
Niraparib Suppresses Cholangiocarcinoma Tumor Growth by Inducing Oxidative and Replication Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Genetic Alterations in DDR Genes in CCA
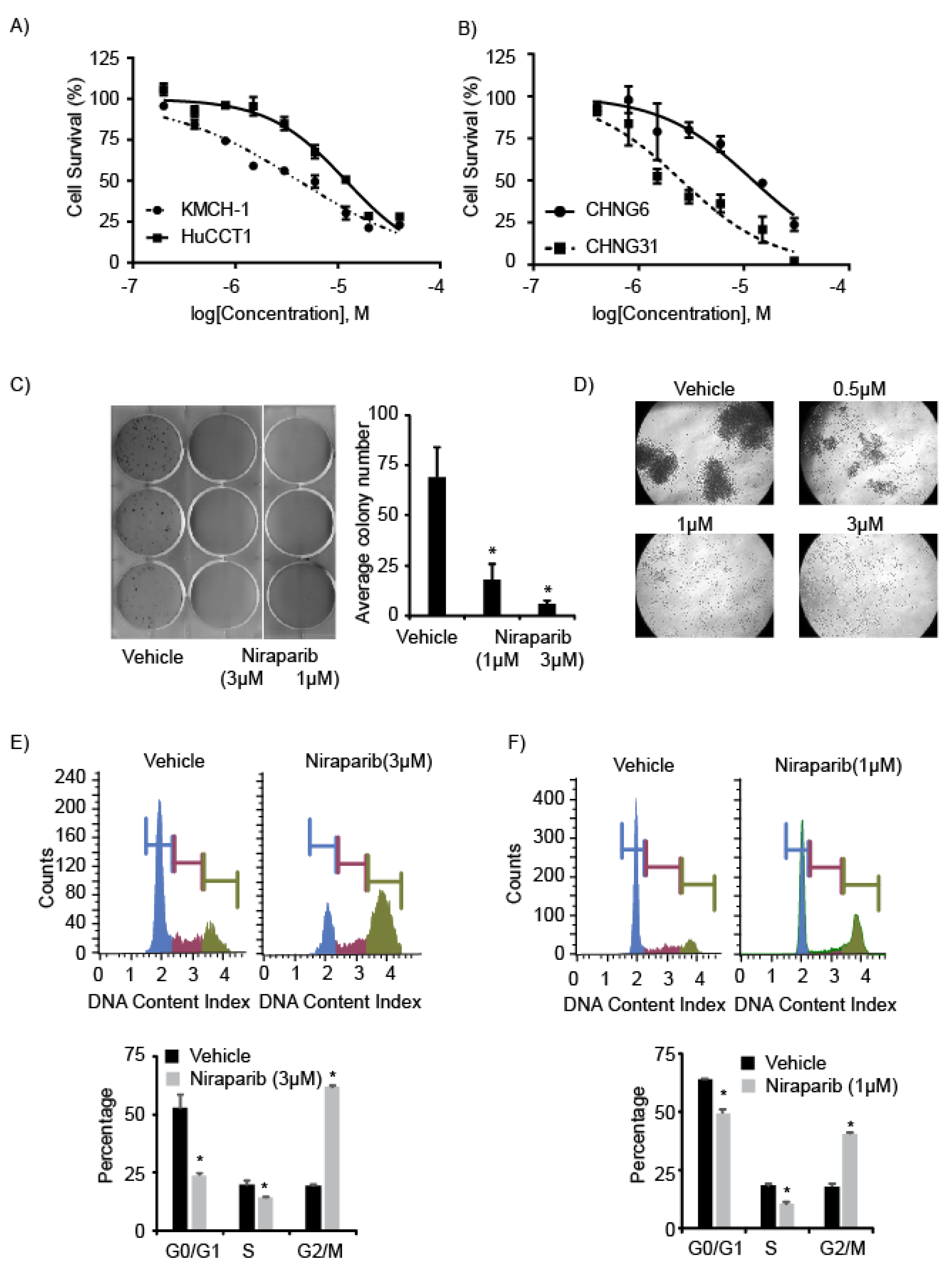
2.2. PARP Inhibitors Suppress Cell Survival and Proliferation
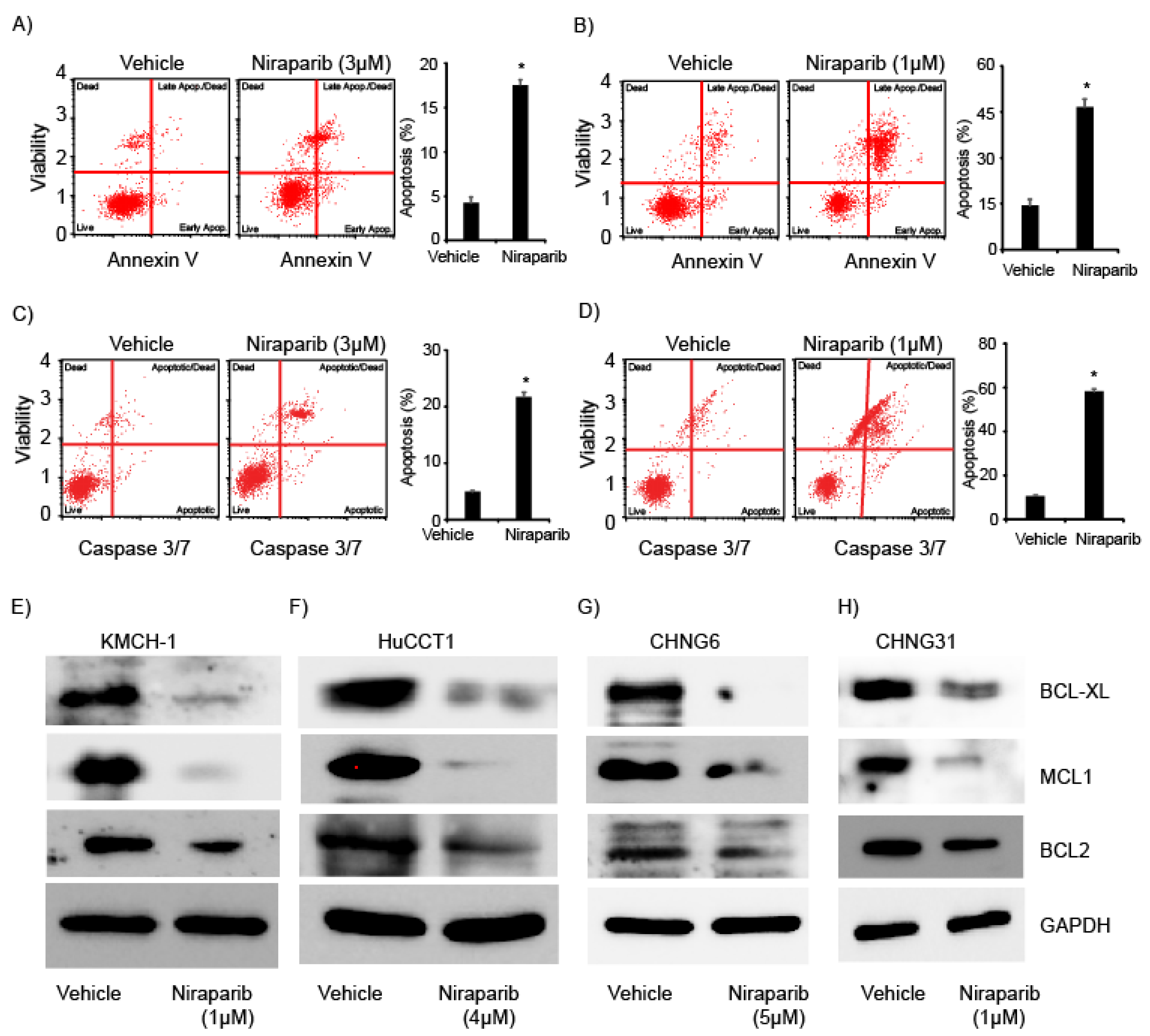
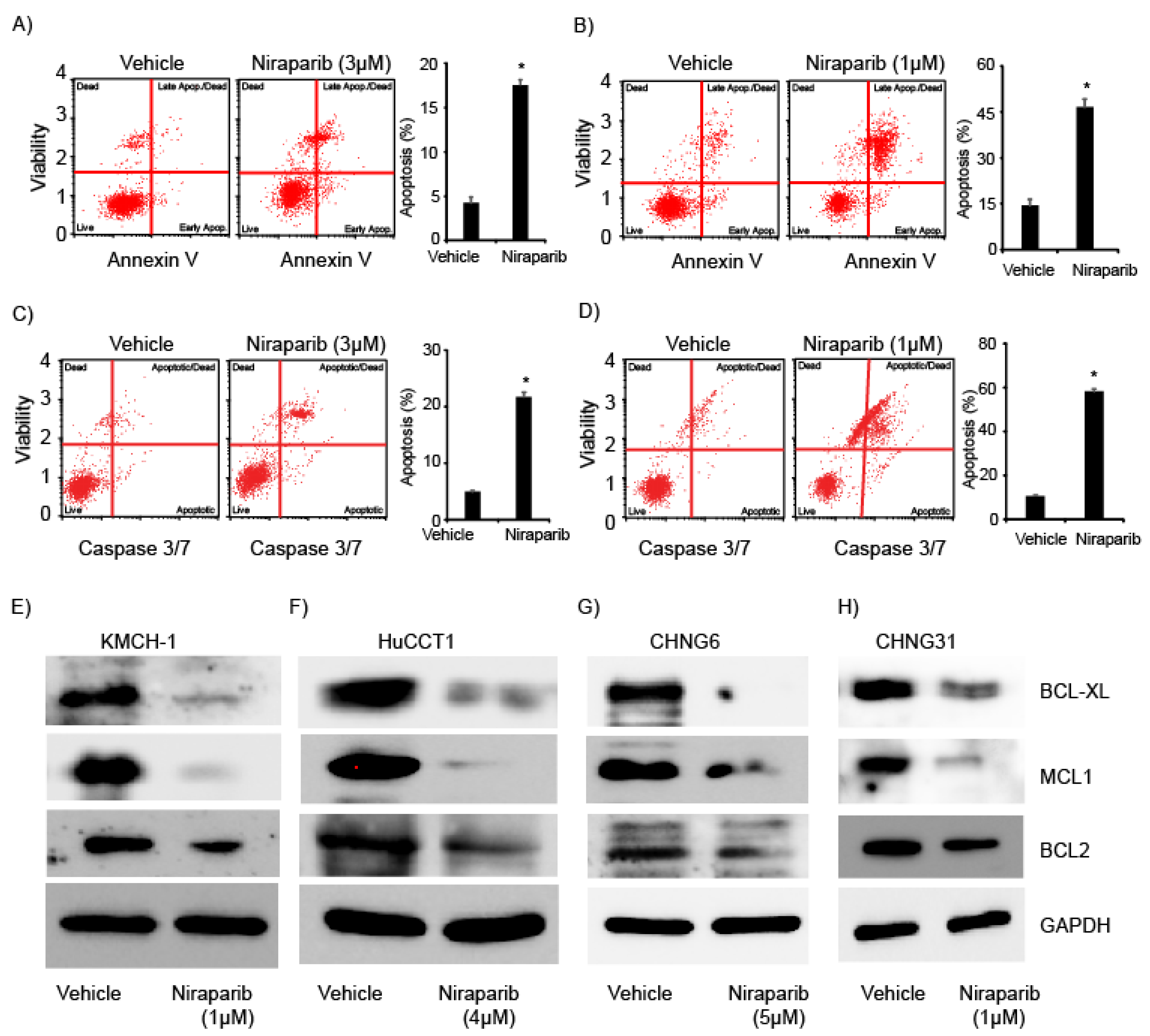
2.3. Niraparib Induces Apoptosis and Suppress Expression of Anti-Apoptotic Proteins
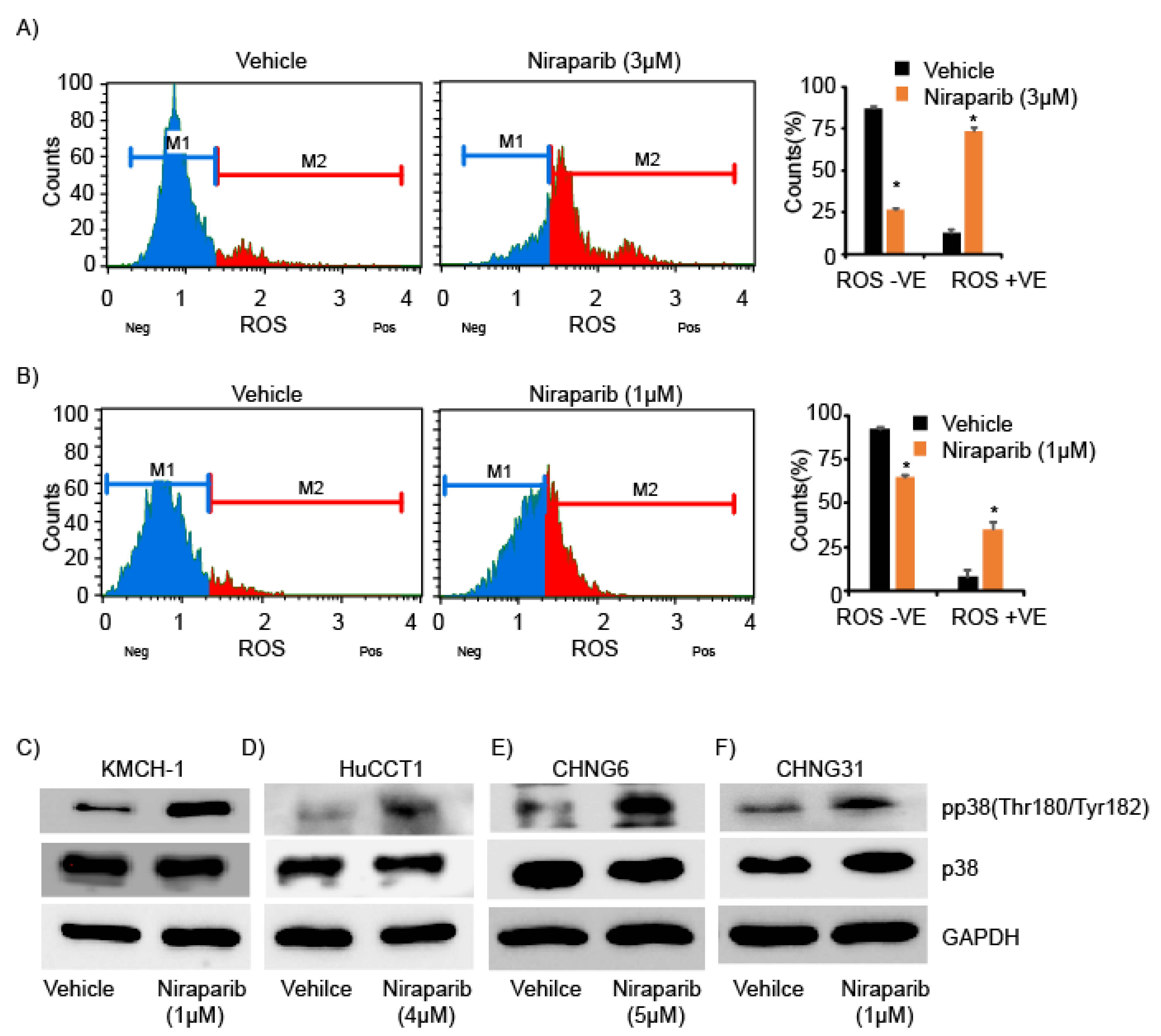
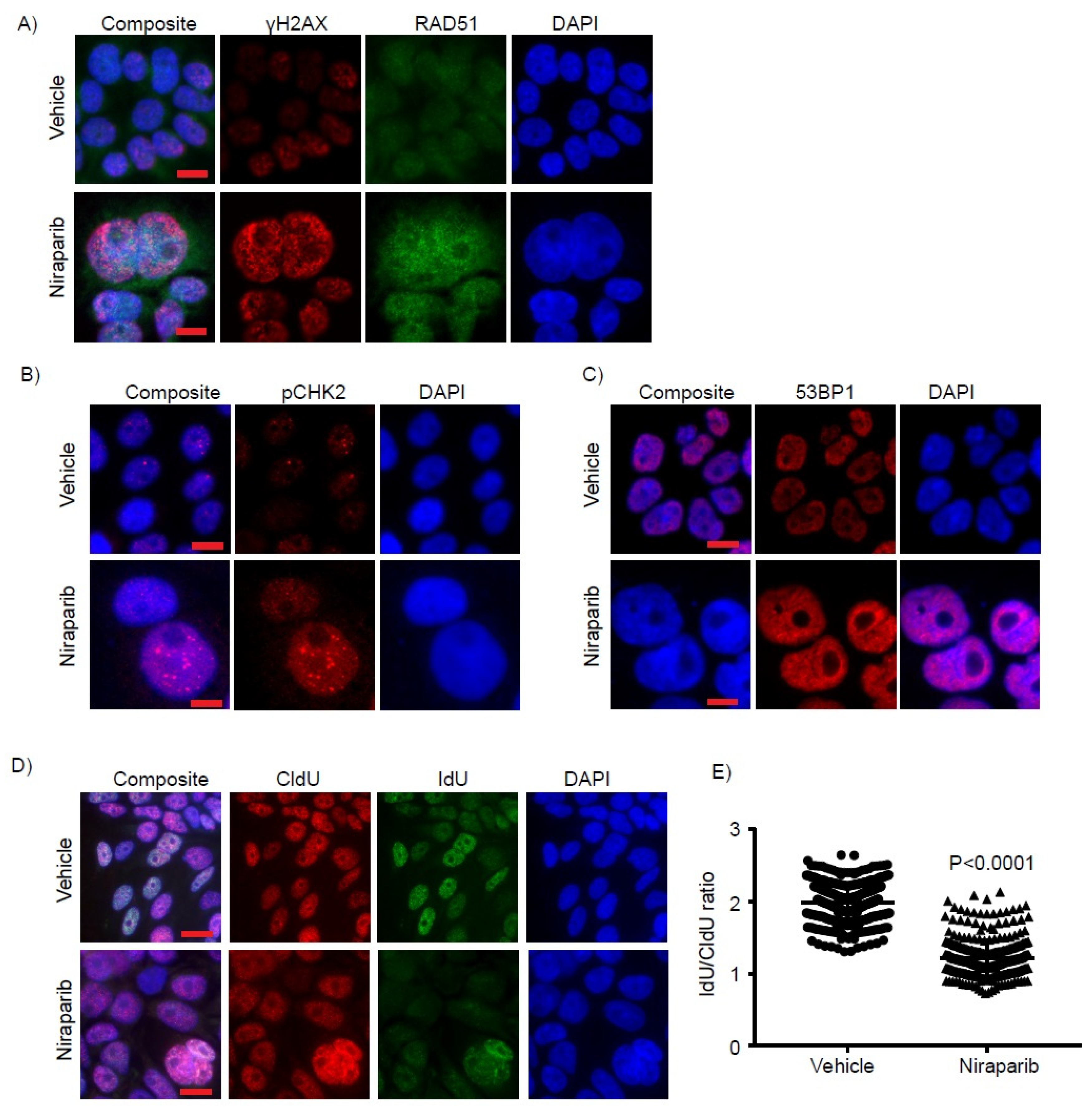
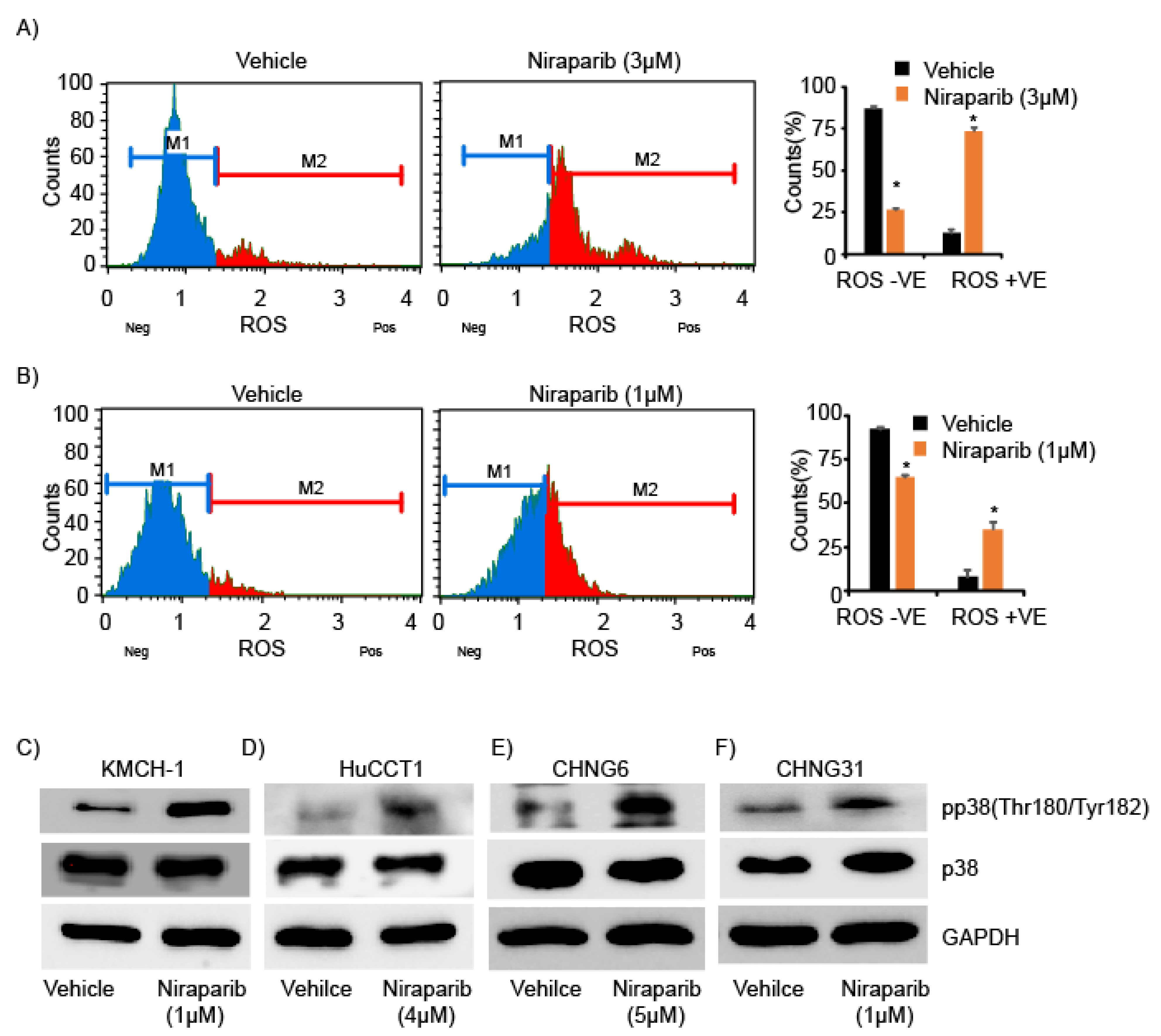
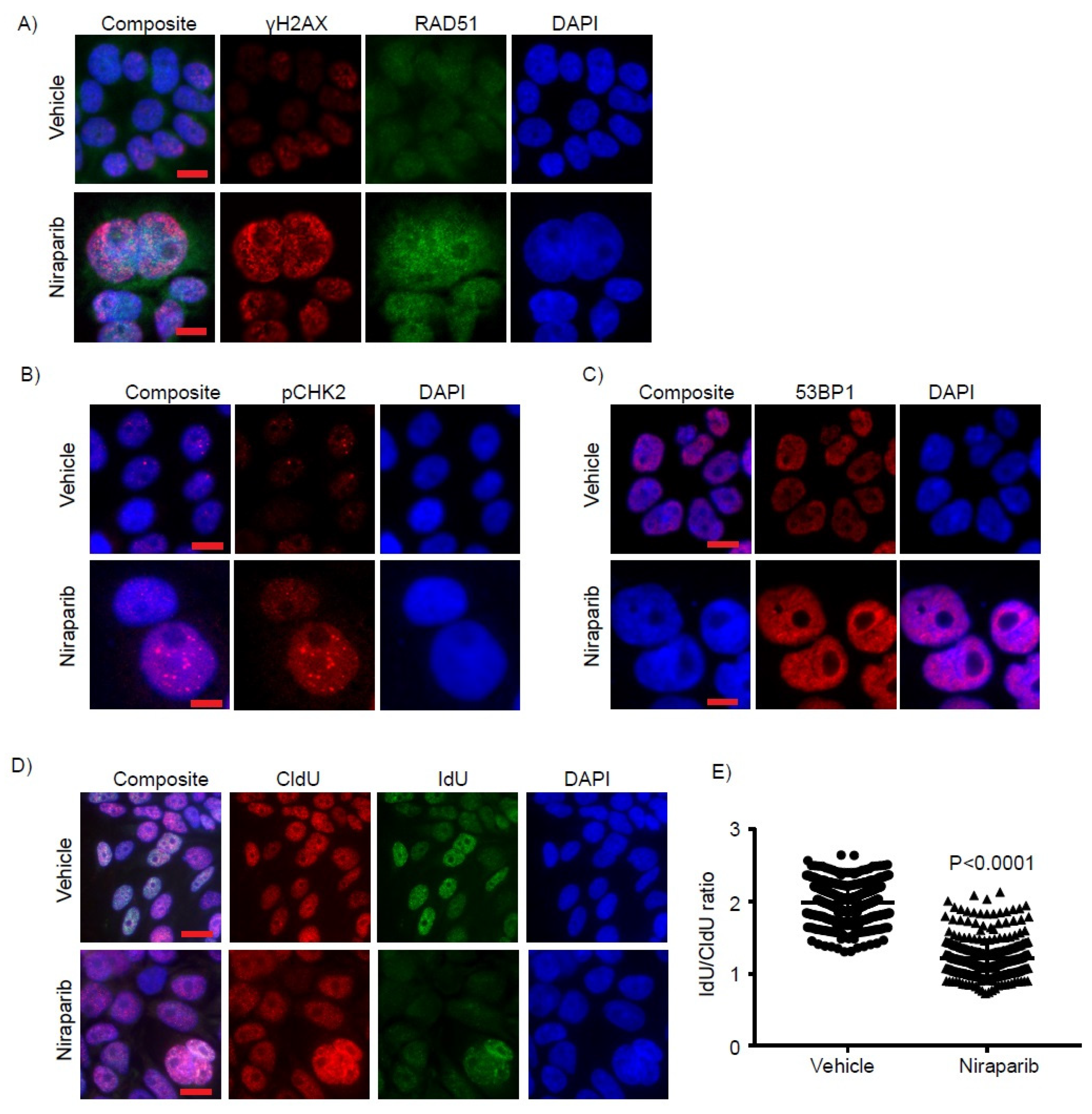
2.4. Niraparib Induces DNA Damage, Oxidative Stress, and Replication Fork Stalling
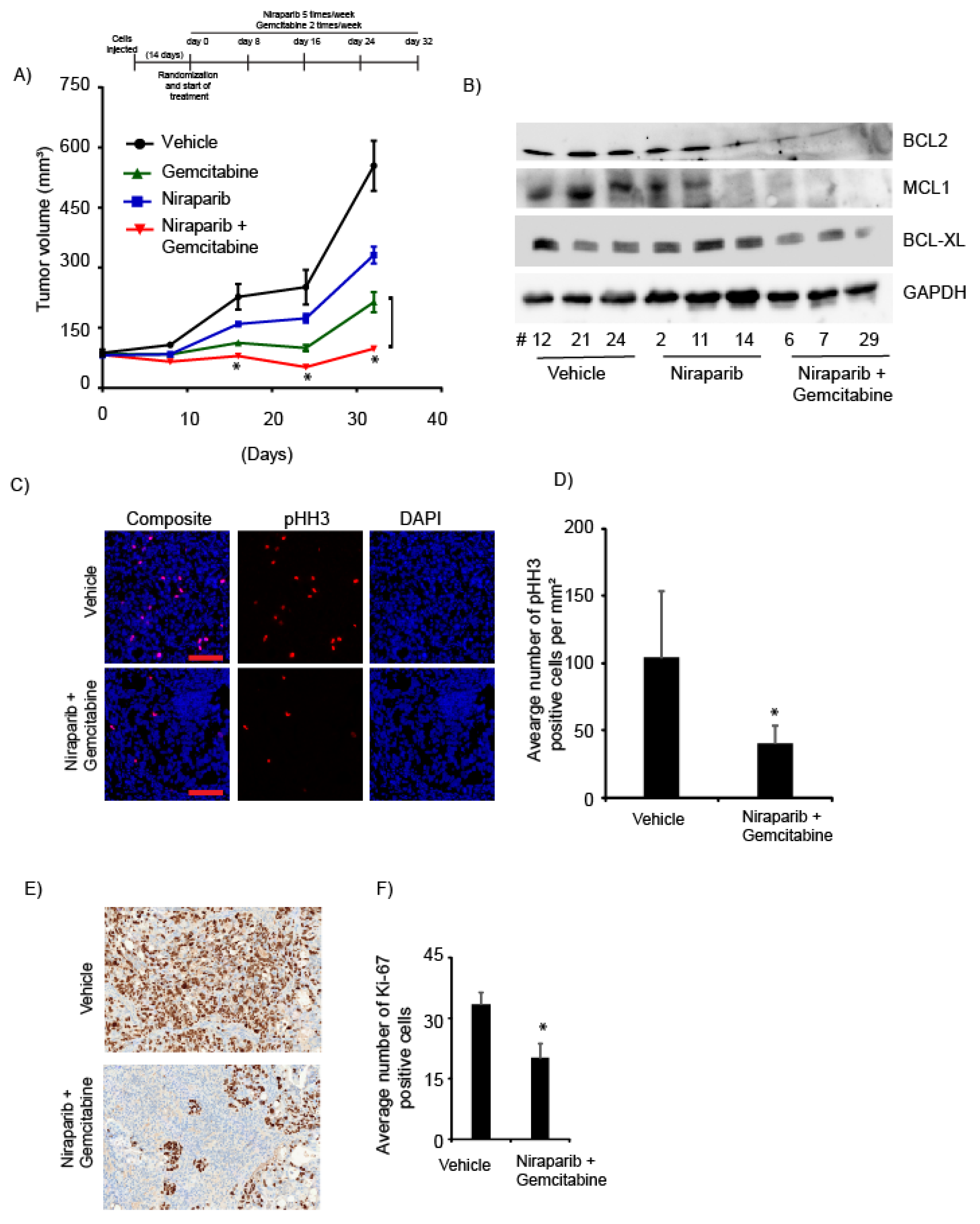
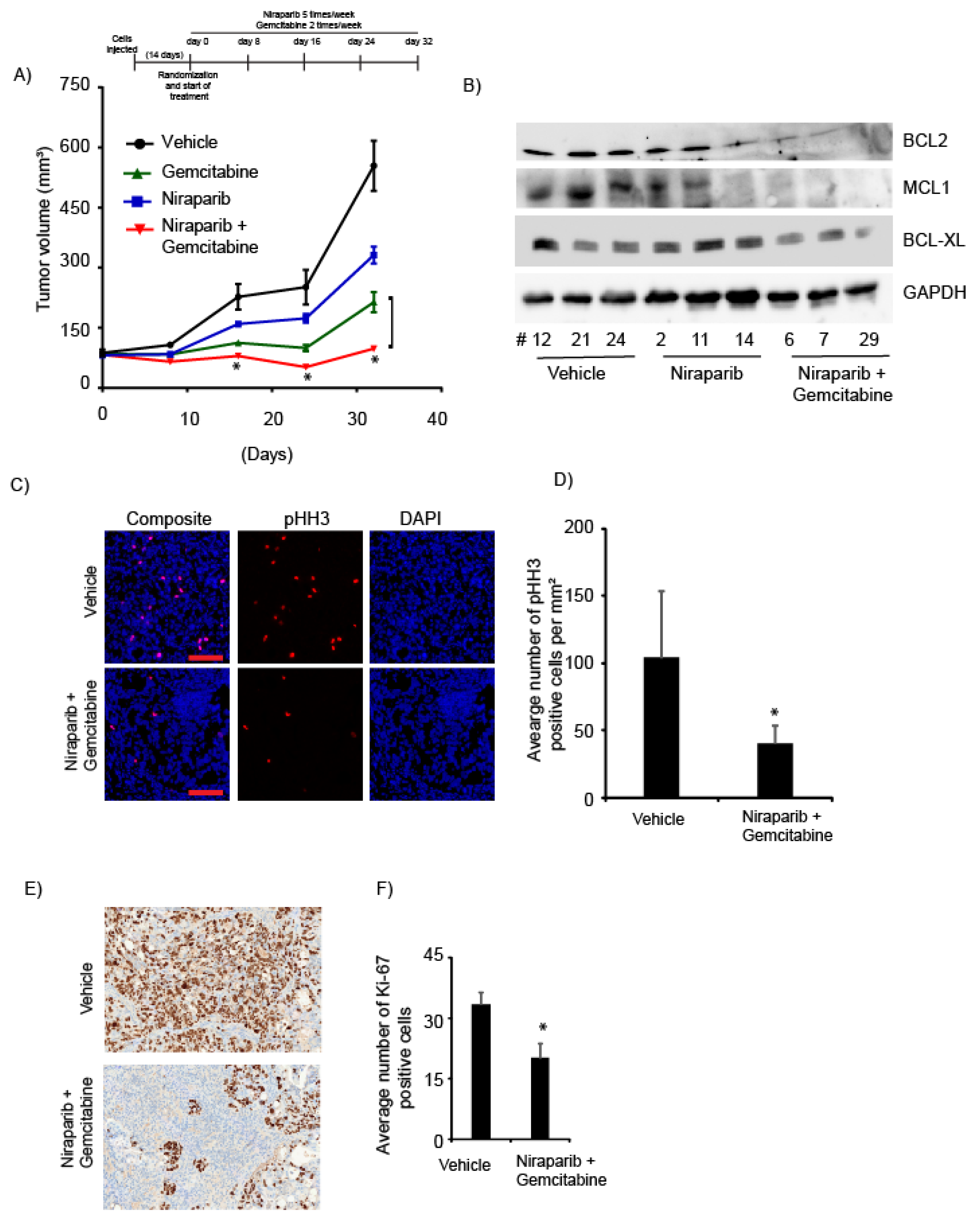
2.5. Niraparib in Combination with Gemcitabine Suppressed In Vivo Tumor Growth
3. Discussion
4. Material and Methods
4.1. Cell Culture and Patient-Derived Xenograft Mouse Models
4.2. TCGA Dataset for DDR Mutation Analysis
4.3. Colony Formation, Spheroid Assay and Drugs
4.4. Cell Survival Assay
4.5. Immunofluorescence and Immunohistochemistry
4.6. Oxidative Stress, Annexin V, Cell Cycle and Caspase 3/7 Assays
4.7. Pulse-Labeling of DNA Replication by CldU and IdU
4.8. Western Blot Analysis
4.9. In Vivo Animal Study
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Njei, B. Changing pattern of epidemiology in intrahepatic cholangiocarcinoma. Hepatology 2014, 60, 1107–1108. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New horizons for precision medicine in biliary tract cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.A.; Taylor-Robinson, S.D.; Toledano, M.B.; Beck, A.; Elliott, P.; Thomas, H.C. Changing international trends in mortality rates for liver, biliary and pancreatic tumours. J. Hepatol. 2002, 37, 806–813. [Google Scholar] [CrossRef]
- von Hahn, T.; Ciesek, S.; Wegener, G.; Plentz, R.R.; Weismuller, T.J.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Malek, N.P. Epidemiological trends in incidence and mortality of hepatobiliary cancers in germany. Scand. J. Gastroenterol. 2011, 46, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Kim, B.; Sanderson, S.O.; Sauver, J.S.; Yawn, B.P.; Larson, J.J.; Therneau, T.M.; Roberts, L.R.; Gores, G.J.; Kim, W.R. Biliary tract cancers in olmsted county, minnesota, 1976–2008. Am. J. Gastroenterol. 2012, 107, 1256–1262. [Google Scholar] [CrossRef] [Green Version]
- Pinter, M.; Hucke, F.; Zielonke, N.; Waldhor, T.; Trauner, M.; Peck-Radosavljevic, M.; Sieghart, W. Incidence and mortality trends for biliary tract cancers in austria. Liver Int. 2014, 34, 1102–1108. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct idh-mutant molecular profiles. Cell Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable fgfr gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, A.; Frega, G.; Ricci, A.D.; Palloni, A.; Abbati, F.; Stefania, D.E.L.; Deserti, M.; Tavolari, S.; Brandi, G. Anti-egfr monoclonal antibodies in advanced biliary tract cancer: A systematic review and meta-analysis. In Vivo 2020, 34, 479–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA repair in cancer: Beyond parp inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P. Biology of poly(adp-ribose) polymerases: The factotums of cell maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of parp1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Wilson, D.M., 3rd. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Caldecott, K.W. Perspectives on parps in s phase. Trends Genet. 2019, 35, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: Parp inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA damaging revolution: Parp inhibitors and beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Paschalis, A.; de Bono, J.S. Prostate cancer 2020: “The times they are a’changing”. Cancer Cell 2020, 38, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Raitses-Gurevich, M.; Kelley, R.K.; Bocobo, A.G.; Borgida, A.; Shroff, R.T.; Holter, S.; Gallinger, S.; Ahn, D.H.; Aderka, D.; et al. Overall survival and clinical characteristics of brca-associated cholangiocarcinoma: A multicenter retrospective study. Oncologist 2017, 22, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance olaparib for germline brca-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Jiang, Y.; Yang, X.B.; Wang, A.Q.; Zheng, Y.C.; Wan, X.S.; Sang, X.T.; Wang, K.; Zhang, D.D.; Xu, J.J.; et al. Response of brca1-mutated gallbladder cancer to olaparib: A case report. World J. Gastroenterol. 2016, 22, 10254–10259. [Google Scholar] [CrossRef]
- Yamada, T.; Egashira, N.; Bando, A.; Nishime, Y.; Tonogai, Y.; Imuta, M.; Yano, T.; Oishi, R. Activation of p38 mapk by oxidative stress underlying epirubicin-induced vascular endothelial cell injury. Free Radic. Biol. Med. 2012, 52, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B.; Gores, G.J. Cholangiocarcinoma: Advances in pathogenesis, diagnosis, and treatment. Hepatology 2008, 48, 308–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeini, A.; Sia, D.; Bardeesy, N.; Mazzaferro, V.; Llovet, J.M. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin. Cancer Res. 2016, 22, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [Green Version]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Biliary tract cancer: State of the art and potential role of DNA damage repair. Cancer Treat. Rev. 2018, 70, 168–177. [Google Scholar] [CrossRef]
- Adeva, J.; Sangro, B.; Salati, M.; Edeline, J.; La Casta, A.; Bittoni, A.; Berardi, R.; Bruix, J.; Valle, J.W. Medical treatment for cholangiocarcinoma. Liver Int. 2019, 39, 123–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Kraus, W.L. On par with parp: Cellular stress signaling through poly(adp-ribose) and parp-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for parp-1 for the assembly or stability of xrcc1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantzer, F.; Schreiber, V.; Niedergang, C.; Trucco, C.; Flatter, E.; De La Rubia, G.; Oliver, J.; Rolli, V.; Menissier-de Murcia, J.; de Murcia, G. Involvement of poly(adp-ribose) polymerase in base excision repair. Biochimie 1999, 81, 69–75. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. Parp inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific parp trapping by bmn 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of parp1 and parp2 by clinical parp inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Halweg, C.J.; Menendez, D.; Resnick, M.A. Differential effects of poly(adp-ribose) polymerase inhibition on DNA break repair in human cells are revealed with epstein-barr virus. Proc. Natl. Acad. Sci. USA 2012, 109, 6590–6595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, H.C.; Kapuriya, N.; Kulp, S.K.; Chen, C.S.; Shapiro, C.L. Differential anti-proliferative activities of poly(adp-ribose) polymerase (parp) inhibitors in triple-negative breast cancer cells. Breast Cancer Res. Treat. 2012, 134, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, D.; Liu, Z.; Xu, X.; Liu, Q.; Zhang, X.; Kong, B.; Wei, J.J.; Gong, Y.; Shao, C. Increased oxidative stress mediates the antitumor effect of parp inhibition in ovarian cancer. Redox. Biol. 2018, 17, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Ursu, S.; Majid, S.; Garger, C.; de Semir, D.; Bezrookove, V.; Desprez, P.Y.; McAllister, S.; Soroceanu, L.; Nosrati, M.; Yimam, K.; et al. Novel tumor suppressor role of mirna-876 in cholangiocarcinoma. Oncogenesis 2019, 8, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moloney, J.N.; Cotter, T.G. Ros signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Davalli, P.; Marverti, G.; Lauriola, A.; D’Arca, D. Targeting oxidatively induced DNA damage response in cancer: Opportunities for novel cancer therapies. Oxid. Med. Cell. Longev. 2018, 2018, 2389523. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Minn, K.; van Deursen, J.; Chen, J. P53 binding protein 53bp1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 2003, 23, 2556–2563. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.H.; Huang, Y.F.; Lin, T.Y.; Shieh, S.Y. The cell cycle checkpoint kinase chk2 mediates DNA damage-induced stabilization of ttk/hmps1. Oncogene 2009, 28, 1366–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, T.; De Nicola, F.; Iezzi, S.; Lecis, D.; D’Angelo, C.; Di Padova, M.; Corbi, N.; Dimiziani, L.; Zannini, L.; Jekimovs, C.; et al. Che-1 phosphorylation by atm/atr and chk2 kinases activates p53 transcription and the g2/m checkpoint. Cancer Cell 2006, 10, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Vikas, P.; Borcherding, N.; Zhang, W. The clinical promise of immunotherapy in triple-negative breast cancer. Cancer Manag. Res. 2018, 10, 6823–6833. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.R.; Bergmann, T.K.; Mau-Sorensen, M.; Christensen, R.D.; Avall-Lundqvist, E.; Birrer, M.J.; Jorgensen, M.; Roed, H.; Malander, S.; Nielsen, F.; et al. A phase i study of the parp inhibitor niraparib in combination with bevacizumab in platinum-sensitive epithelial ovarian cancer: Nsgo avanova1/engot-ov24. Cancer Chemother. Pharmacol. 2019, 84, 791–798. [Google Scholar] [CrossRef]
- Friedlander, M.; Meniawy, T.; Markman, B.; Mileshkin, L.; Harnett, P.; Millward, M.; Lundy, J.; Freimund, A.; Norris, C.; Mu, S.; et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: Results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 2019, 20, 1306–1315. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, J.; Li, R.; Han, Z.; Li, L.; Li, G.; Yang, B.; Yin, Q.; Wang, Y.; Ke, Y.; et al. Effectiveness of olaparib treatment in a patient with gallbladder cancer with an atm-inactivating mutation. Oncologist 2020, 25, 375–379. [Google Scholar] [CrossRef] [Green Version]
- Sabbatino, F.; Liguori, L.; Malapelle, U.; Schiavi, F.; Tortora, V.; Conti, V.; Filippelli, A.; Tortora, G.; Ferrone, C.R.; Pepe, S. Case report: Bap1 mutation and rad21 amplification as predictive biomarkers to parp inhibitor in metastatic intrahepatic cholangiocarcinoma. Front. Oncol. 2020, 10, 567289. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Majid, S.; Bezrookove, V.; Phan, B.; Ursu, S.; Nosrati, M.; De Semir, D.; Sagebiel, R.W.; Miller, J.R., 3rd; Debs, R.; et al. Bptf transduces mitf-driven prosurvival signals in melanoma cells. Proc. Natl. Acad. Sci. USA 2016, 113, 6254–6258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saqub, H.; Proetsch-Gugerbauer, H.; Bezrookove, V.; Nosrati, M.; Vaquero, E.M.; de Semir, D.; Ice, R.J.; McAllister, S.; Soroceanu, L.; Kashani-Sabet, M.; et al. Dinaciclib, a cyclin-dependent kinase inhibitor, suppresses cholangiocarcinoma growth by targeting cdk2/5/9. Sci. Rep. 2020, 10, 18489. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Nosrati, M.; Bezrookove, V.; de Semir, D.; Majid, S.; Thummala, S.; Sun, V.; Tong, S.; Leong, S.P.; Minor, D.; et al. The role of bptf in melanoma progression and in response to braf-targeted therapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezrookove, V.; Patino, J.M.; Nosrati, M.; Desprez, P.-Y.; McAllister, S.; Soroceanu, L.; Baron, A.; Osorio, R.; Kashani-Sabet, M.; Dar, A.A. Niraparib Suppresses Cholangiocarcinoma Tumor Growth by Inducing Oxidative and Replication Stress. Cancers 2021, 13, 4405. https://doi.org/10.3390/cancers13174405
Bezrookove V, Patino JM, Nosrati M, Desprez P-Y, McAllister S, Soroceanu L, Baron A, Osorio R, Kashani-Sabet M, Dar AA. Niraparib Suppresses Cholangiocarcinoma Tumor Growth by Inducing Oxidative and Replication Stress. Cancers. 2021; 13(17):4405. https://doi.org/10.3390/cancers13174405
Chicago/Turabian StyleBezrookove, Vladimir, John M. Patino, Mehdi Nosrati, Pierre-Yves Desprez, Sean McAllister, Liliana Soroceanu, Ari Baron, Robert Osorio, Mohammed Kashani-Sabet, and Altaf A. Dar. 2021. "Niraparib Suppresses Cholangiocarcinoma Tumor Growth by Inducing Oxidative and Replication Stress" Cancers 13, no. 17: 4405. https://doi.org/10.3390/cancers13174405
APA StyleBezrookove, V., Patino, J. M., Nosrati, M., Desprez, P.-Y., McAllister, S., Soroceanu, L., Baron, A., Osorio, R., Kashani-Sabet, M., & Dar, A. A. (2021). Niraparib Suppresses Cholangiocarcinoma Tumor Growth by Inducing Oxidative and Replication Stress. Cancers, 13(17), 4405. https://doi.org/10.3390/cancers13174405