
Targeting CD82/KAI1 for Precision Therapeutics in Surmounting Metastatic Potential in Breast Cancer

 ,
,
Abstract
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
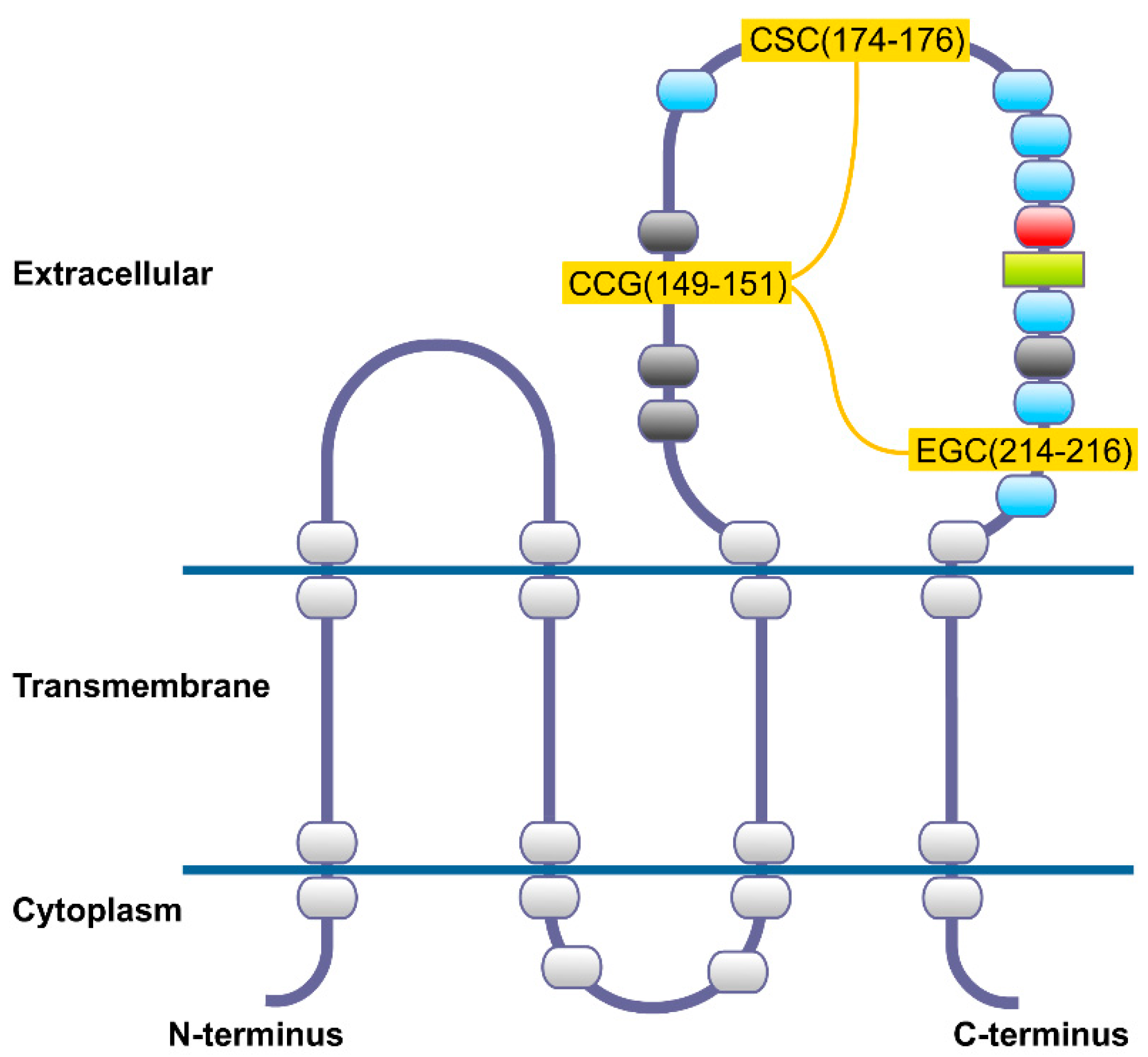
2. CD82 Glycoprotein
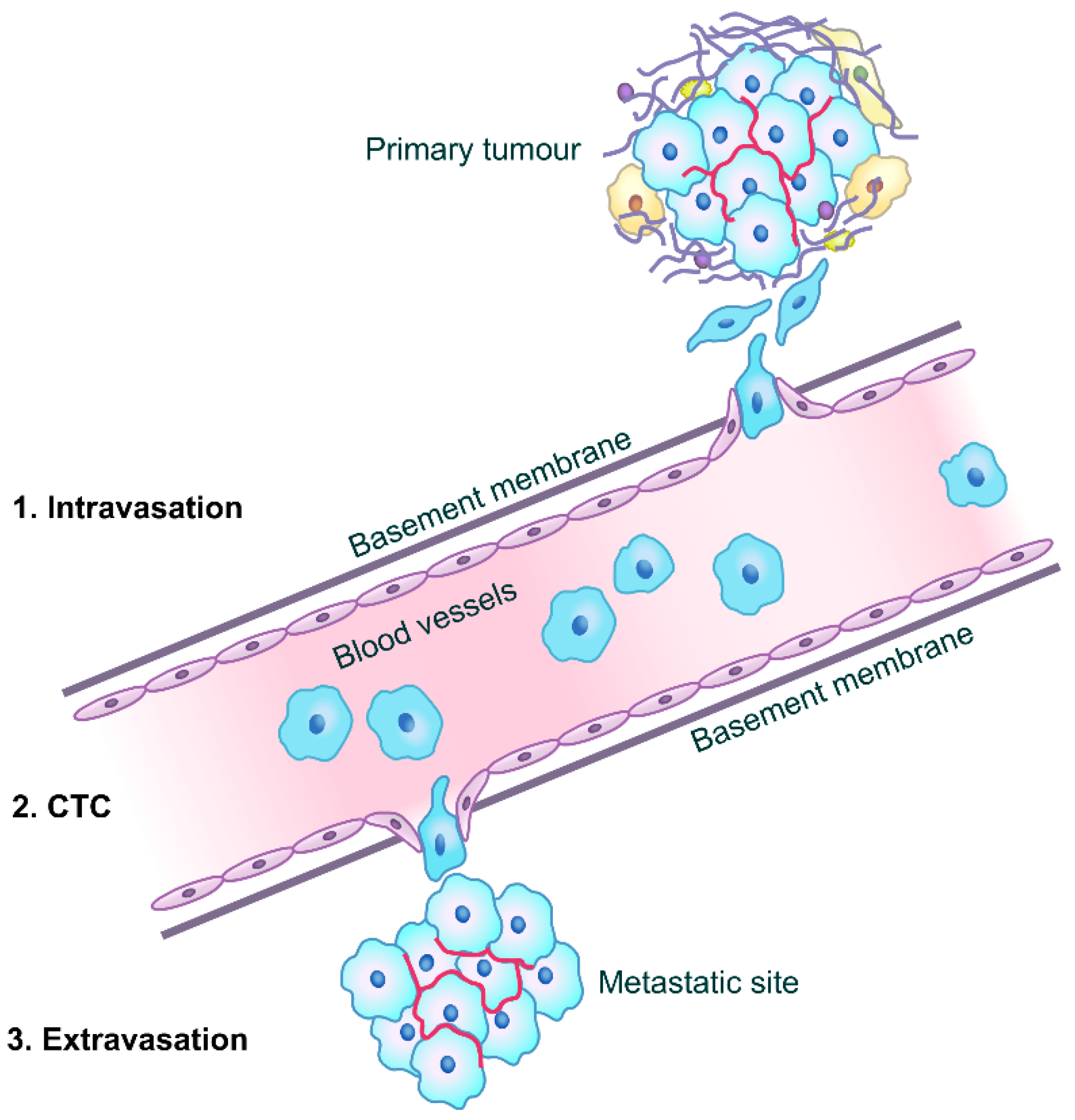
3. CD82 and Cancer Metastasis
4. Significance of CD82 Expression as a Metastasis Suppressor in Different Types of Cancer
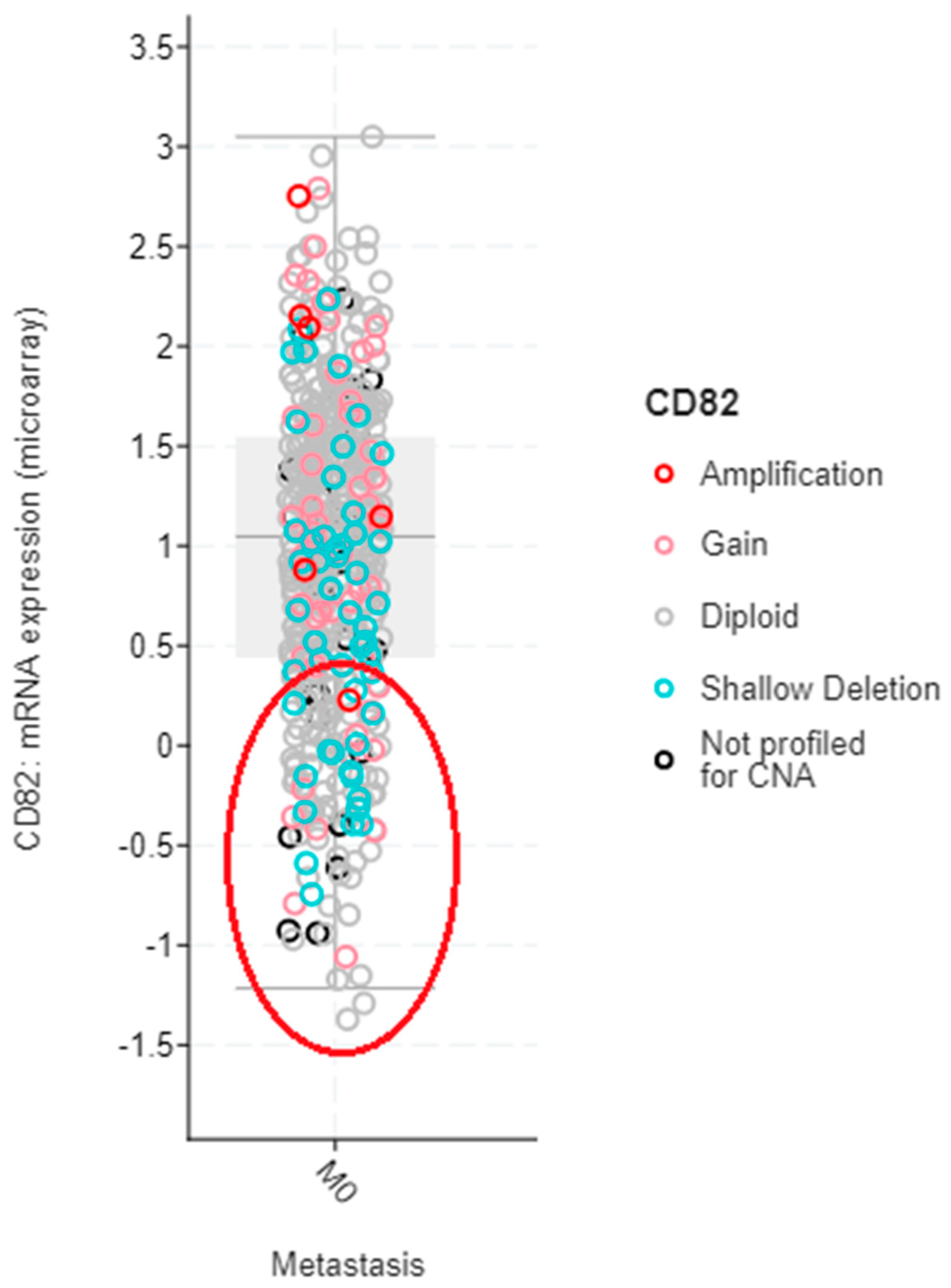
4.1. Breast Cancer
4.2. Other Cancers
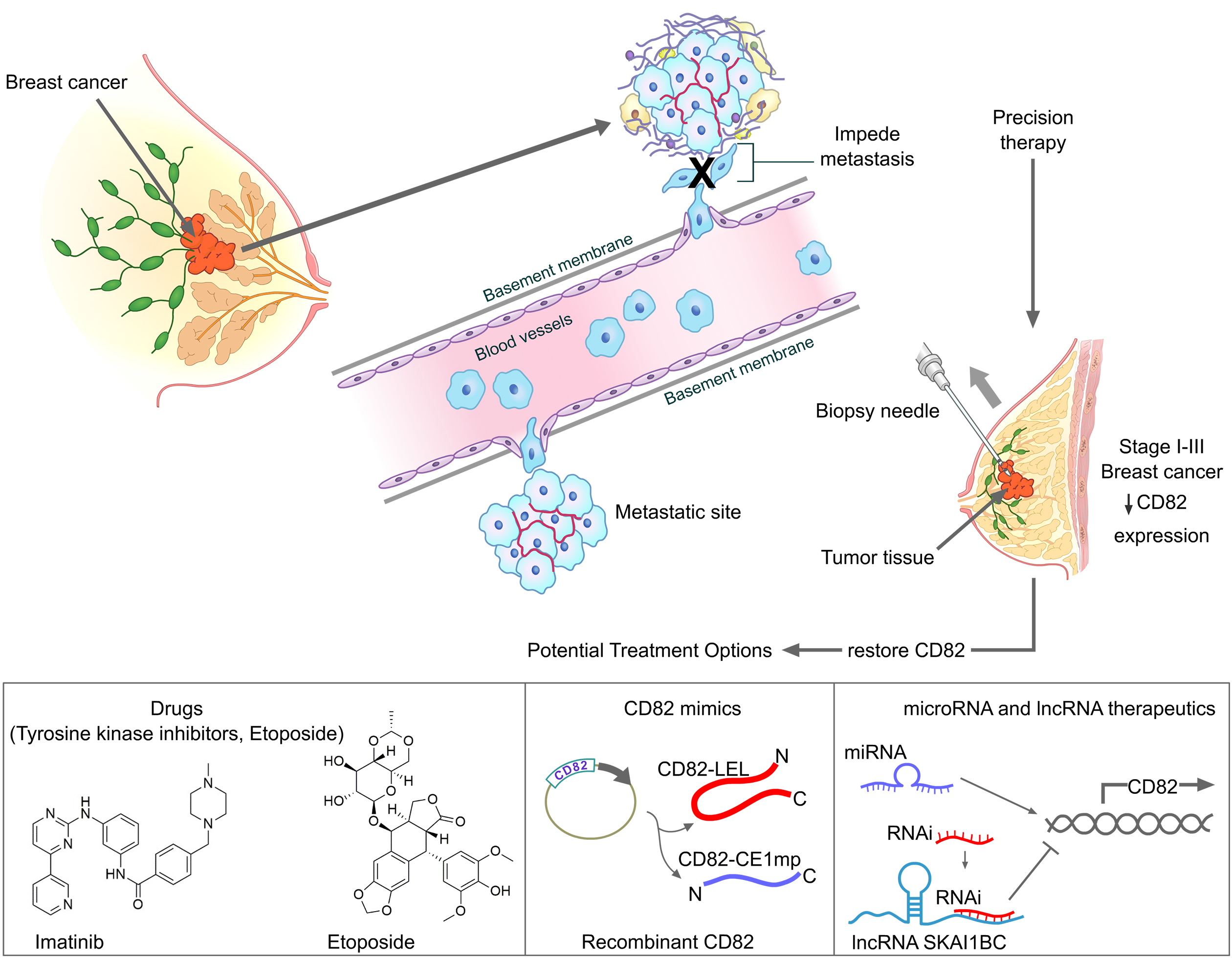
5. CD82 as a Therapeutic Target for Personalized Therapy in Breast Cancer
5.1. Selection of Suitable Breast Cancer Patients for Therapy
5.2. Potential Therapeutic Options for Upregulating CD82 in Breast Cancer
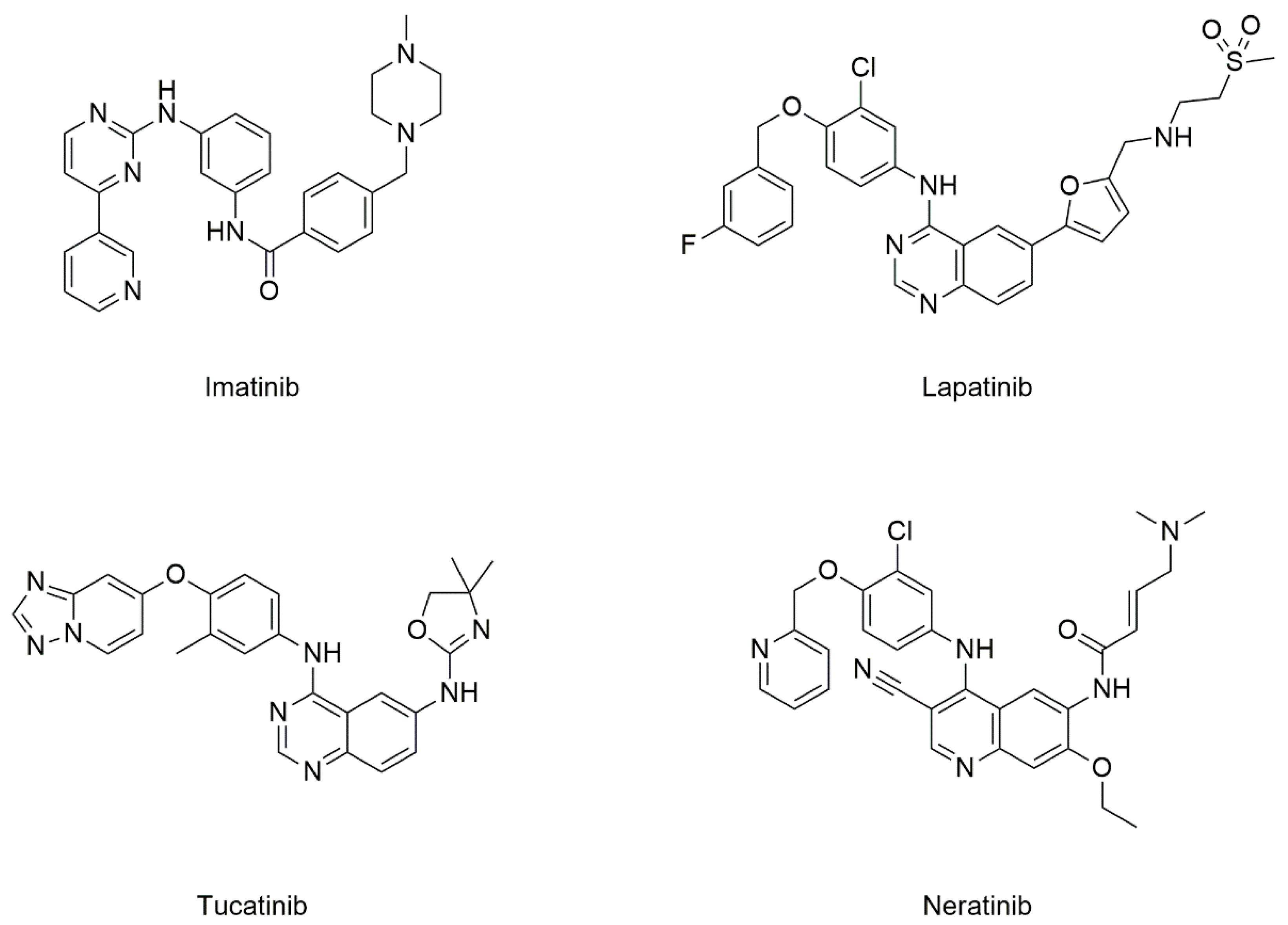
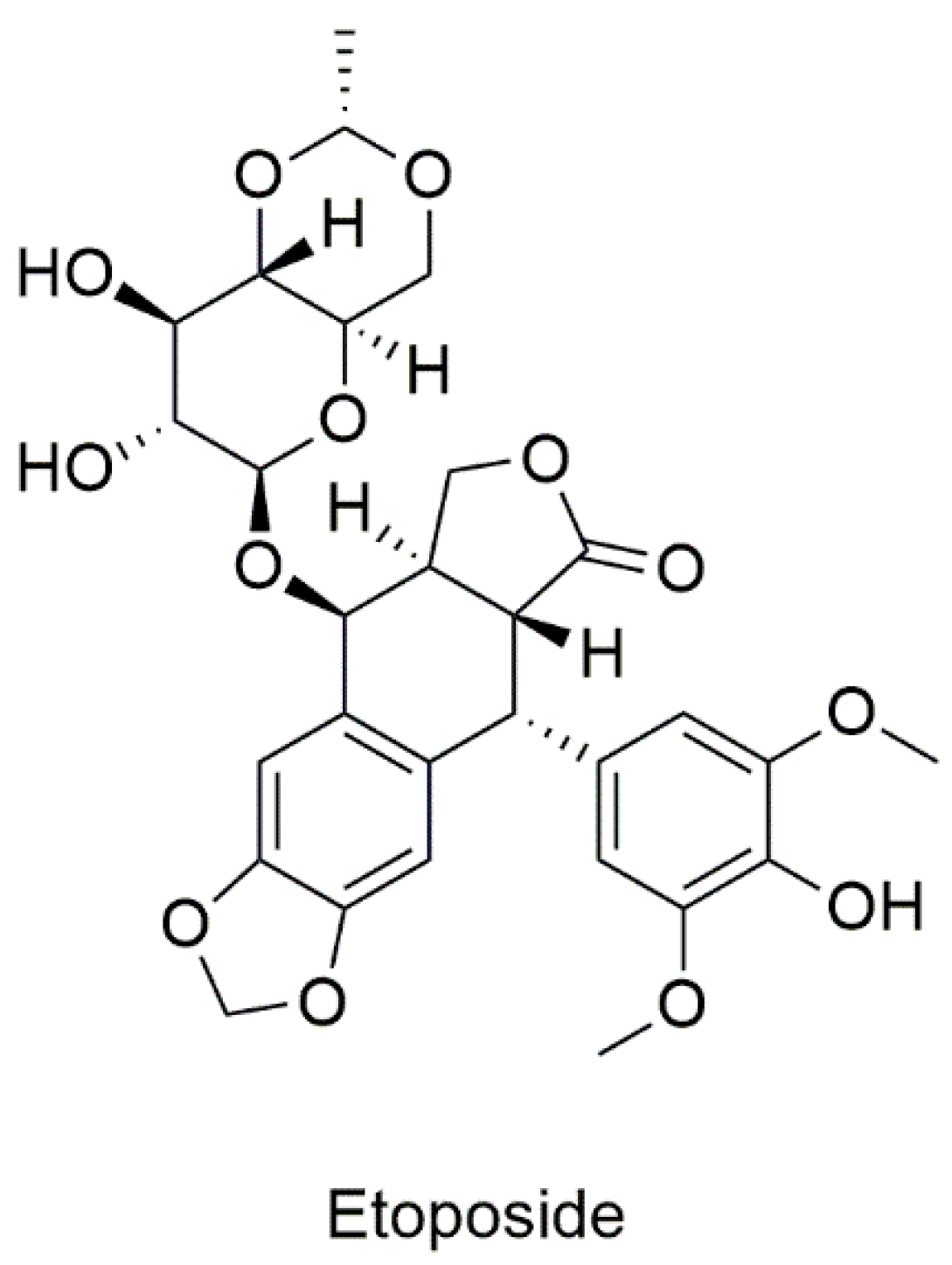
5.2.1. Drugs Known to Target CD82
Tyrosine Kinase Inhibitors (TKIs)
Etoposide
5.2.2. CD82 Mimics
5.2.3. Epigenetic Drugs for Treatment of Breast Cancer
Long Non-Coding RNA (lncRNA)-Based Therapy
MicroRNA (miRNA) Therapeutics
6. Challenges and Future Directions
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed]
- Guan, X. Cancer metastases: Challenges and opportunities. Acta. Pharm. Sin. B 2015, 5, 402–418. [Google Scholar] [CrossRef]
- Fidler, I.; Kripke, M. Metastasis results from preexisting variant cells within a malignant tumor. Science 1977, 197, 893–895. [Google Scholar] [CrossRef]
- Navin, N.E.; Hicks, J. Tracing the tumor lineage. Mol. Oncol. 2010, 4, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Bernards, R.; Weinberg, R.A. Metastasis genes: A progression puzzle. Nature 2002, 418, 823. [Google Scholar] [CrossRef] [PubMed]
- van ’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Skhinas, J.N.; Cox, T.R. The interplay between extracellular matrix remodelling and kinase signalling in cancer progression and metastasis. Cell Adh. Migr. 2018, 12, 529–537. [Google Scholar] [CrossRef]
- Kalita, B.; Coumar, M.S. Deciphering molecular mechanisms of metastasis: Novel insights into targets and therapeutics. Cell. Oncol. 2021. [Google Scholar] [CrossRef]
- Gloushankova, N.A.; Rubtsova, S.N.; Zhitnyak, I.Y. Cadherin-mediated cell-cell interactions in normal and cancer cells. Tissue Barriers 2017, 5, e1356900. [Google Scholar] [CrossRef]
- Gkretsi, V.; Stylianopoulos, T. Cell adhesion and matrix stiffness: Coordinating cancer cell invasion and metastasis. Front. Oncol. 2018, 8, 145. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Scully, O.J.; Bay, B.H.; Yip, G.; Yu, Y. Breast cancer metastasis. Cancer Genom. Proteom. 2012, 9, 311–320. [Google Scholar]
- Jin, L.; Han, B.; Siegel, E.; Cui, Y.; Giuliano, A.; Cui, X. Breast cancer lung metastasis: Molecular biology and therapeutic implications. Cancer Biol. Ther. 2018, 19, 858–868. [Google Scholar] [CrossRef]
- Vignoli, A.; Risi, E.; McCartney, A.; Migliaccio, I.; Moretti, E.; Malorni, L.; Luchinat, C.; Biganzoli, L.; Tenori, L. Precision oncology via NMR-based metabolomics: A review on breast cancer. Int. J. Mol. Sci. 2021, 22, 4687. [Google Scholar] [CrossRef]
- Dong, J.-T.; Isaacs, W.B.; Barrett, J.C.; Isaacs, J.T. Genomic organization of the human KAI1 metastasis-suppressor gene. Genomics 1997, 41, 25–32. [Google Scholar] [CrossRef]
- Bienstock, R.J.; Barrett, J.C. KAI1, A prostate metastasis suppressor: Prediction of solvated structure and interactions with binding partners; integrins, cadherins, and cell-surface receptor proteins†. Mol. Carcinog. 2001, 32, 139–153. [Google Scholar] [CrossRef]
- Liu, W.M.; Zhang, X.A. KAI1/CD82, a tumor metastasis suppressor. Cancer Lett. 2006, 240, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.; Marreiros, A.; Russell, P.J. KAI1 tetraspanin and metastasis suppressor. Int. J. Biochem. Cell Biol. 2005, 37, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sato, Y.; Isaji, T.; Fukuda, T.; Matsumoto, A.; Miyoshi, E.; Gu, J.; Taniguchi, N. Branched N-glycans regulate the biological functions of integrins and cadherins. FEBS J. 2008, 275, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Kuroki, Y.; Ohtsubo, K.; Taniguchi, N. Core fucose and bisecting GlcNAc, the direct modifiers of the N-glycan core: Their functions and target proteins. Carbohydr. Res. 2009, 344, 1387–1390. [Google Scholar] [CrossRef]
- Bari, R.; Zhang, Y.H.; Zhang, F.; Wang, N.X.; Stipp, C.S.; Zheng, J.J.; Zhang, X.A. Transmembrane interactions are needed for KAI1/CD82-mediated suppression of cancer invasion and metastasis. Am. J. Pathol. 2009, 174, 647–660. [Google Scholar] [CrossRef]
- Kovalenko, O.V.; Metcalf, D.G.; DeGrado, W.F.; Hemler, M.E. Structural organization and interactions of transmembrane domains in tetraspanin proteins. BMC Struct. Biol. 2005, 5, 11. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, L.; Reddivari, M.; Zhang, X.A. The palmitoylation of metastasis suppressor KAI1/CD82 is important for its motility- and invasiveness-inhibitory activity. Cancer Res. 2004, 64, 7455–7463. [Google Scholar] [CrossRef]
- Ma, X.; He, X.; Wang, C.; Huang, X.; Li, Y.; Ma, K. Small extracellular ring domain is necessary for CD82/KAI1’anti-metastasis function. Biochem. Biophys. Res. Commun. 2021, 557, 110–116. [Google Scholar] [CrossRef]
- Odintsova, E.; van Niel, G.; Conjeaud, H.; Raposo, G.; Iwamoto, R.; Mekada, E.; Berditchevski, F. Metastasis suppressor tetraspanin CD82/KAI1 regulates ubiquitylation of epidermal growth factor receptor. J. Biol. Chem. 2013, 288, 26323–26334. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Weissman, A.M. Dissecting the diverse functions of the metastasis suppressor CD82/KAI1. FEBS Lett. 2011, 585, 3166–3173. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.M.; Zhang, F.; Moshiach, S.; Zhou, B.; Huang, C.; Srinivasan, K.; Khurana, S.; Zheng, Y.; Lahti, J.M.; Zhang, X.A. Tetraspanin CD82 inhibits protrusion and retraction in cell movement by attenuating the plasma membrane-dependent actin organization. PLoS ONE 2012, 7, e51797. [Google Scholar] [CrossRef]
- He, B.; Liu, L.; Cook, G.A.; Grgurevich, S.; Jennings, L.K.; Zhang, X.A. Tetraspanin CD82 attenuates cellular morphogenesis through down-regulating integrin α6-mediated cell adhesion. J. Biol. Chem. 2005, 280, 3346–3354. [Google Scholar] [CrossRef]
- Odintsova, E.; Sugiura, T.; Berditchevski, F. Attenuation of EGF receptor signaling by a metastasis suppressor, the tetraspanin CD82/KAI-1. Curr. Biol. 2000, 10, 1009–1012. [Google Scholar] [CrossRef]
- Huang, C.; Fu, C.; Wren, J.D.; Wang, X.; Zhang, F.; Zhang, Y.H.; Connel, S.A.; Chen, T.; Zhang, X.A. Tetraspanin-enriched microdomains regulate digitation junctions. Cell Mol. Life Sci. 2018, 75, 3423–3439. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.A.; Park, I.; Byun, H.J.; Jeoung, D.; Kim, Y.M.; Lee, H. Metastasis suppressor KAI1/CD82 attenuates the matrix adhesion of human prostate cancer cells by suppressing fibronectin expression and β1 integrin activation. Cell Physiol. Biochem. 2011, 27, 575–586. [Google Scholar] [CrossRef]
- Pehkonen, H.; Lento, M.; von Nandelstadh, P.; Filippou, A.; Grénman, R.; Lehti, K.; Monni, O. Liprin-α1 modulates cancer cell signaling by transmembrane protein CD82 in adhesive membrane domains linked to cytoskeleton. Cell Commun. Signal 2018, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Hays, F.A.; Tomasek, J.J.; Benyajati, S.; Zhang, X.A. Tetraspanin CD82 interaction with cholesterol promotes extracellular vesicle-mediated release of ezrin to inhibit tumour cell movement. J. Extracell Vesicles 2020, 9, 1692417. [Google Scholar] [CrossRef]
- Khan, N.S.; Lukason, D.P.; Feliu, M.; Ward, R.A.; Lord, A.K.; Reedy, J.L.; Ramirez-Ortiz, Z.G.; Tam, J.M.; Kasperkovitz, P.V.; Negoro, P.E.; et al. CD82 controls CpG-dependent TLR9 signaling. FASEB J. 2019, 33, 12500–12514. [Google Scholar] [CrossRef]
- Yang, X.; Welch, D.R.; Phillips, K.K.; Weissman, B.E.; Wei, L.L. KAI1, a putative marker for metastatic potential in human breast cancer. Cancer Lett. 1997, 119, 149–155. [Google Scholar] [CrossRef]
- Yang, X.; Wei, L.; Tang, C.; Slack, R.; Montgomery, E.; Lippman, M. KAI1 protein is down-regulated during the progression of human breast cancer. Clin. Cancer Res. 2000, 6, 3424–3429. [Google Scholar]
- Mooez, S.; Malik, F.A.; Kayani, M.A.; Rashid, R.; Zahid, A.; Khan, A. Expressional alterations and transcript isoforms of metastasis suppressor genes (KAI1 and KiSS1) in breast cancer patients. Asian Pac. J. Cancer Prev. 2011, 12, 2785–2791. [Google Scholar] [PubMed]
- Malik, F.A.; Sanders, A.J.; Jones, A.D.; Mansel, R.E.; Jiang, W.G. Transcriptional and translational modulation of KAI1 expression in ductal carcinoma of the breast and the prognostic significance. Int. J. Mol. Med. 2009, 23, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.M.; Tongers, K.; Maass, N.; Mehdorn, H.M.; Held-Feindt, J. Reduced metastasis-suppressor gene mRNA-expression in breast cancer brain metastases. J. Cancer Res. Clin. Oncol. 2005, 131, 191–198. [Google Scholar] [CrossRef]
- Krishna Latha, T.; Verma, A.; Thakur, G.K.; Banerjee, B.; Kaur, N.; Singh, U.R.; Sharma, S. Down regulation of KAI1/CD82 in lymph node positive and advanced T-stage group in breast cancer patients. Asian Pac. J. Cancer Prev. 2019, 20, 3321–3329. [Google Scholar] [CrossRef]
- Han, Z.; Chen, Z.; Zheng, R.; Cheng, Z.; Gong, X.; Wang, D. Clinicopathological significance of CD133 and CD44 expression in infiltrating ductal carcinoma and their relationship to angiogenesis. World J. Surg. Oncol. 2015, 13, 56. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.I.; Kohno, N.; Ogawa, E.; Adachi, M.; Taki, T.; Miyake, M. Correlation of reduction in MRP-1/CD9 and KAI1/CD82 expression with recurrences in breast cancer patients. Am. J. Pathol. 1998, 153, 973–983. [Google Scholar] [CrossRef]
- Christgen, M.; Christgen, H.; Heil, C.; Krech, T.; Länger, F.; Kreipe, H.; Lehmann, U. Expression of KAI1/CD82 in distant metastases from estrogen receptor-negative breast cancer. Cancer Sci. 2009, 100, 1767–1771. [Google Scholar] [CrossRef]
- Malik, F.A.; Sanders, A.J.; Kayani, M.A.; Jiang, W.G. Effect of expressional alteration of KAI1 on breast cancer cell growth, adhesion, migration and invasion. Cancer Genom. Proteom. 2009, 6, 205–213. [Google Scholar]
- Odintsova, E.; Voortman, J.; Gilbert, E.; Berditchevski, F. Tetraspanin CD82 regulates compartmentalisation and ligand-induced dimerization of EGFR. J. Cell Sci. 2003, 116, 4557–4566. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, J.; Dreyer, T.F.; Bächer, A.S.; Sinner, E.K.; Heinrich, C.; Benge, A.; Gross, E.; Preis, S.; Rother, J.; Roberts, A.; et al. Differential tumor biological role of the tumor suppressor KAI1 and its splice variant in human breast cancer cells. Oncotarget 2018, 9, 6369–6390. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, J.H.; Seo, Y.W.; Park, S.R.; Kim, Y.J.; Kim, K.K. Expression of a splice variant of KAI1, a tumor metastasis suppressor gene, influences tumor invasion and progression. Cancer Res. 2003, 63, 7247–7255. [Google Scholar]
- Zhu, C.; He, L.; Zhou, X.; Nie, X.; Gu, Y. Sulfatase 2 promotes breast cancer progression through regulating some tumor-related factors. Oncol. Rep. 2016, 35, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Christgen, M.; Bruchhardt, H.; Ballmaier, M.; Krech, T.; Länger, F.; Kreipe, H.; Lehmann, U. KAI1/CD82 is a novel target of estrogen receptor-mediated gene repression and downregulated in primary human breast cancer. Int. J. Cancer 2008, 123, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhong, W.; Bu, J.; Li, Y.; Li, R.; Nie, R.; Xiao, C.; Ma, K.; Huang, X.; Li, Y. Exosomal protein CD82 as a diagnostic biomarker for precision medicine for breast cancer. Mol. Carcinog. 2019, 58, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Lamb, P.; Rinker-Schaeffer, C.; Vukanovic, J.; Ichikawa, T.; Isaacs, J.; Barrett, J. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science 1995, 268, 884–886. [Google Scholar] [CrossRef]
- Ma, Z.; Gao, Y.; Liu, W.; Zheng, L.; Jin, B.; Duan, B.; Xie, H.; Guo, P.; Zeng, J.; Wang, K.; et al. CD82 suppresses ADAM17-dependent E-cadherin cleavage and cell migration in prostate cancer. Dis. Markers 2020, 2020, 8899924. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Devaraj, S.N. KAI1/CD82, metastasis suppressor gene as a therapeutic target for non-small-cell lung carcinoma. J. Environ. Pathol. Toxicol. Oncol. 2017, 36, 269–275. [Google Scholar] [CrossRef]
- Ci, H.; Xu, Z.; Xu, J.; Wang, Y.; Wu, S. Expressions of KAI1 and E-cadherin in nonsmall cell lung cancer and their correlation with vasculogenic mimicry. Medicine 2018, 97, e12293. [Google Scholar] [CrossRef]
- Guo, X.; Friess, H.; Graber, H.U.; Kashiwagi, M.; Zimmermann, A.; Korc, M.; Büchler, M.W. KAI1 expression is up-regulated in early pancreatic cancer and decreased in the presence of metastases. Cancer Res. 1996, 56, 4876–4880. [Google Scholar]
- Friess, H.; Guo, X.Z.; Berberat, P.; Graber, H.U.; Zimmermann, A.; Korc, M.; Büchler, M.W. Reduced KAI1 expression in pancreatic cancer is associated with lymph node and distant metastases. Int. J. Cancer 1998, 79, 349–355. [Google Scholar] [CrossRef]
- Liu, X.; Guo, X.Z.; Li, H.Y.; Chen, J. KAI1 reverses the epithelial-mesenchymal transition in human pancreatic cancer cells. Hepatobiliary Pancreat. Dis. Int. 2019, 18, 471–477. [Google Scholar] [CrossRef]
- Zhou, X.L.; Wang, M. Expression levels of survivin, Bcl-2, and KAI1 proteins in cervical cancer and their correlation with metastasis. Genet. Mol. Res. 2015, 14, 17059–17067. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Cheng, Y.; Martinka, M.; Ong, C.J.; Li, G. Prognostic significance of KAI1/CD82 in human melanoma and its role in cell migration and invasion through the regulation of ING4. Carcinogenesis 2013, 35, 86–95. [Google Scholar] [CrossRef]
- Nishioka, C.; Ikezoe, T.; Takeuchi, A.; Nobumoto, A.; Tsuda, M.; Yokoyama, A. The novel function of CD82 and its impact on BCL2L12 via AKT/STAT5 signal pathway in acute myelogenous leukemia cells. Leukemia 2015, 29, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Du, L.; Ju, J.; Ma, C.; Shen, Z.; Yang, X.; Liang, L.; Ni, Q.; Sun, M. Overexpression of KAI1/CD82 suppresses in vitro cell growth, migration, invasion and xenograft growth in oral cancer. Mol. Med. Rep. 2017, 15, 1527–1532. [Google Scholar] [CrossRef]
- Zeng, T.D.; Zheng, B.; Zheng, W.; Chen, C. CD82/KAI1 inhibits invasion and metastasis of esophageal squamous cell carcinoma via TGF-β1. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5928–5937. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, Y.; He, X.; Chen, Y.; Wei, W.; Yang, X.; Ma, K. Gangliosides and CD82 inhibit the motility of colon cancer by downregulating the phosphorylation of EGFR at different tyrosine sites and signaling pathways. Mol. Med. Rep. 2020, 22, 3994–4002. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Bergin, A.R.T.; Loi, S. Triple-negative breast cancer: Recent treatment advances. F1000Res 2019, 8. [Google Scholar] [CrossRef]
- Jackson, S.E.; Chester, J.D. Personalised cancer medicine. Int. J. Cancer 2015, 137, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Alimirzaie, S.; Bagherzadeh, M.; Akbari, M.R. Liquid biopsy in breast cancer: A comprehensive review. Clin. Genet. 2019, 95, 643–660. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Morandi, P.; Krishnamurthy, S.; Reuben, J.M.; Lee, B.N.; Francis, D.; Booser, D.J.; Green, M.C.; Arun, B.K.; Pusztai, L.; et al. Imatinib mesylate (Gleevec) in advanced breast cancer-expressing C-Kit or PDGFR-beta: Clinical activity and biological correlations. Ann. Oncol. 2008, 19, 1713–1719. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]
- Keung, E.Z.; Raut, C.P.; Rutkowski, P. The landmark series: Systemic therapy for resectable gastrointestinal stromal tumors. Ann. Surg. Oncol. 2020, 27, 3659–3671. [Google Scholar] [CrossRef] [PubMed]
- Shandiz, S.A.S.; Khosravani, M.; Mohammadi, S.; Noorbazargan, H.; Mirzaie, A.; Inanlou, D.N.; Jalali, M.D.; Jouzaghkar, H.; Baghbani-Arani, F.; Keshavarz-Pakseresht, B. Evaluation of imatinib mesylate (Gleevec) on KAI1/CD82 gene expression in breast cancer MCF-7 cells using quantitative real-time PCR. Asian Pac. J. Trop. Biomed. 2016, 6, 159–163. [Google Scholar] [CrossRef]
- Ryan, Q.; Ibrahim, A.; Cohen, M.H.; Johnson, J.; Ko, C.W.; Sridhara, R.; Justice, R.; Pazdur, R. FDA drug approval summary: Lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 2008, 13, 1114–1119. [Google Scholar] [CrossRef]
- Abraham, J.; Montero, A.J.; Jankowitz, R.C.; Salkeni, M.A.; Beumer, J.H.; Kiesel, B.F.; Piette, F.; Adamson, L.M.; Nagy, R.J.; Lanman, R.B.; et al. Safety and efficacy of T-DM1 plus neratinib in patients with metastatic HER2-positive breast cancer: NSABP Foundation trial FB-10. J. Clin. Oncol. 2019, 37, 2601–2609. [Google Scholar] [CrossRef]
- Duchnowska, R.; Loibl, S.; Jassem, J. Tyrosine kinase inhibitors for brain metastases in HER2-positive breast cancer. Cancer Treat. Rev. 2018, 67, 71–77. [Google Scholar] [CrossRef]
- FDA. FDA Approves Neratinib for Metastatic HER2-Positive Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-neratinib-metastatic-her2-positive-breast-cancer (accessed on 23 May 2021).
- Martin, M.; Holmes, F.A.; Ejlertsen, B.; Delaloge, S.; Moy, B.; Iwata, H.; von Minckwitz, G.; Chia, S.K.L.; Mansi, J.; Barrios, C.H.; et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1688–1700. [Google Scholar] [CrossRef]
- FDA. FDA Approves Tucatinib for Patients with HER2-Positive Metastatic Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tucatinib-patients-her2-positive-metastatic-breast-cancer (accessed on 23 May 2021).
- Liu, J.; Pandya, P.; Afshar, S. Therapeutic Advances in Oncology. Int. J. Mol. Sci. 2021, 22, 2008. [Google Scholar] [CrossRef]
- Hande, K.R. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef]
- Giannone, G.; Milani, A.; Ghisoni, E.; Genta, S.; Mittica, G.; Montemurro, F.; Valabrega, G. Oral etoposide in heavily pre-treated metastatic breast cancer: A retrospective series. Breast 2018, 38, 160–164. [Google Scholar] [CrossRef]
- Yuan, P.; Di, L.; Zhang, X.; Yan, M.; Wan, D.; Li, L.; Zhang, Y.; Cai, J.; Dai, H.; Zhu, Q.; et al. Efficacy of Oral Etoposide in Pretreated Metastatic Breast Cancer: A Multicenter Phase 2 Study. Medicine 2015, 94, e774. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Zhu, A.; Si, Y.; Yue, J.; Wang, X.; Wang, J.; Ma, F.; Xu, B.; Yuan, P. A phase II, single-arm study of apatinib and oral etoposide in heavily pre-treated metastatic breast cancer. Front. Oncol. 2020, 10, 565384. [Google Scholar] [CrossRef] [PubMed]
- Segar, J.M.; Reed, D.; Stopeck, A.; Livingston, R.B.; Chalasani, P. A phase II study of irinotecan and etoposide as treatment for refractory metastatic breast cancer. Oncologist 2019, 24, 1512-e1267. [Google Scholar] [CrossRef]
- Saphner, T.; Weller, E.A.; Tormey, D.C.; Pandya, K.J.; Falkson, C.I.; Stewart, J.; Robert, N.J. 21-day oral etoposide for metastatic breast cancer: A phase II study and review of the literature. Am. J. Clin. Oncol. 2000, 23, 258–262. [Google Scholar] [CrossRef]
- Mashimo, T.; Bandyopadhyay, S.; Goodarzi, G.; Watabe, M.; Pai, S.K.; Gross, S.C.; Watabe, K. Activation of the tumor metastasis suppressor gene, KAI1, by etoposide is mediated by p53 and c-Jun genes. Biochem. Biophys. Res. Commun. 2000, 274, 370–376. [Google Scholar] [CrossRef]
- Khedri, A.; Khaghani, S.; Kheirollah, A.; Babaahmadi-Rezaei, H.; Shadboorestan, A.; Zangooei, M.; Afra, H.S.; Meshkani, R.; Ghahremani, M.H. Signaling Crosstalk of FHIT, p53, and p38 in etoposide-induced apoptosis in MCF-7 cells. J. Cell. Biochem. 2019, 120, 9125–9137. [Google Scholar] [CrossRef]
- Zou, Z.; Gao, C.; Nagaich, A.K.; Connell, T.; Saito, S.; Moul, J.W.; Seth, P.; Appella, E.; Srivastava, S. p53 regulates the expression of the tumor suppressor gene maspin. J. Biol. Chem. 2000, 275, 6051–6054. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. A systematic review and pooled analysis of studies of oral etoposide in metastatic breast cancer. Eur. J. Breast Health 2018, 14, 10–16. [Google Scholar] [CrossRef]
- Ho, S.-H.; Martin, F.; Higginbottom, A.; Partridge, L.J.; Parthasarathy, V.; Moseley, G.W.; Lopez, P.; Cheng-Mayer, C.; Monk, P.N. Recombinant extracellular domains of tetraspanin proteins are potent inhibitors of the infection of macrophages by Human Immunodeficiency Virus type 1. J. Virol. 2006, 80, 6487–6496. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Ma, X.; Wang, C.; Luan, M.; Li, Y.; Huang, X.; Ma, K. The peptide mimicking small extracellular ring domain of CD82 inhibits tumor cell migration in vitro and metastasis in vivo. J. Cancer. Res. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- He, X.; Huang, X.; Wang, C.; Luan, M.; Li, Y.; Ma, X.; Ma, K. The peptide mimicking small extracellular ring domain of CD82 inhibits epithelial-mesenchymal transition by downregulating Wnt pathway and upregulating hippo pathway. Biochem. Biophys. Res. Commun. 2020, 533, 338–345. [Google Scholar] [CrossRef]
- Fang, Y.; Fullwood, M.J. Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Aram, R.; Dotan, I.; Hotz-Wagenblatt, A.; Canaani, D. Identification of a novel metastasis inducing lncRNA which suppresses the KAI1/CD82 metastasis suppressor gene and is upregulated in triple-negative breast cancer. Oncotarget 2017, 8, 67538–67552. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, W.; Guan, X.; Tang, J. The long non-coding RNA landscape in triple-negative breast cancer. Cell Prolif. 2021, 54, e12966. [Google Scholar] [CrossRef]
- Macfarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, function and role in cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef]
- Bouchie, A. First microRNA mimic enters clinic. Nat. Biotechnol. 2013, 31, 577. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S. Posttranscriptional upregulation by microRNAs. WIREs RNA 2012, 3, 311–330. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug. Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Baumann, V.; Winkler, J. miRNA-based therapies: Strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med. Chem. 2014, 6, 1967–1984. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.S.; Cheah, Y.K. Potential miRNAs for miRNA-based therapeutics in breast cancer. Non-Coding RNA 2020, 6, 29. [Google Scholar] [CrossRef]
- Liang, Y.J.; Wang, Q.Y.; Zhou, C.X.; Yin, Q.Q.; He, M.; Yu, X.T.; Cao, D.X.; Chen, G.Q.; He, J.R.; Zhao, Q. MiR-124 targets Slug to regulate epithelial-mesenchymal transition and metastasis of breast cancer. Carcinogenesis 2013, 34, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Sang, M.; Liu, F.; Ai, N.; Geng, C. miR-124 regulates EMT based on ZEB2 target to inhibit invasion and metastasis in triple-negative breast cancer. Pathol. Res. Pract. 2019, 215, 697–704. [Google Scholar] [CrossRef]
- Shi, P.; Chen, C.; Li, X.; Wei, Z.; Liu, Z.; Liu, Y. MicroRNA-124 suppresses cell proliferation and invasion of triple negative breast cancer cells by targeting STAT3. Mol. Med. Rep. 2019, 19, 3667–3675. [Google Scholar] [CrossRef]
- Hsieh, T.H.; Hsu, C.Y.; Tsai, C.F.; Long, C.Y.; Chai, C.Y.; Hou, M.F.; Lee, J.N.; Wu, D.C.; Wang, S.C.; Tsai, E.M. miR-125a-5p is a prognostic biomarker that targets HDAC4 to suppress breast tumorigenesis. Oncotarget 2015, 6, 494–509. [Google Scholar] [CrossRef]
- Liang, Z.; Pan, Q.; Zhang, Z.; Huang, C.; Yan, Z.; Zhang, Y.; Li, J. MicroRNA-125a-5p controls the proliferation, apoptosis, migration and PTEN/MEK1/2/ERK1/2 signaling pathway in MCF-7 breast cancer cells. Mol. Med. Rep. 2019, 20, 4507–4514. [Google Scholar] [CrossRef]
- Ninio-Many, L.; Hikri, E.; Burg-Golani, T.; Stemmer, S.M.; Shalgi, R.; Ben-Aharon, I. miR-125a induces HER2 expression and sensitivity to trastuzumab in triple-negative breast cancer lines. Front Oncol. 2020, 10, 191. [Google Scholar] [CrossRef]
- Cheng, S.; Huang, Y.; Lou, C.; He, Y.; Zhang, Y.; Zhang, Q. FSTL1 enhances chemoresistance and maintains stemness in breast cancer cells via integrin β3/Wnt signaling under miR-137 regulation. Cancer Biol. Ther. 2019, 20, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Yu, L.; Wu, Y.; Wang, S.; Yao, J.; Zheng, X.; Xie, S.; Zhang, S.; Lu, X.; Liu, Y.; et al. miR-137 alleviates doxorubicin resistance in breast cancer through inhibition of epithelial-mesenchymal transition by targeting DUSP4. Cell Death Dis. 2019, 10, 922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.D.; Sun, D.W.; Mao, L.; Zhang, J.; Jiang, L.H.; Li, J.; Wu, Y.; Ji, H.; Chen, W.; Wang, J.; et al. MiR-139-5p inhibits the biological function of breast cancer cells by targeting Notch1 and mediates chemosensitivity to docetaxel. Biochem. Biophys. Res. Commun. 2015, 465, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Pajic, M.; Froio, D.; Daly, S.; Doculara, L.; Millar, E.; Graham, P.H.; Drury, A.; Steinmann, A.; de Bock, C.E.; Boulghourjian, A.; et al. miR-139-5p modulates radiotherapy resistance in breast cancer by repressing multiple gene networks of DNA repair and ROS defense. Cancer Res. 2018, 78, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef]
- García-García, F.; Salinas-Vera, Y.M.; García-Vázquez, R.; Marchat, L.A.; Rodríguez-Cuevas, S.; López-González, J.S.; Carlos-Reyes, Á.; Ramos-Payán, R.; Aguilar-Medina, M.; Pérez-Plasencia, C.; et al. miR-145-5p is associated with pathological complete response to neoadjuvant chemotherapy and impairs cell proliferation by targeting TGFβR2 in breast cancer. Oncol. Rep. 2019, 41, 3527–3534. [Google Scholar] [CrossRef]
- Tan, X.; Li, Z.; Ren, S.; Rezaei, K.; Pan, Q.; Goldstein, A.T.; Macri, C.J.; Cao, D.; Brem, R.F.; Fu, S.W. Dynamically decreased miR-671-5p expression is associated with oncogenic transformation and radiochemoresistance in breast cancer. Breast Cancer Res. 2019, 21, 89. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Fu, Y.; Chen, L.; Lee, W.; Lai, Y.; Rezaei, K.; Tabbara, S.; Latham, P.; Teal, C.B.; Man, Y.G.; et al. miR-671-5p inhibits epithelial-to-mesenchymal transition by downregulating FOXM1 expression in breast cancer. Oncotarget 2016, 7, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhao, Y.; Shao, N.; Ye, R.; Lin, Y.; Zhang, N.; Li, W.; Zhang, Y.; Wang, S. Overexpression of microRNA-96-5p inhibits autophagy and apoptosis and enhances the proliferation, migration and invasiveness of human breast cancer cells. Oncol. Lett. 2017, 13, 4402–4412. [Google Scholar] [CrossRef]
- Moazzeni, H.; Najafi, A.; Khani, M. Identification of direct target genes of miR-7, miR-9, miR-96, and miR-182 in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol. Cell Probes 2017, 34, 45–52. [Google Scholar] [CrossRef]
- Huang, L.; Liu, X. microRNA-370 promotes cell Growth by targeting WNK2 in breast cancer. DNA Cell Biol. 2019, 38, 501–509. [Google Scholar] [CrossRef]
- Lv, J.; Xia, K.; Xu, P.; Sun, E.; Ma, J.; Gao, S.; Zhou, Q.; Zhang, M.; Wang, F.; Chen, F.; et al. miRNA expression patterns in chemoresistant breast cancer tissues. Biomed. Pharmacother. 2014, 68, 935–942. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wang, C.; Wang, F.; Wang, Y.; Shen, M.; Chen, K.; Cheng, P.; Zhang, Y.; Yang, J.; Zhu, R.; et al. Anti-miR-197 inhibits migration in HCC cells by targeting KAI 1/CD82. Biochem. Biophys. Res. Commun. 2014, 446, 541–548. [Google Scholar] [CrossRef]
- Long, J.; Luo, J.; Yin, X. MiR-338-5p promotes the growth and metastasis of malignant melanoma cells via targeting CD82. Biomed. Pharmacother. 2018, 102, 1195–1202. [Google Scholar] [CrossRef]
- Xu, L.; Hou, Y.; Tu, G.; Chen, Y.; Du, Y.E.; Zhang, H.; Wen, S.; Tang, X.; Yin, J.; Lang, L.; et al. Nuclear Drosha enhances cell invasion via an EGFR-ERK1/2-MMP7 signaling pathway induced by dysregulated miRNA-622/197 and their targets LAMC2 and CD82 in gastric cancer. Cell Death Dis. 2017, 8, e2642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.H.; Yao, Y.L.; Wu, X.Y.; Wu, J.H.; Gu, T.; Chen, L.; Gu, J.H.; Liu, Y.; Xu, L. Anti-miR-362-3p inhibits migration and invasion of human gastric cancer cells by its target CD82. Dig. Dis. Sci. 2015, 60, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Huang, F.; Yao, Y.; Wang, J.; Wei, J.; Wu, Q.; Xiang, S.; Xu, L. Interaction of transforming growth factor-β-Smads/microRNA-362-3p/CD82 mediated by M2 macrophages promotes the process of epithelial-mesenchymal transition in hepatocellular carcinoma cells. Cancer Sci. 2019, 110, 2507–2519. [Google Scholar] [CrossRef]
- Rahman, M.M.; Brane, A.C.; Tollefsbol, T.O. MicroRNAs and epigenetics strategies to reverse breast cancer. Cells 2019, 8, 1214. [Google Scholar] [CrossRef]
- Grimaldi, A.M.; Salvatore, M.; Incoronato, M. miRNA-based therapeutics in breast cancer: A systematic review. Front Oncol. 2021, 11, 668464. [Google Scholar] [CrossRef] [PubMed]
- Rivenbark, A.G.; O’Connor, S.M.; Coleman, W.B. Molecular and cellular heterogeneity in breast cancer: Challenges for personalized medicine. Am. J. Pathol. 2013, 183, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Vagia, E.; Mahalingam, D.; Cristofanilli, M. The landscape of targeted therapies in TNBC. Cancers 2020, 12, 916. [Google Scholar] [CrossRef] [PubMed]
- Ávalos-Moreno, M.; López-Tejada, A.; Blaya-Cánovas, J.L.; Cara-Lupiañez, F.E.; González-González, A.; Lorente, J.A.; Sánchez-Rovira, P.; Granados-Principal, S. Drug repurposing for triple-negative breast cancer. J. Pers. Med. 2020, 10, 200. [Google Scholar] [CrossRef]
- Stipp, C.S.; Kolesnikova, T.V.; Hemler, M.E. Functional domains in tetraspanin proteins. Trends Biochem. Sci. 2003, 28, 106–112. [Google Scholar] [CrossRef]
- Forterre, A.; Komuro, H.; Aminova, S.; Harada, M. A comprehensive review of cancer microRNA therapeutic delivery strategies. Cancers 2020, 12, 1852. [Google Scholar] [CrossRef]
- Lum, L.G.; Tushir-Singh, J. Arming “old guards” with “new dual-targeting weapons”. Cancer Cell 2021, 39, 604–606. [Google Scholar] [CrossRef]
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef] [PubMed]
- Odle, T.G. Precision medicine in breast cancer. Radiol. Technol. 2017, 88, 401M–421M. [Google Scholar] [PubMed]
- Nandy, A.; Gangopadhyay, S.; Mukhopadhyay, A. Individualizing breast cancer treatment - The dawn of personalized medicine. Exp. Cell Res. 2014, 320, 1–11. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viera, M.; Yip, G.W.C.; Shen, H.-M.; Baeg, G.H.; Bay, B.H. Targeting CD82/KAI1 for Precision Therapeutics in Surmounting Metastatic Potential in Breast Cancer. Cancers 2021, 13, 4486. https://doi.org/10.3390/cancers13174486
Viera M, Yip GWC, Shen H-M, Baeg GH, Bay BH. Targeting CD82/KAI1 for Precision Therapeutics in Surmounting Metastatic Potential in Breast Cancer. Cancers. 2021; 13(17):4486. https://doi.org/10.3390/cancers13174486
Chicago/Turabian StyleViera, Maximillian, George Wai Cheong Yip, Han-Ming Shen, Gyeong Hun Baeg, and Boon Huat Bay. 2021. "Targeting CD82/KAI1 for Precision Therapeutics in Surmounting Metastatic Potential in Breast Cancer" Cancers 13, no. 17: 4486. https://doi.org/10.3390/cancers13174486
APA StyleViera, M., Yip, G. W. C., Shen, H.-M., Baeg, G. H., & Bay, B. H. (2021). Targeting CD82/KAI1 for Precision Therapeutics in Surmounting Metastatic Potential in Breast Cancer. Cancers, 13(17), 4486. https://doi.org/10.3390/cancers13174486