
Spherical Nucleic Acids as Precision Therapeutics for the Treatment of Cancer—From Bench to Bedside

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
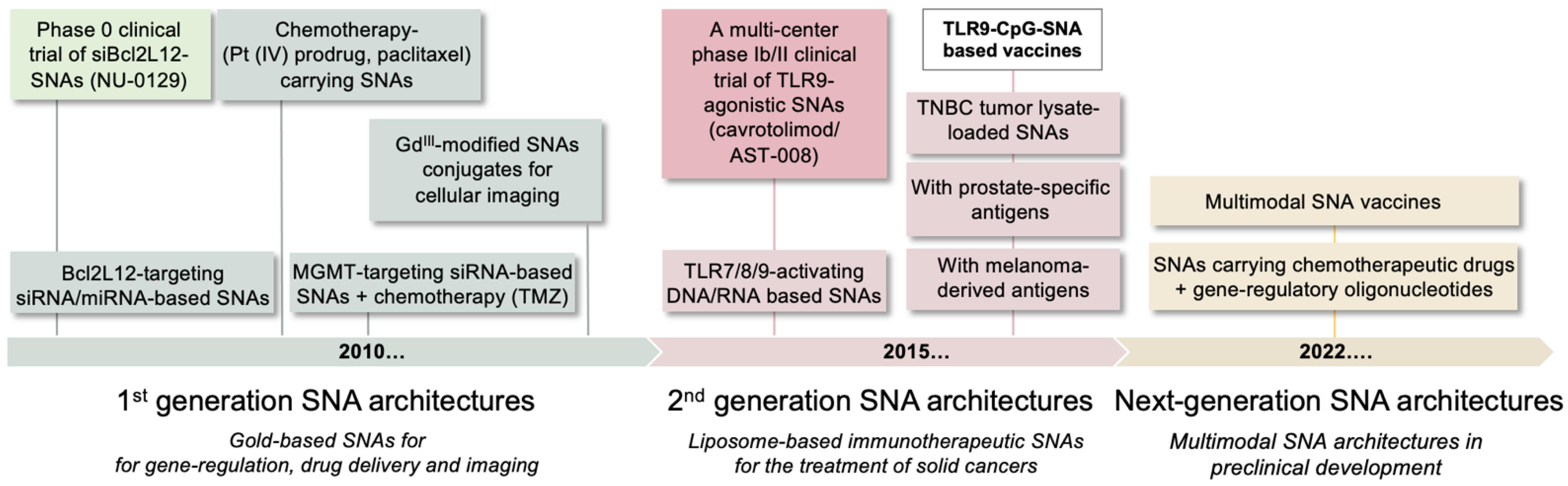
1. Introduction
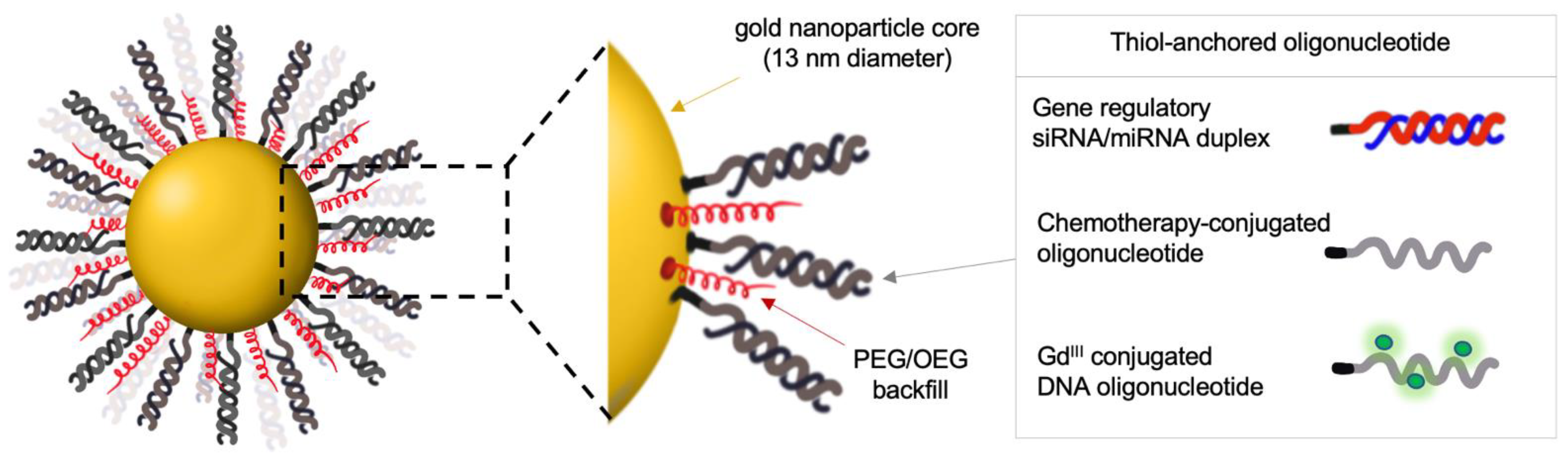
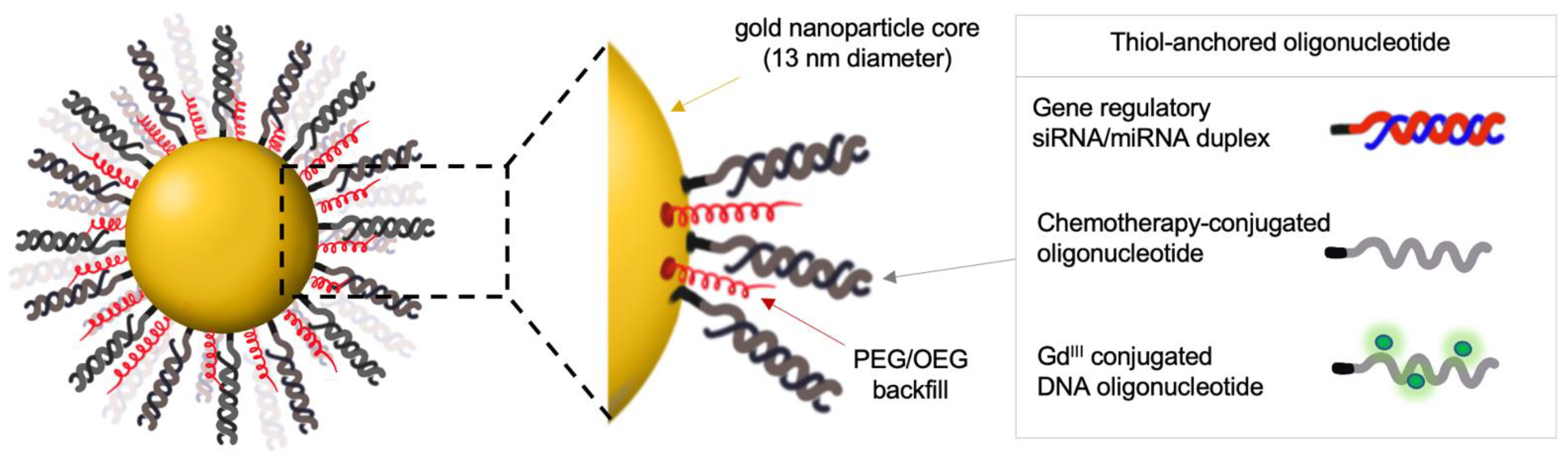
2. Gene-Regulatory SNAs to Reactivate p53 Tumor Suppression in Glioblastoma
2.1. Preclinical Evaluation of Gene-Regulatory SNAs for the Treatment of Glioblastoma
2.2. A Phase 0 Clinical Trial of Bcl2L12-Targeting SNAs in Patients with Recurrent Glioblastoma
3. SNAs as a Novel Immunotherapeutic Modality for Solid Cancers
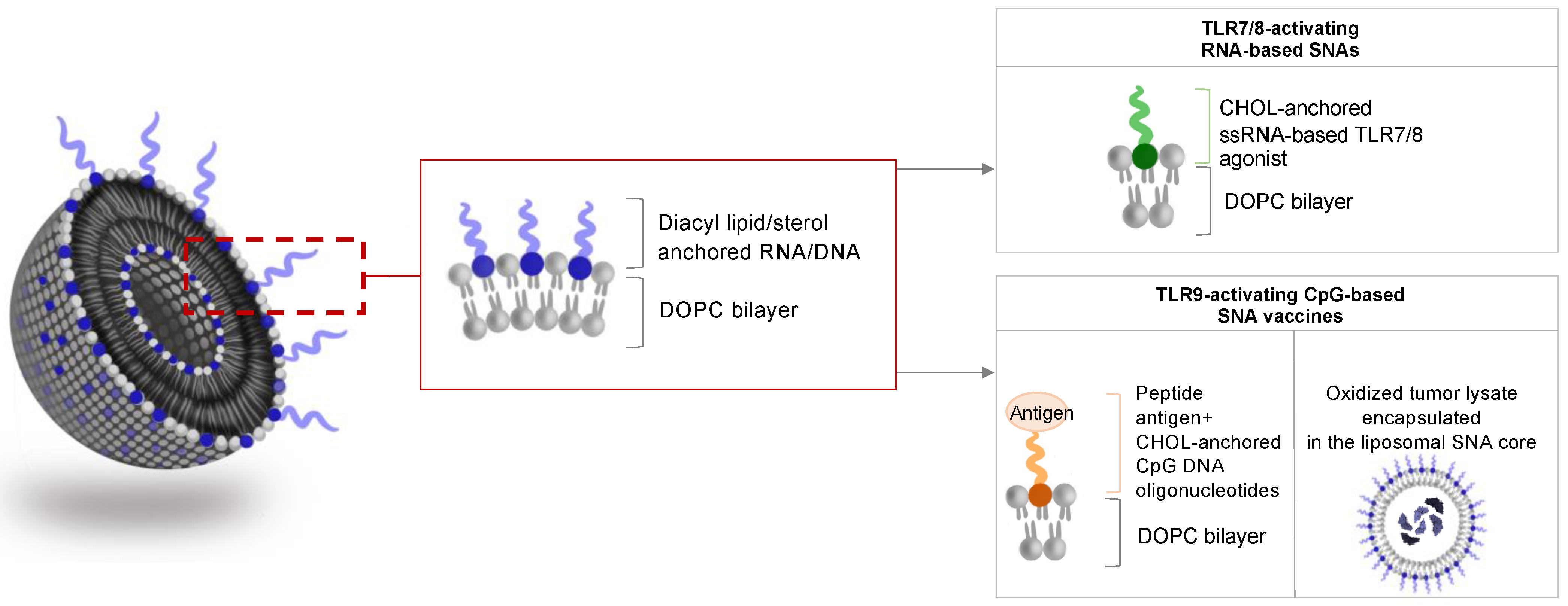
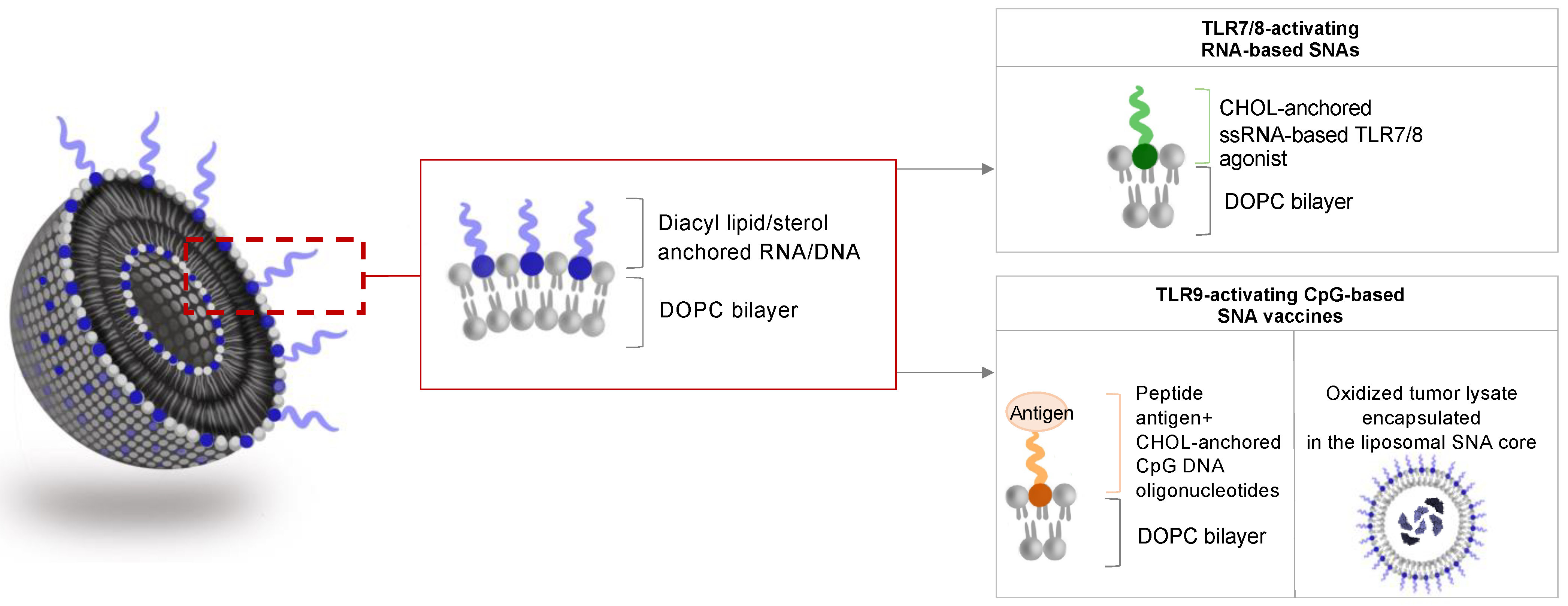
3.1. SNAs to Activate TLR Signaling
3.2. SNA-Based Vaccines Carrying Oligonucleotide Adjuvant and Antigenic Peptide
3.3. The Concept of Rational SNA Vaccinology
3.4. Tumor-Lysate Loaded SNAs as Cancer Vaccines
3.5. High-Throughput Screening to Define SNA Structure-Activity Relationships
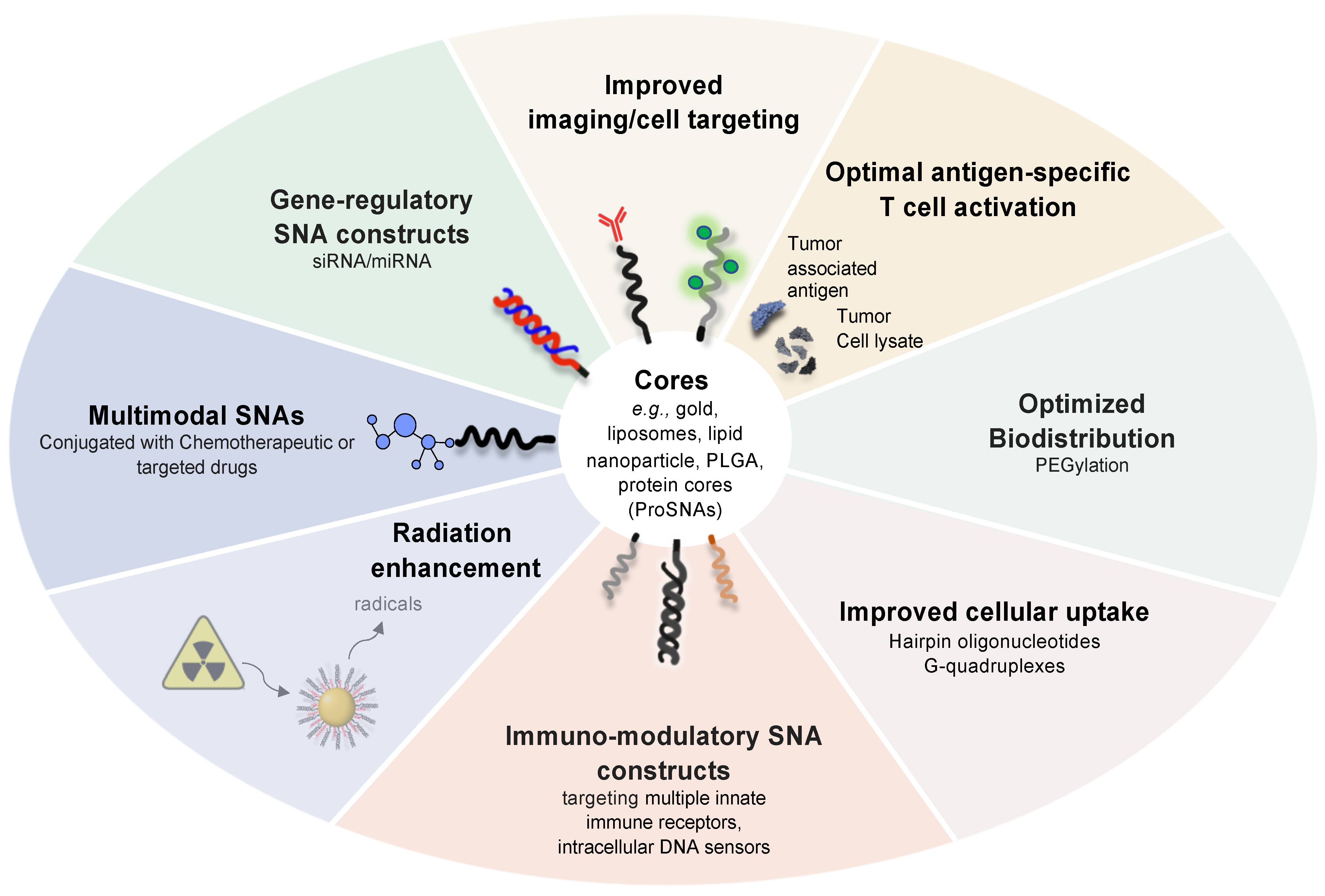
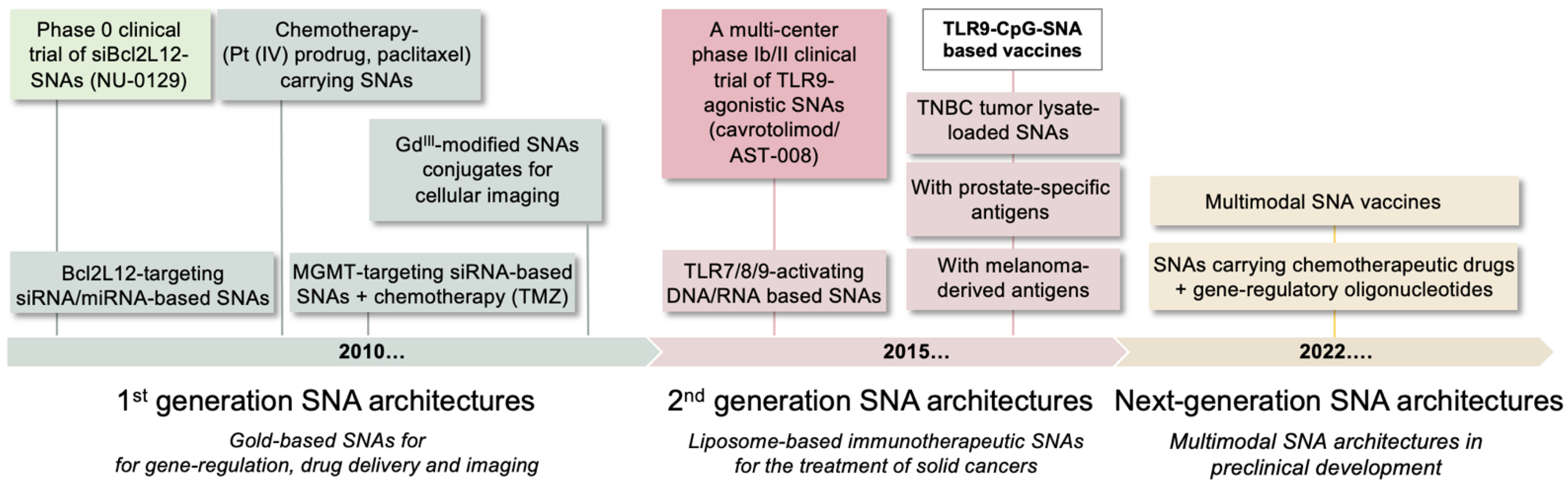
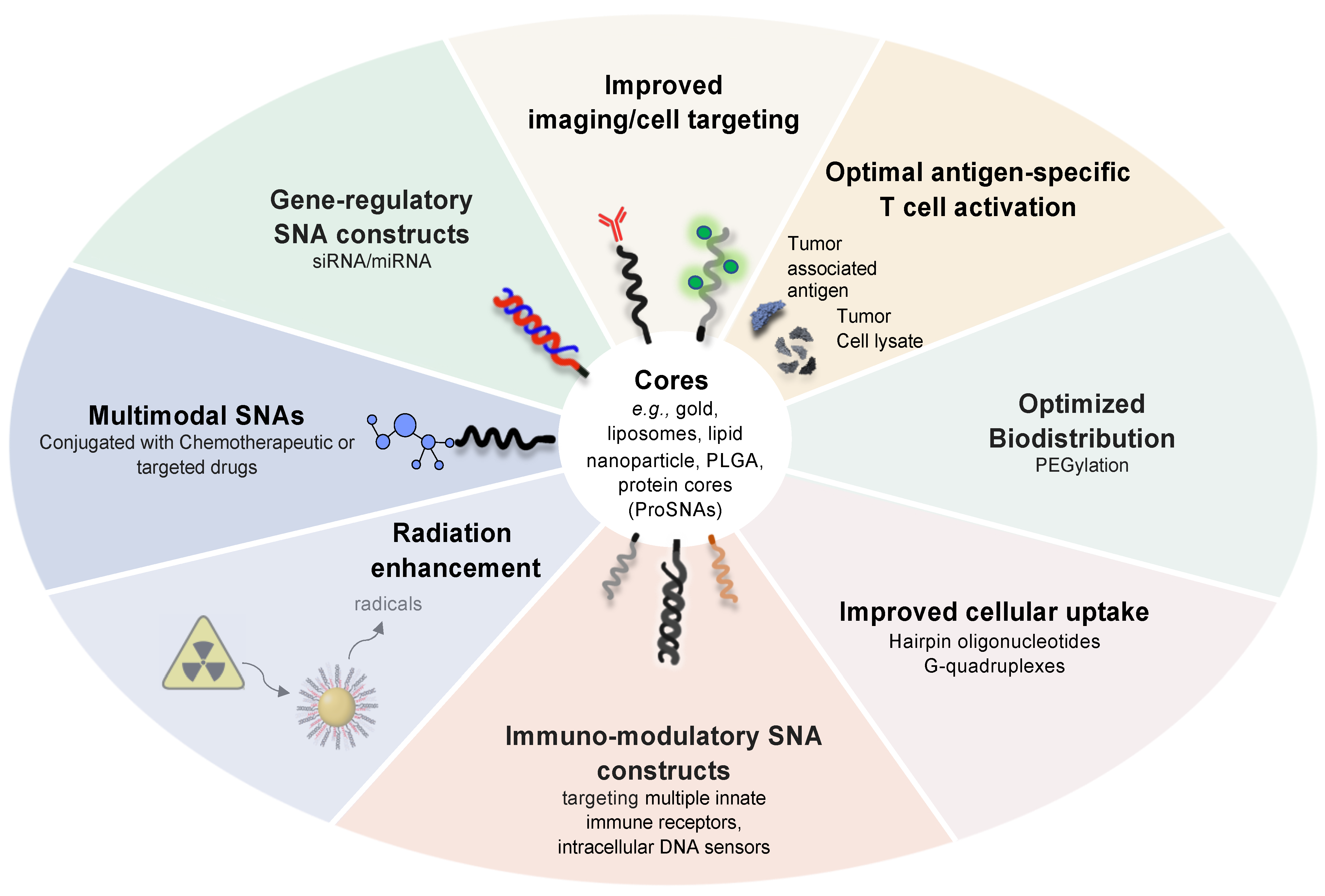
4. Other Multimodal SNA Architectures in Preclinical Development
5. Conclusions and Outlook
6. Patents
Author Contributions
Funding
Conflicts of Interest
References
- Naghizadeh, S.; Mansoori, B.; Mohammadi, A.; Sakhinia, E.; Baradaran, B. Gene silencing strategies in cancer therapy: An update for drug resistance. Curr. Med. Chem. 2019, 26, 6282–6303. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.J.; Zhang, Y. An update on RNA interference-mediated gene silencing in cancer therapy. Expert Opin. Biol. Ther. 2014, 14, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.Q.; Zhang, Y. RNAi-mediated gene silencing in cancer therapy. Expert Opin. Biol. Ther. 2012, 12, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef]
- Stegh, A.H. Toward personalized cancer nanomedicine—Past, present, and future. Integr. Biol. 2013, 5, 48–65. [Google Scholar] [CrossRef]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [Google Scholar] [CrossRef]
- Narayan, S.P.; Choi, C.H.; Hao, L.; Calabrese, C.M.; Auyeung, E.; Zhang, C.; Goor, O.J.; Mirkin, C.A. The Sequence-Specific Cellular Uptake of Spherical Nucleic Acid Nanoparticle Conjugates. Small 2015, 11, 4173–4182. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.H.; Hao, L.; Narayan, S.P.; Auyeung, E.; Mirkin, C.A. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. Proc. Natl. Acad. Sci. USA 2013, 110, 7625–7630. [Google Scholar] [CrossRef] [Green Version]
- Chinen, A.B.; Guan, C.M.; Ko, C.H.; Mirkin, C.A. The Impact of Protein Corona Formation on the Macrophage Cellular Uptake and Biodistribution of Spherical Nucleic Acids. Small 2017, 13, 1603847. [Google Scholar] [CrossRef] [Green Version]
- Chinen, A.B.; Ferrer, J.R.; Merkel, T.J.; Mirkin, C.A. Relationships between Poly(ethylene glycol) Modifications on RNA-Spherical Nucleic Acid Conjugates and Cellular Uptake and Circulation Time. Bioconjug. Chem. 2016, 27, 2715–2721. [Google Scholar] [CrossRef]
- Seferos, D.S.; Giljohann, D.A.; Rosi, N.L.; Mirkin, C.A. Locked nucleic acid-nanoparticle conjugates. Chembiochem 2007, 8, 1230–1232. [Google Scholar] [CrossRef] [PubMed]
- Pokorski, J.K.; Nam, J.M.; Vega, R.A.; Mirkin, C.A.; Appella, D.H. Cyclopentane-modified PNA improves the sensitivity of nanoparticle-based scanometric DNA detection. Chem. Commun. 2005, 2005, 2101–2103. [Google Scholar] [CrossRef] [PubMed]
- Giljohann, D.A.; Seferos, D.S.; Prigodich, A.E.; Patel, P.C.; Mirkin, C.A. Gene regulation with polyvalent siRNA-nanoparticle conjugates. J. Am. Chem. Soc. 2009, 131, 2072–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, S.A.; Day, E.S.; Ko, C.H.; Hurley, L.A.; Luciano, J.P.; Kouri, F.M.; Merkel, T.J.; Luthi, A.J.; Patel, P.C.; Cutler, J.I.; et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci. Transl. Med. 2013, 5, 209ra152. [Google Scholar] [CrossRef] [Green Version]
- Randeria, P.S.; Seeger, M.A.; Wang, X.Q.; Wilson, H.; Shipp, D.; Mirkin, C.A.; Paller, A.S. siRNA-based spherical nucleic acids reverse impaired wound healing in diabetic mice by ganglioside GM3 synthase knockdown. Proc. Natl. Acad. Sci. USA 2015, 112, 5573–5578. [Google Scholar] [CrossRef] [Green Version]
- Sita, T.L.; Kouri, F.M.; Hurley, L.A.; Merkel, T.J.; Chalastanis, A.; May, J.L.; Ghelfi, S.T.; Cole, L.E.; Cayton, T.C.; Barnaby, S.N.; et al. Dual bioluminescence and near-infrared fluorescence monitoring to evaluate spherical nucleic acid nanoconjugate activity in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, 4129–4134. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Patel, P.C.; Alhasan, A.H.; Giljohann, D.A.; Mirkin, C.A. Nucleic acid-gold nanoparticle conjugates as mimics of microRNA. Small 2011, 7, 3158–3162. [Google Scholar] [CrossRef]
- Kouri, F.M.; Hurley, L.A.; Daniel, W.L.; Day, E.S.; Hua, Y.; Hao, L.; Peng, C.Y.; Merkel, T.J.; Queisser, M.A.; Ritner, C.; et al. miR-182 integrates apoptosis, growth, and differentiation programs in glioblastoma. Genes Dev. 2015, 29, 732–745. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hao, L.; Bu, H.F.; Scott, A.W.; Tian, K.; Liu, F.; De Plaen, I.G.; Liu, Y.; Mirkin, C.A.; Tan, X.D. Spherical nucleic acid targeting microRNA-99b enhances intestinal MFG-E8 gene expression and restores enterocyte migration in lipopolysaccharide-induced septic mice. Sci. Rep. 2016, 6, 31687. [Google Scholar] [CrossRef] [Green Version]
- Barnaby, S.N.; Thaner, R.V.; Ross, M.B.; Brown, K.A.; Schatz, G.C.; Mirkin, C.A. Modular and Chemically Responsive Oligonucleotide “Bonds” in Nanoparticle Superlattices. J. Am. Chem. Soc. 2015, 137, 13566–13571. [Google Scholar] [CrossRef] [Green Version]
- Rouge, J.L.; Sita, T.L.; Hao, L.; Kouri, F.M.; Briley, W.E.; Stegh, A.H.; Mirkin, C.A. Ribozyme-Spherical Nucleic Acids. J. Am. Chem. Soc. 2015, 137, 10528–10531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Lytton-Jean, A.K.; Hurst, S.J.; Mirkin, C.A. Silver nanoparticle-oligonucleotide conjugates based on DNA with triple cyclic disulfide moieties. Nano Lett. 2007, 7, 2112–2115. [Google Scholar] [CrossRef] [Green Version]
- Cutler, J.I.; Zheng, D.; Xu, X.; Giljohann, D.A.; Mirkin, C.A. Polyvalent oligonucleotide iron oxide nanoparticle “click” conjugates. Nano Lett. 2010, 10, 1477–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Macfarlane, R.J.; Young, K.L.; Choi, C.H.; Hao, L.; Auyeung, E.; Liu, G.; Zhou, X.; Mirkin, C.A. A general approach to DNA-programmable atom equivalents. Nat. Mater. 2013, 12, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Young, K.L.; Scott, A.W.; Hao, L.; Mirkin, S.E.; Liu, G.; Mirkin, C.A. Hollow spherical nucleic acids for intracellular gene regulation based upon biocompatible silica shells. Nano Lett. 2012, 12, 3867–3871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banga, R.J.; Chernyak, N.; Narayan, S.P.; Nguyen, S.T.; Mirkin, C.A. Liposomal spherical nucleic acids. J. Am. Chem. Soc. 2014, 136, 9866–9869. [Google Scholar] [CrossRef]
- Sinegra, A.J.; Evangelopoulos, M.; Park, J.; Huang, Z.; Mirkin, C.A. Lipid Nanoparticle Spherical Nucleic Acids for Intracellular DNA and RNA Delivery. Nano Lett. 2021, 21, 6584–6591. [Google Scholar] [CrossRef]
- Zhu, S.; Xing, H.; Gordiichuk, P.; Park, J.; Mirkin, C.A. PLGA Spherical Nucleic Acids. Adv. Mater 2018, 30, e1707113. [Google Scholar] [CrossRef]
- Banga, R.J.; Meckes, B.; Narayan, S.P.; Sprangers, A.J.; Nguyen, S.T.; Mirkin, C.A. Cross-Linked Micellar Spherical Nucleic Acids from Thermoresponsive Templates. J. Am. Chem. Soc. 2017, 139, 4278–4281. [Google Scholar] [CrossRef]
- Ebrahimi, S.B.; Samanta, D.; Kusmierz, C.D.; Mirkin, C.A. Protein transfection via spherical nucleic acids. Nat. Protoc. 2022, 17, 327–357. [Google Scholar] [CrossRef]
- Li, H.; Zhang, B.; Lu, X.; Tan, X.; Jia, F.; Xiao, Y.; Cheng, Z.; Li, Y.; Silva, D.O.; Schrekker, H.S.; et al. Molecular spherical nucleic acids. Proc. Natl. Acad. Sci. USA 2018, 115, 4340–4344. [Google Scholar] [CrossRef] [Green Version]
- Barnaby, S.N.; Sita, T.L.; Petrosko, S.H.; Stegh, A.H.; Mirkin, C.A. Therapeutic applications of spherical nucleic acids. Cancer Treat. Res. 2015, 166, 23–50. [Google Scholar] [PubMed]
- Zheng, D.; Giljohann, D.A.; Chen, D.L.; Massich, M.D.; Wang, X.Q.; Iordanov, H.; Mirkin, C.A.; Paller, A.S. Topical delivery of siRNA-based spherical nucleic acid nanoparticle conjugates for gene regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 11975–11980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprangers, A.J.; Hao, L.; Banga, R.J.; Mirkin, C.A. Liposomal Spherical Nucleic Acids for Regulating Long Noncoding RNAs in the Nucleus. Small 2017, 13, 1602753. [Google Scholar] [CrossRef] [PubMed]
- Yamankurt, G.; Stawicki, R.J.; Posadas, D.M.; Nguyen, J.Q.; Carthew, R.W.; Mirkin, C.A. The effector mechanism of siRNA spherical nucleic acids. Proc. Natl. Acad. Sci. USA 2020, 117, 1312–1320. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Schultz, N.; Mischel, P.S.; Cloughesy, T.F. Will kinase inhibitors make it as glioblastoma drugs? Curr. Top. Microbiol. Immunol. 2012, 355, 135–169. [Google Scholar]
- Mischel, P.S.; Cloughesy, T.F. Targeted molecular therapy of GBM. Brain Pathol. 2003, 13, 52–61. [Google Scholar] [CrossRef]
- Stegh, A.H.; Brennan, C.; Mahoney, J.A.; Forloney, K.L.; Jenq, H.T.; Luciano, J.P.; Protopopov, A.; Chin, L.; Depinho, R.A. Glioma oncoprotein Bcl2L12 inhibits the p53 tumor suppressor. Genes Dev. 2010, 24, 2194–2204. [Google Scholar] [CrossRef] [Green Version]
- Stegh, A.H.; Chin, L.; Louis, D.N.; DePinho, R.A. What drives intense apoptosis resistance and propensity for necrosis in glioblastoma? A role for Bcl2L12 as a multifunctional cell death regulator. Cell Cycle 2008, 7, 2833–2839. [Google Scholar] [CrossRef]
- Stegh, A.H.; Kesari, S.; Mahoney, J.E.; Jenq, H.T.; Forloney, K.L.; Protopopov, A.; Louis, D.N.; Chin, L.; DePinho, R.A. Bcl2L12-mediated inhibition of effector caspase-3 and caspase-7 via distinct mechanisms in glioblastoma. Proc. Natl. Acad. Sci. USA 2008, 105, 10703–10708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegh, A.H.; Kim, H.; Bachoo, R.M.; Forloney, K.L.; Zhang, J.; Schulze, H.; Park, K.; Hannon, G.J.; Yuan, J.; Louis, D.N.; et al. Bcl2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma. Genes Dev. 2007, 21, 98–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fendri, A.; Kontos, C.K.; Khabir, A.; Mokdad-Gargouri, R.; Scorilas, A. BCL2L12 is a novel biomarker for the prediction of short-term relapse in nasopharyngeal carcinoma. Mol. Med. 2010, 17, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Florou, D.; Papadopoulos, I.N.; Scorilas, A. Molecular analysis and prognostic impact of the novel apoptotic gene BCL2L12 in gastric cancer. Biochem. Biophys. Res. Commun. 2010, 391, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.K.; Papadopoulos, I.N.; Scorilas, A. Quantitative expression analysis and prognostic significance of the novel apoptosis-related gene BCL2L12 in colon cancer. Biol. Chem. 2008, 389, 1467–1475. [Google Scholar] [CrossRef]
- Goti, D.; Hrzenjak, A.; Levak-Frank, S.; Frank, S.; van der Westhuyzen, D.R.; Malle, E.; Sattler, W. Scavenger receptor class B, type I is expressed in porcine brain capillary endothelial cells and contributes to selective uptake of HDL-associated vitamin E. J. Neurochem. 2001, 76, 498–508. [Google Scholar] [CrossRef]
- Mackic, J.B.; Stins, M.; McComb, J.G.; Calero, M.; Ghiso, J.; Kim, K.S.; Yan, S.D.; Stern, D.; Schmidt, A.M.; Frangione, B.; et al. Human blood-brain barrier receptors for Alzheimer’s amyloid-beta 1- 40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J. Clin. Investig. 1998, 102, 734–743. [Google Scholar] [CrossRef]
- Kumthekar, P.; Ko, C.H.; Paunesku, T.; Dixit, K.; Sonabend, A.M.; Bloch, O.; Tate, M.; Schwartz, M.; Zuckerman, L.; Lezon, R.; et al. A first-in-human phase 0 clinical study of RNA interference-based spherical nucleic acids in patients with recurrent glioblastoma. Sci. Transl. Med. 2021, 13, eabb3945. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Wang, X.; Zhang, D. TLRs as a Promise Target Along With Immune Checkpoint Against Gastric Cancer. Front. Cell Dev. Biol. 2020, 8, 611444. [Google Scholar] [CrossRef]
- Keshavarz, A.; Pourbagheri-Sigaroodi, A.; Zafari, P.; Bagheri, N.; Ghaffari, S.H.; Bashash, D. Toll-like receptors (TLRs) in cancer; with an extensive focus on TLR agonists and antagonists. IUBMB Life 2021, 73, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Pahlavanneshan, S.; Sayadmanesh, A.; Ebrahimiyan, H.; Basiri, M. Toll-Like Receptor-Based Strategies for Cancer Immunotherapy. J. Immunol. Res. 2021, 2021, 9912188. [Google Scholar] [CrossRef] [PubMed]
- Radovic-Moreno, A.F.; Chernyak, N.; Mader, C.C.; Nallagatla, S.; Kang, R.S.; Hao, L.; Walker, D.A.; Halo, T.L.; Merkel, T.J.; Rische, C.H.; et al. Immunomodulatory spherical nucleic acids. Proc. Natl. Acad. Sci. USA 2015, 112, 3892–3897. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Huang, Z.N.; Mirkin, C.A.; Odom, T.W. Endosomal Organization of CpG Constructs Correlates with Enhanced Immune Activation. Nano Lett. 2020, 20, 6170–6175. [Google Scholar] [CrossRef]
- Yue, J.; Pallares, R.M.; Cole, L.E.; Coughlin, E.E.; Mirkin, C.A.; Lee, A.; Odom, T.W. Smaller CpG-Conjugated Gold Nanoconstructs Achieve Higher Targeting Specificity of Immune Activation. ACS Appl. Mater. Interfaces 2018, 10, 21920–21926. [Google Scholar] [CrossRef]
- Meckes, B.; Banga, R.J.; Nguyen, S.T.; Mirkin, C.A. Enhancing the Stability and Immunomodulatory Activity of Liposomal Spherical Nucleic Acids through Lipid-Tail DNA Modifications. Small 2018, 14, 1702909. [Google Scholar] [CrossRef]
- Blazar, B.R.; Krieg, A.M.; Taylor, P.A. Synthetic unmethylated cytosine-phosphate-guanosine oligodeoxynucleotides are potent stimulators of antileukemia responses in naive and bone marrow transplant recipients. Blood 2001, 98, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.N.; Cole, L.E.; Callmann, C.E.; Wang, S.; Mirkin, C.A. Sequence Multiplicity within Spherical Nucleic Acids. ACS Nano 2020, 14, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Chernyak, N.; Dominguez, D.; Cole, L.; Zhang, B.; Mirkin, C.A. RNA-Based Immunostimulatory Liposomal Spherical Nucleic Acids as Potent TLR7/8 Modulators. Small 2019, 15, e1903338. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.N.; Callmann, C.E.; Cole, L.E.; Wang, S.; Mirkin, C.A. Synergistic Immunostimulation through the Dual Activation of Toll-like Receptor 3/9 with Spherical Nucleic Acids. ACS Nano 2021, 15, 13329–13338. [Google Scholar] [CrossRef] [PubMed]
- Skakuj, K.; Wang, S.; Qin, L.; Lee, A.; Zhang, B.; Mirkin, C.A. Conjugation Chemistry-Dependent T-Cell Activation with Spherical Nucleic Acids. J. Am. Chem. Soc. 2018, 140, 1227–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skakuj, K.; Teplensky, M.H.; Wang, S.; Dittmar, J.W.; Mirkin, C.A. Chemically Tuning the Antigen Release Kinetics from Spherical Nucleic Acids Maximizes Immune Stimulation. ACS Cent. Sci. 2021, 7, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qin, L.; Yamankurt, G.; Skakuj, K.; Huang, Z.; Chen, P.C.; Dominguez, D.; Lee, A.; Zhang, B.; Mirkin, C.A. Rational vaccinology with spherical nucleic acids. Proc. Natl. Acad. Sci. USA 2019, 116, 10473–10481. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Wang, S.; Dominguez, D.; Long, A.; Chen, S.; Fan, J.; Ahn, J.; Skakuj, K.; Huang, Z.; Lee, A.; et al. Development of Spherical Nucleic Acids for Prostate Cancer Immunotherapy. Front. Immunol. 2020, 11, 1333. [Google Scholar] [CrossRef]
- Teplensky, M.H.; Dittmar, J.W.; Qin, L.; Wang, S.; Evangelopoulos, M.; Zhang, B.; Mirkin, C.A. Spherical Nucleic Acid Vaccine Structure Markedly Influences Adaptive Immune Responses of Clinically Utilized Prostate Cancer Targets. Adv. Healthc. Mater. 2021, 10, e2101262. [Google Scholar] [CrossRef]
- Chiang, C.L.; Benencia, F.; Coukos, G. Whole tumor antigen vaccines. Semin. Immunol. 2010, 22, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Callmann, C.E.; Cole, L.E.; Kusmierz, C.D.; Huang, Z.; Horiuchi, D.; Mirkin, C.A. Tumor cell lysate-loaded immunostimulatory spherical nucleic acids as therapeutics for triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 17543–17550. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Chain, B.M.; Olszowska, E.; Olszowski, S.; Zgliczynski, J.M. Enhancement of immunogenic properties of ovalbumin as a result of its chlorination. Int. J. Biochem. 1991, 23, 1393–1395. [Google Scholar] [CrossRef]
- Allison, M.E.; Fearon, D.T. Enhanced immunogenicity of aldehyde-bearing antigens: A possible link between innate and adaptive immunity. Eur. J. Immunol. 2000, 30, 2881–2887. [Google Scholar] [CrossRef]
- Callmann, C.E.; Kusmierz, C.D.; Dittmar, J.W.; Broger, L.; Mirkin, C.A. Impact of Liposomal Spherical Nucleic Acid Structure on Immunotherapeutic Function. ACS Cent. Sci. 2021, 7, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Yamankurt, G.; Berns, E.J.; Xue, A.; Lee, A.; Bagheri, N.; Mrksich, M.; Mirkin, C.A. Exploration of the nanomedicine-design space with high-throughput screening and machine learning. Nat. Biomed. Eng. 2019, 3, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. Cytoplasmic DNA innate immune pathways. Immunol. Rev. 2011, 243, 99–108. [Google Scholar] [CrossRef]
- Barber, G.N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014, 35, 88–93. [Google Scholar] [CrossRef]
- Gurard-Levin, Z.A.; Mrksich, M. Combining self-assembled monolayers and mass spectrometry for applications in biochips. Annu. Rev. Anal. Chem. 2008, 1, 767–800. [Google Scholar] [CrossRef] [Green Version]
- Mrksich, M. Mass spectrometry of self-assembled monolayers: A new tool for molecular surface science. ACS Nano 2008, 2, 7–18. [Google Scholar] [CrossRef]
- Mrksich, M.; Whitesides, G.M. Using self-assembled monolayers to understand the interactions of man-made surfaces with proteins and cells. Annu. Rev. Biophys. Biomol. Struct. 1996, 25, 55–78. [Google Scholar] [CrossRef]
- Zhao, Q.; Temsamani, J.; Iadarola, P.L.; Jiang, Z.; Agrawal, S. Effect of different chemically modified oligodeoxynucleotides on immune stimulation. Biochem. Pharmacol. 1996, 51, 173–182. [Google Scholar] [CrossRef]
- Akinc, A.; Zumbuehl, A.; Goldberg, M.; Leshchiner, E.S.; Busini, V.; Hossain, N.; Bacallado, S.A.; Nguyen, D.N.; Fuller, J.; Alvarez, R.; et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.G.; Lynn, D.M.; Langer, R. Semi-automated synthesis and screening of a large library of degradable cationic polymers for gene delivery. Angew. Chem. 2003, 42, 3153–3158. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Daniel, W.L.; Giljohann, D.A.; Mirkin, C.A.; Lippard, S.J. Polyvalent oligonucleotide gold nanoparticle conjugates as delivery vehicles for platinum(IV) warheads. J. Am. Chem. Soc. 2009, 131, 14652–14653. [Google Scholar] [CrossRef]
- Zheng, J.; Zhu, G.; Li, Y.; Li, C.; You, M.; Chen, T.; Song, E.; Yang, R.; Tan, W. A spherical nucleic acid platform based on self-assembled DNA biopolymer for high-performance cancer therapy. ACS Nano 2013, 7, 6545–6554. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Hao, L.; Hurst, S.J.; Mirkin, C.A. Antibody-linked spherical nucleic acids for cellular targeting. J. Am. Chem. Soc. 2012, 134, 16488–16491. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Xu, X.; MacRenaris, K.W.; Zhang, X.Q.; Mirkin, C.A.; Meade, T.J. Multimodal gadolinium-enriched DNA-gold nanoparticle conjugates for cellular imaging. Angew. Chem. Int. Ed. Engl. 2009, 48, 9143–9147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Xu, X.; Lam, R.; Giljohann, D.; Ho, D.; Mirkin, C.A. Strategy for increasing drug solubility and efficacy through covalent attachment to polyvalent DNA-nanoparticle conjugates. ACS Nano 2011, 5, 6962–6970. [Google Scholar] [CrossRef]
- Vasher, M.K.; Yamankurt, G.; Mirkin, C.A. Hairpin-like siRNA-Based Spherical Nucleic Acids. J. Am. Chem. Soc. 2022, 144, 3174–3181. [Google Scholar] [CrossRef]
- Doi, T.; Higashino, K.; Kurihara, Y.; Wada, Y.; Miyazaki, T.; Nakamura, H.; Uesugi, S.; Imanishi, T.; Kawabe, Y.; Itakura, H.; et al. Charged collagen structure mediates the recognition of negatively charged macromolecules by macrophage scavenger receptors. J. Biol. Chem. 1993, 268, 2126–2133. [Google Scholar] [CrossRef]
- Pearson, A.M.; Rich, A.; Krieger, M. Polynucleotide binding to macrophage scavenger receptors depends on the formation of base-quartet-stabilized four-stranded helices. J. Biol. Chem. 1993, 268, 3546–3554. [Google Scholar] [CrossRef]
- Suzuki, K.; Doi, T.; Imanishi, T.; Kodama, T.; Tanaka, T. Oligonucleotide aggregates bind to the macrophage scavenger receptor. Eur. J. Biochem. 1999, 260, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.D.; Cheng, N.N.; Qu, Y.; Suarez, G.D.; Guo, T. Nanoscale energy deposition by X-ray absorbing nanostructures. J. Phys. Chem. B 2007, 111, 11622–11625. [Google Scholar] [CrossRef] [PubMed]
- Brodin, J.D.; Sprangers, A.J.; McMillan, J.R.; Mirkin, C.A. DNA-Mediated Cellular Delivery of Functional Enzymes. J. Am. Chem. Soc. 2015, 137, 14838–14841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusmierz, C.D.; Bujold, K.E.; Callmann, C.E.; Mirkin, C.A. Defining the Design Parameters for in Vivo Enzyme Delivery Through Protein Spherical Nucleic Acids. ACS Cent. Sci. 2020, 6, 815–822. [Google Scholar] [CrossRef]
- Samanta, D.; Ebrahimi, S.B.; Kusmierz, C.D.; Cheng, H.F.; Mirkin, C.A. Protein Spherical Nucleic Acids for Live-Cell Chemical Analysis. J. Am. Chem. Soc. 2020, 142, 13350–13355. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahajan, A.S.; Stegh, A.H. Spherical Nucleic Acids as Precision Therapeutics for the Treatment of Cancer—From Bench to Bedside. Cancers 2022, 14, 1615. https://doi.org/10.3390/cancers14071615
Mahajan AS, Stegh AH. Spherical Nucleic Acids as Precision Therapeutics for the Treatment of Cancer—From Bench to Bedside. Cancers. 2022; 14(7):1615. https://doi.org/10.3390/cancers14071615
Chicago/Turabian StyleMahajan, Akanksha S., and Alexander H. Stegh. 2022. "Spherical Nucleic Acids as Precision Therapeutics for the Treatment of Cancer—From Bench to Bedside" Cancers 14, no. 7: 1615. https://doi.org/10.3390/cancers14071615
APA StyleMahajan, A. S., & Stegh, A. H. (2022). Spherical Nucleic Acids as Precision Therapeutics for the Treatment of Cancer—From Bench to Bedside. Cancers, 14(7), 1615. https://doi.org/10.3390/cancers14071615