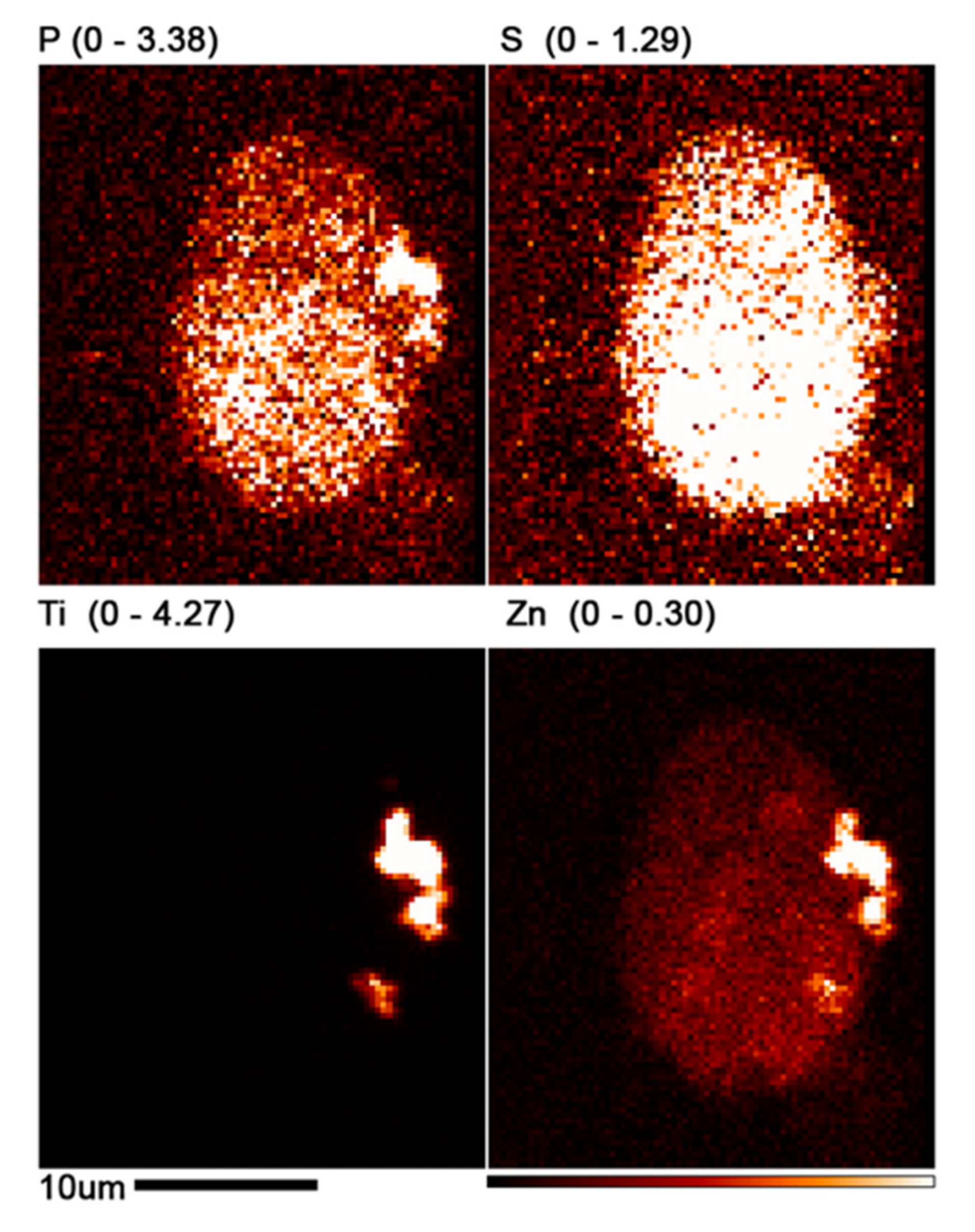
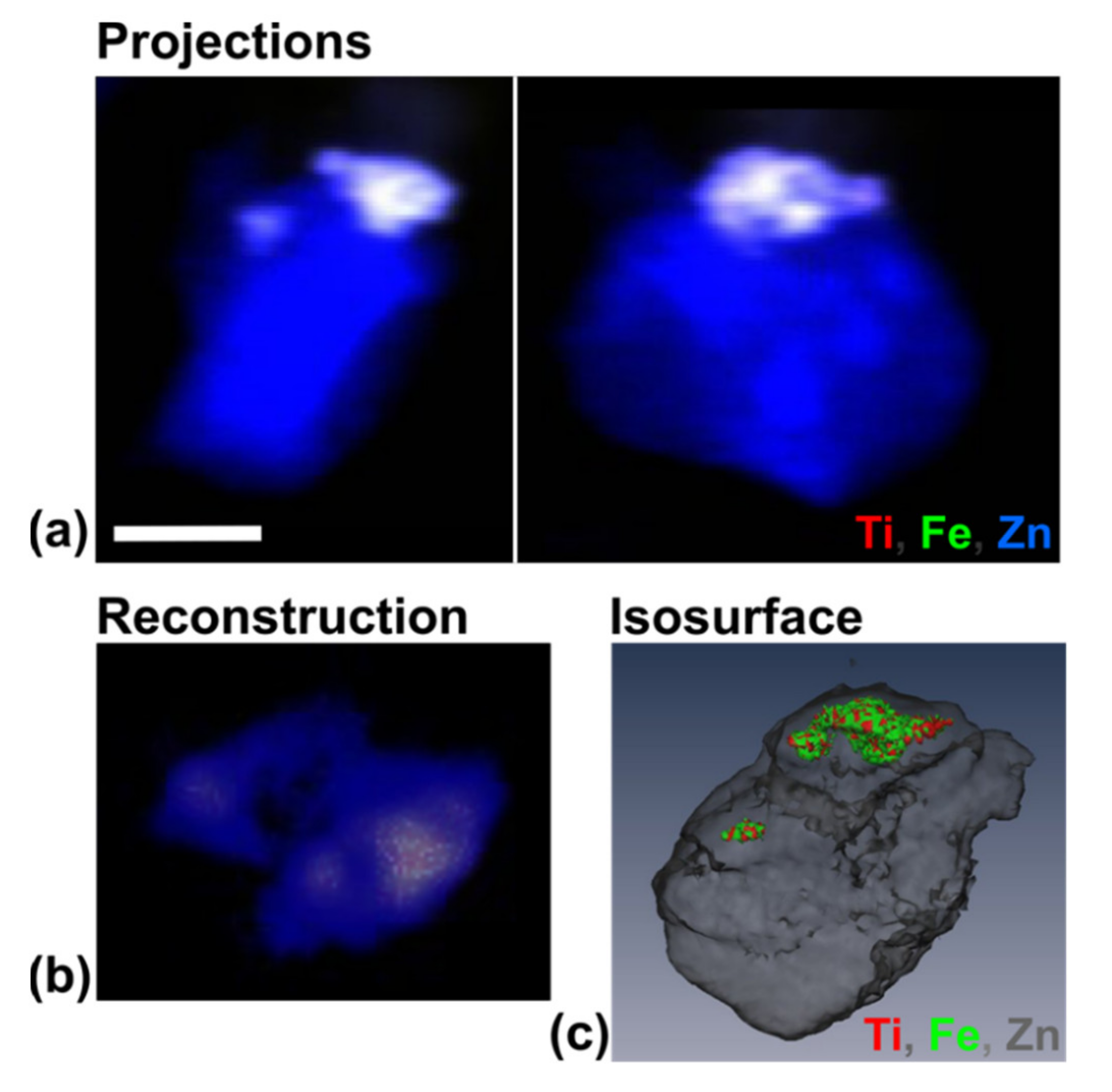
3.4. Use of X-ray Fluorescence Tomography to Evaluate Heterogeneity of Nanocomposite Distribution in Cell Clusters and Cells by “Low-Resolution Tomography” and “High-Resolution Tomography”
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Papademetriou, I.; Tsinas, Z.; Hsu, J.; Muro, S. Combination-targeting to multiple endothelial cell adhesion molecules modulates binding, endocytosis, and in vivo biodistribution of drug nanocarriers and their therapeutic cargoes. J. Control. Release 2014, 188, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Chen, S.; Paunesku, T.; Gleber, S.C.; Liu, W.C.; Doty, C.B.; Mak, R.; Deng, J.; Jin, Q.; Lai, B.; et al. Epidermal growth factor receptor targeted nuclear delivery and high-resolution whole cell X-ray imaging of Fe3O4@TiO2 nanoparticles in cancer cells. ACS Nano 2013, 7, 10502–10517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumková, J.; Smutná, T.; Vrlíková, L.; Le Coustumer, P.; Večeřa, Z.; Dočekal, B.; Mikuška, P.; Čapka, L.; Fictum, P.; Hampl, A.; et al. Sub-chronic inhalation of lead oxide nanoparticles revealed their broad distribution and tissue-specific subcellular localization in target organs. Part. Fibre Toxicol. 2017, 14, 55. [Google Scholar] [CrossRef] [Green Version]
- Fraga, S.; Faria, H.; Soares, M.E.; Duarte, J.A.; Soares, L.; Pereira, E.; Costa-Pereira, C.; Teixeira, J.P.; de Lourdes Bastos, M.; Carmo, H. Influence of the surface coating on the cytotoxicity, genotoxicity and uptake of gold nanoparticles in human HepG2 cells. J. Appl. Toxicol. 2013, 33, 1111–1119. [Google Scholar] [CrossRef]
- Thurn, K.T.; Arora, H.; Paunesku, T.; Wu, A.; Brown, E.M.; Doty, C.; Kremer, J.; Woloschak, G. Endocytosis of titanium dioxide nanoparticles in prostate cancer PC-3M cells. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastra, R.; Paunesku, T.; Gutama, B.; Reyes, F., Jr.; François, J.; Martinez, S.; Xin, L.; Brown, K.; Zander, A.; Raha, S.; et al. Protein binding effects of dopamine coated titanium dioxide shell nanoparticles. Precis. Nanomed. 2019, 2, 393–438. [Google Scholar] [CrossRef]
- Falaschetti, C.A.; Paunesku, T.; Kurepa, J.; Nanavati, D.; Chou, S.S.; De, M.; Song, M.; Jang, J.T.; Wu, A.; Dravid, V.P.; et al. Negatively charged metal oxide nanoparticles interact with the 20S proteasome and differentially modulate its biologic functional effects. ACS Nano 2013, 7, 7759–7772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paunesku, T.; Gutiontov, S.; Brown, K.; Woloschak, G.E. Radiosensitization and nanoparticles. Cancer Treat. Res. 2015, 166, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Arora, H.C.; Jensen, M.P.; Yuan, Y.; Wu, A.; Vogt, S.; Paunesku, T.; Woloschak, G.E. Nanocarriers enhance Doxorubicin uptake in drug-resistant ovarian cancer cells. Cancer Res. 2012, 72, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourassa, D.; Gleber, S.C.; Vogt, S.; Yi, H.; Will, F.; Richter, H.; Shin, C.H.; Fahrni, C.J. 3D imaging of transition metals in the zebrafish embryo by X-ray fluorescence microtomography. Metallomics 2014, 6, 1648–1655. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.A.; Punshon, T.; Lanzirotti, A.; Li, L.; Alonso, J.M.; Ecker, J.R.; Kaplan, J.; Guerinot, M.L. Localization of iron in Arabidopsis seed requires the vacuolar membrane transporter VIT1. Science 2006, 314, 1295–1298. [Google Scholar] [CrossRef]
- McColl, G.; James, S.A.; Mayo, S.; Howard, D.L.; Ryan, C.G.; Kirkham, R.; Moorhead, G.F.; Paterson, D.; De Jonge, M.D.; Bush, A.I. Caenorhabditis elegans maintains highly compartmentalized cellular distribution of metals and steep concentration gradients of manganese. PLoS ONE 2012, 7, e32685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonge, M.D.; Holzner, C.; Baines, S.B.; Twining, B.S.; Ignatyev, K.; Diaz, J.; Howard, D.L.; Legnini, D.; Miceli, A.; McNulty, I.; et al. Quantitative 3D elemental microtomography of Cyclotella meneghiniana at 400-nm resolution. Proc. Natl. Acad. Sci. USA 2010, 107, 15676–15680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Que, E.L.; Bleher, R.; Duncan, F.E.; Kong, B.Y.; Gleber, S.C.; Vogt, S.; Chen, S.; Garwin, S.A.; Bayer, A.R.; Dravid, V.P.; et al. Quantitative mapping of zinc fluxes in the mammalian egg reveals the origin of fertilization-induced zinc sparks. Nat. Chem. 2015, 7, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Victor, T.W.; O’Toole, K.H.; Easthon, L.M.; Ge, M.; Smith, R.J.; Huang, X.; Yan, H.; Chu, Y.S.; Chen, S.; Gursoy, D.; et al. Lanthanide-binding tags for 3D X-ray imaging of proteins in cells at nanoscale resolution. J. Am. Chem. Soc. 2020, 142, 2145–2149. [Google Scholar] [CrossRef] [PubMed]
- Victor, T.W.; Easthon, L.M.; Ge, M.; O’Toole, K.H.; Smith, R.J.; Huang, X.; Yan, H.; Allen, K.N.; Chu, Y.S.; Miller, L.M. X-ray fluorescence nanotomography of single bacteria with a sub-15 nm beam. Sci. Rep. 2015, 8, 13415. [Google Scholar] [CrossRef] [Green Version]
- Fus, F.; Yang, Y.; Lee, H.Z.S.; Top, S.; Carriere, M.; Bouron, A.; Pacureanu, A.; Silva, J.C.A.; Salmain, M.; Vessières, A.; et al. Intracellular localization of an osmocenyl-tamoxifen derivative in breast cancer cells revealed by synchrotron radiation X-ray fluorescence nanoimaging. Angew. Chem. 2019, 131, 3499–3503. [Google Scholar] [CrossRef]
- Holmberg, J.P.; Ahlberg, E.; Bergenholtz, J.; Hassellov, M.; Abbas, Z. Surface charge and interfacial potential of titanium dioxide nanoparticles: Experimental and theoretical investigations. J. Colloid Interface Sci. 2013, 407, 168–176. [Google Scholar] [CrossRef]
- Abbas, Z.; Holmberg, J.P.; Hellstrom, A.K.; Hagstrom, M.; Bergenholtz, J.; Hassellov, M.; Ahlberg, E. Synthesis, characterization and particle size distribution of TiO2 colloidal nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 2011, 384, 254–261. [Google Scholar] [CrossRef]
- Paunesku, T.; Rajh, T.; Wiederrecht, G.; Maser, J.; Vogt, S.; Stojicevic, N.; Protic, M.; Lai, B.; Oryhon, J.; Thurnauer, M.; et al. Biology of TiO2-oligonucleotide nanocomposites. Nat. Mater. 2003, 2, 343–346. [Google Scholar] [CrossRef]
- Rajh, T.; Chen, L.X.; Lukas, K.; Liu, T.; Thurnauer, M.C.; Tiede, D.M. Surface restructuring of nanoparticles: An efficient route for ligand−metal oxide crosstalk. J. Phys. Chem. B 2002, 106, 10543–10552. [Google Scholar] [CrossRef]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Vogt, S. MAPS: A set of software tools for analysis and visualization of 3D X-ray fluorescence data sets. J. Phys. IV France 2003, 104, 635–638. [Google Scholar] [CrossRef]
- Hong, Y.P.; Chen, S.; Jacobsen, C. A new workflow for X-ray fluorescence tomography: MapstoTomoPy. Proc. SPIE Int. Soc. Opt. Eng. 2015, 9592, 9592W. [Google Scholar]
- Marin, F.; Chen, S.; De Carlo, F.; Glowacki, A.; Antipova, O.; Li, L.X. Processing X-ray fluorescence tomography data with XRFtomo. Microsc. Microanal. 2020, 26 (Suppl. 2), 1282–1284. [Google Scholar] [CrossRef]
- Paunesku, T.; Vogt, S.; Lai, B.; Maser, J.; Stojicevic, N.; Thurn, K.T.; Osipo, C.; Liu, H.; Legnini, D.; Wang, Z.; et al. Intracellular distribution of TiO2-DNA oligonucleotide nanoconjugates directed to nucleolus and mitochondria indicates sequence specificity. Nano Lett. 2007, 7, 596–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refaat, T.; West, D.; El Achy, S.; Parimi, V.; May, J.; Xin, L.; Harris, K.R.; Liu, W.; Wanzer, M.B.; Finney, L.; et al. Distribution of iron oxide core-titanium dioxide shell nanoparticles in VX2 tumor bearing rabbits introduced by two different delivery modalities. Nanomaterials 2016, 6, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darzynkiewicz, Z.; Bedner, E.; Smolewski, P. Flow cytometry in analysis of cell cycle and apoptosis. Semin. Hematol. 2001, 38, 179–193. [Google Scholar] [CrossRef]
- Adida, C.; Crotty, P.L.; McGrath, J.; Berrebi, D.; Diebold, J.; Altieri, D.C. Developmentally regulated expression of the novel cancer anti-apoptosis gene Survivin in human and mouse differentiation. Am. J. Pathol. 1998, 152, 43–49. [Google Scholar]
- Uren, A.G.; Wong, L.; Pakusch, M.; Fowler, K.J.; Burrows, F.J.; Vaux, D.L.; Choo, K.H. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr. Biol. 2000, 10, 1319–1328. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Ackermann, E.J.; Bennett, C.F.; Rothermel, A.L.; Plescia, J.; Tognin, S.; Villa, A.; Marchisio, P.C.; Altieri, D.C. Pleiotropic cell-division defects and apoptosis induced by interference with Survivin function. Nat. Cell Biol. 1999, 1, 461–466. [Google Scholar] [CrossRef]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by Survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef]
- Altieri, D.C. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003, 22, 8581–8589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, S.; Pelus, L.M. Survivin, a cancer target with an emerging role in normal adult tissues. Mol. Cancer Ther. 2006, 5, 1087–1098. [Google Scholar] [CrossRef] [Green Version]
- LaCasse, E.C.; Baird, S.; Korneluk, R.G.; MacKenzie, A.E. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 1998, 17, 3247–3259. [Google Scholar] [CrossRef] [Green Version]
- Pavlyukov, M.S.; Antipova, N.V.; Balashova, M.V.; Vinogradova, T.V.; Kopantzev, E.P.; Shakhparonov, M.I. Survivin monomer plays an essential role in apoptosis regulation. J. Biol. Chem. 2011, 286, 23296–23307. [Google Scholar] [CrossRef] [Green Version]
- Tsuneki, M.; Madri, J.A. CD44 regulation of endothelial cell proliferation and apoptosis via modulation of CD31 and VE-cadherin expression. J. Biol. Chem. 2014, 289, 5357–5370. [Google Scholar] [CrossRef] [Green Version]
- Tsuneki, M.; Madri, J.A. CD44 influences fibroblast behaviors via modulation of cell-cell and cell-matrix interactions, affecting Survivin and hippo pathways. J. Cell. Physiol. 2016, 231, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, B.; Pellegrino, M.; Baldini, F. Human dermal fibroblasts HDFa can be used as an appropriate healthy control for PMMA nanoparticles-Survivin molecular beacon cellular uptake studies. Biomed. Pharmacother. 2015, 69, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Fortugno, P.; Wall, N.R.; Giodini, A.; O’Connor, D.S.; Plescia, J.; Padgett, K.M.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Survivin exists in immunochemically distinct subcellular pools and is involved in spindle microtubule function. J. Cell Sci. 2002, 115, 575–585. [Google Scholar] [CrossRef]
- Brown, K.; Thurn, T.; Xin, L.; Liu, W.; Bazak, R.; Chen, S.; Lai, B.; Vogt, S.; Jacobsen, C.; Paunesku, T.; et al. Intracellular in situ labeling of TiO2 nanoparticles for fluorescence microscopy detection. Nano Res. 2018, 11, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somogyi, A.; Medjoubi, K.; Baranton, G.; Le Roux, V.; Ribbens, M.; Polack, F.; Philippot, P.; Samama, J.P. Optical design and multi-length-scale scanning spectro-microscopy possibilities at the Nanoscopium beamline of Synchrotron Soleil. J. Synchrotron Radiat. 2015, 22, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Zijno, A.; De Angelis, I.; De Berardis, B.; Andreoli, C.; Russo, M.T.; Pietraforte, D.; Scorza, G.; Degan, P.; Ponti, J.; Rossi, F.; et al. Different mechanisms are involved in oxidative DNA damage and genotoxicity induction by ZnO and TiO2 nanoparticles in human colon carcinoma cells. Toxicol. Vitr. 2015, 29, 1503–1512. [Google Scholar] [CrossRef]
- Fortugno, P.; Beltrami, E.; Plescia, J.; Fontana, J.; Pradhan, D.; Marchisio, P.C.; Sessa, W.C.; Altieri, D.C. Regulation of Survivin function by Hsp90. Proc. Natl. Acad. Sci. USA 2003, 100, 13791–13796. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wu, W.; Tahir, S.K.; Kroeger, P.E.; Rosenberg, S.H.; Cowsert, L.M.; Bennett, F.; Krajewski, S.; Krajewska, M.; Welsh, K.; et al. Down-regulation of Survivin by antisense oligonucleotides increases apoptosis, inhibits cytokinesis and anchorage-independent growth. Neoplasia 2000, 2, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abad, M.A.; Ruppert, J.G.; Buzuk, L.; Wear, M.; Zou, J.; Webb, K.M.; Kelly, D.A.; Voigt, P.; Rappsilber, J.; Earnshaw, W.C.; et al. Borealin-nucleosome interaction secures chromosome association of the chromosomal passenger complex. J. Cell Biol. 2019, 218, 3912–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosini, G.; Adida, C.; Sirugo, G.; Altieri, D.C. Induction of apoptosis and inhibition of cell proliferation by Survivin gene targeting. J. Biol. Chem. 1998, 273, 11177–11182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beghein, E.; Van Audenhove, I.; Zwaenepoel, O.; Verhelle, A.; De Ganck, A.; Gettemans, J. A new Survivin tracer tracks, delocalizes and captures endogenous Survivin at different subcellular locations and in distinct organelles. Sci. Rep. 2016, 6, 31177. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.; Carmena, M.; Sambade, C.; Earnshaw, W.C.; Wheatley, S.P. Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J. Cell Sci. 2003, 116, 2987–2998. [Google Scholar] [CrossRef] [Green Version]
- Dohi, T.; Beltrami, E.; Wall, N.R.; Plescia, J.; Altieri, D.C. Mitochondrial Survivin inhibits apoptosis and promotes tumorigenesis. J. Clin. Investig. 2004, 114, 1117–1127. [Google Scholar] [CrossRef]
- Dunajová, L.; Cash, E.; Markus, R.; Rochette, S.; Townley, A.R.; Wheatley, S.P. The N-terminus of Survivin is a mitochondrial-targeting sequence and Src regulator. J. Cell Sci. 2016, 129, 2707–2712. [Google Scholar] [CrossRef] [Green Version]
- Giodini, A.; Kallio, M.J.; Wall, N.R.; Gorbsky, G.J.; Tognin, S.; Marchisio, P.C.; Symons, M.; Altieri, D.C. Regulation of microtubule stability and mitotic progression by Survivin. Cancer Res. 2002, 62, 2462–2467. [Google Scholar]
- Han, T.L.; Sha, H.; Ji, J.; Li, Y.T.; Wu, D.S.; Lin, H.; Hu, B.; Jiang, Z.X. Depletion of Survivin suppresses docetaxel-induced apoptosis in HeLa cells by facilitating mitotic slippage. Sci. Rep. 2021, 11, 2283. [Google Scholar] [CrossRef]
- Hao, F.; Lee, R.J.; Yang, C.; Zhong, L.; Sun, Y.; Dong, S.; Cheng, Z.; Teng, L.; Meng, Q.; Lu, J.; et al. Targeted Co-delivery of siRNA and methotrexate for tumor therapy via mixed micelles. Pharmaceutics 2019, 11, 92. [Google Scholar] [CrossRef] [Green Version]
- Kallio, M.J.; Nieminen, M.; Eriksson, J.E. Human inhibitor of apoptosis protein (IAP) Survivin participates in regulation of chromosome segregation and mitotic exit. FASEB J. 2001, 15, 2721–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Jutzy, J.M.; Aspe, J.R.; McGregor, D.W.; Neidigh, J.W.; Wall, N.R. Survivin is released from cancer cells via exosomes. Apoptosis 2011, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lear, T.; Iannone, O.; Shiva, S.; Corey, C.; Rajbhandari, S.; Jerome, J.; Chen, B.B.; Mallampalli, R.K. The proapoptotic F-box protein Fbxl7 regulates mitochondrial function by mediating the ubiquitylation and proteasomal degradation of Survivin. J. Biol. Chem. 2015, 290, 11843–11852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, M.M.; Ferguson Bennit, H.R.; Gonda, A.; Diaz Osterman, C.J.; Hibma, A.; Khan, S.; Wall, N.R. Exosomes secreted from human cancer cell lines contain inhibitors of apoptosis (IAP). Cancer Microenviron. 2015, 8, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vong, Q.P.; Cao, K.; Li, H.Y.; Iglesias, P.A.; Zheng, Y. Chromosome alignment and segregation regulated by ubiquitination of Survivin. Science 2005, 310, 1499–1504. [Google Scholar] [CrossRef]
- Wang, H.; Holloway, M.P.; Ma, L.; Cooper, Z.A.; Riolo, M.; Samkari, A.; Elenitoba-Johnson, K.S.; Chin, Y.E.; Altura, R.A. Acetylation directs Survivin nuclear localization to repress STAT3 oncogenic activity. J. Biol. Chem. 2010, 285, 36129–36137. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, Y.; Honda, T.; Tanno, Y.; Watanabe, Y. Two histone marks establish the inner centromere and chromosome bi-orientation. Science 2010, 330, 239–243. [Google Scholar] [CrossRef]
- AlShamaileh, H.; Wang, T.; Xiang, D.; Yin, W.; Tran, P.H.; Barrero, R.A.; Zhang, P.Z.; Li, Y.; Kong, L.; Liu, K.; et al. Aptamer-mediated Survivin RNAi enables 5-fluorouracil to eliminate colorectal cancer stem cells. Sci. Rep. 2017, 7, 5898. [Google Scholar] [CrossRef] [PubMed]
- Fenstermaker, R.A.; Figel, S.A.; Qiu, J.; Barone, T.A.; Dharma, S.S.; Winograd, E.K.; Galbo, P.M.; Wiltsie, L.M.; Ciesielski, M.J. Survivin monoclonal antibodies detect Survivin cell surface expression and inhibit tumor growth in vivo. Clin. Cancer Res. 2018, 24, 2642–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanhueza, C.; Wehinger, S.; Bennett, J.C.; Valenzuela, M.; Owen, G.I.; Quest, A.F. The twisted Survivin connection to angiogenesis. Mol. Cancer 2015, 14, 198. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, N.; Kanwar, J.R.; Deepa, P.R.; Navaneethakrishnan, S.; Joseph, C.; Krishnakumar, S. Targeting HSP90/Survivin using a cell permeable structure based peptido-mimetic shepherdin in retinoblastoma. Chem. Biol. Interact. 2016, 252, 141–149. [Google Scholar] [CrossRef]
- Warrier, N.M.; Agarwal, P.; Kumar, P. Emerging importance of Survivin in stem cells and cancer: The development of new cancer therapeutics. Stem Cell Rev. Rep. 2020, 16, 828–852. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, C.; Xiong, M.; Yang, Y.; Zhang, Q.; Liu, X.; Zou, B.; Gou, M. Self-assembled nanoparticle mediated Survivin-T34A for ovarian cancer therapy. J. Biomed. Nanotechnol. 2018, 14, 2092–2101. [Google Scholar] [CrossRef]
- Tang, Q.; Efe, G.; Chiarella, A.M.; Leung, J.; Chen, M.; Yamazoe, T.; Su, Z.; Pitarresi, J.R.; Li, J.; Islam, M.; et al. Mutant p53 regulates Survivin to foster lung metastasis. Genes Dev. 2021, 35, 528–541. [Google Scholar] [CrossRef]
- Yusufu, A.; Tuerdi, R.; Redati, D.; Rehemutula, A.; Zhao, Z.L.; Wang, H.J. Expression and clinical correlation of Survivin and PTEN in gastric cancer patients. Oncol. Lett. 2020, 20, 297. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tomography Data Collection Parameters | ||||||
|---|---|---|---|---|---|---|
| Beamline | Incident X-ray Energy (Kev) | Flux (Counts/s) | Dwell/Pixel (ms) | Step Size (mm) | # Projections | Angular Coverage (°) |
| 2-ID-E | 10 | ~109 | 30 | 3 | 60 | 180 |
| Bionanoprobe | 10 | 3 × 109 | 200 | 0.08 | 48 | 141 |
| 2D Scan Parameters | ||||||
| Beamline | Incident X-ray Energy (Kev) | Flux (Counts/s) | Dwell/Pixel (ms) | Step Size (mm) | ||
| 2-ID-E | 10 | ~109 | 50 | 0.3 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Lastra, R.O.; Paunesku, T.; Antipova, O.; Li, L.; Deng, J.; Luo, Y.; Wanzer, M.B.; Popovic, J.; Li, Y.; et al. Development of Multi-Scale X-ray Fluorescence Tomography for Examination of Nanocomposite-Treated Biological Samples. Cancers 2021, 13, 4497. https://doi.org/10.3390/cancers13174497
Chen S, Lastra RO, Paunesku T, Antipova O, Li L, Deng J, Luo Y, Wanzer MB, Popovic J, Li Y, et al. Development of Multi-Scale X-ray Fluorescence Tomography for Examination of Nanocomposite-Treated Biological Samples. Cancers. 2021; 13(17):4497. https://doi.org/10.3390/cancers13174497
Chicago/Turabian StyleChen, Si, Ruben Omar Lastra, Tatjana Paunesku, Olga Antipova, Luxi Li, Junjing Deng, Yanqi Luo, Michael Beau Wanzer, Jelena Popovic, Ya Li, and et al. 2021. "Development of Multi-Scale X-ray Fluorescence Tomography for Examination of Nanocomposite-Treated Biological Samples" Cancers 13, no. 17: 4497. https://doi.org/10.3390/cancers13174497
APA StyleChen, S., Lastra, R. O., Paunesku, T., Antipova, O., Li, L., Deng, J., Luo, Y., Wanzer, M. B., Popovic, J., Li, Y., Glasco, A. D., Jacobsen, C., Vogt, S., & Woloschak, G. E. (2021). Development of Multi-Scale X-ray Fluorescence Tomography for Examination of Nanocomposite-Treated Biological Samples. Cancers, 13(17), 4497. https://doi.org/10.3390/cancers13174497