
Inhibition of the Eukaryotic 80S Ribosome as a Potential Anticancer Therapy: A Structural Perspective

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
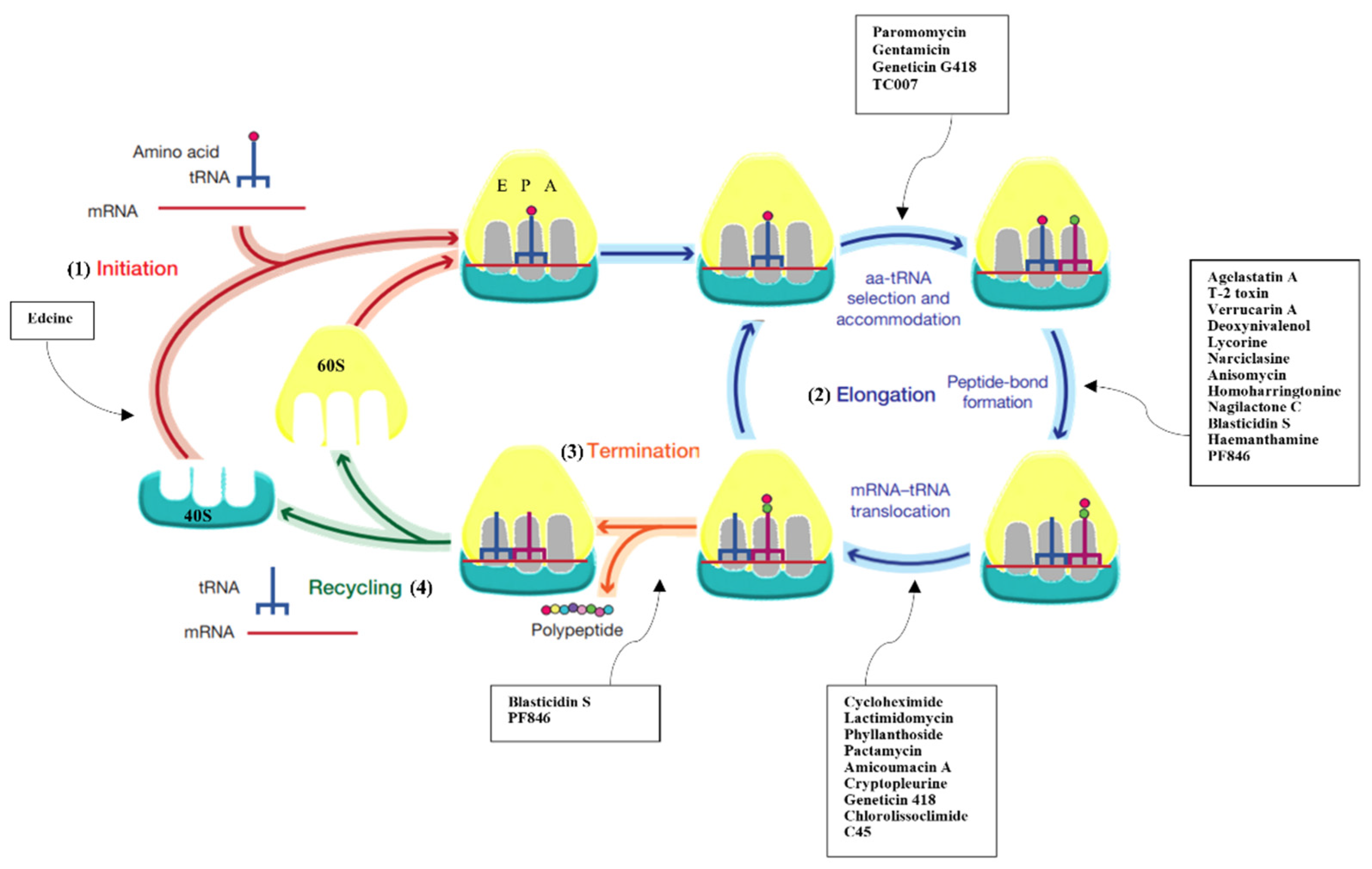
1. Introduction
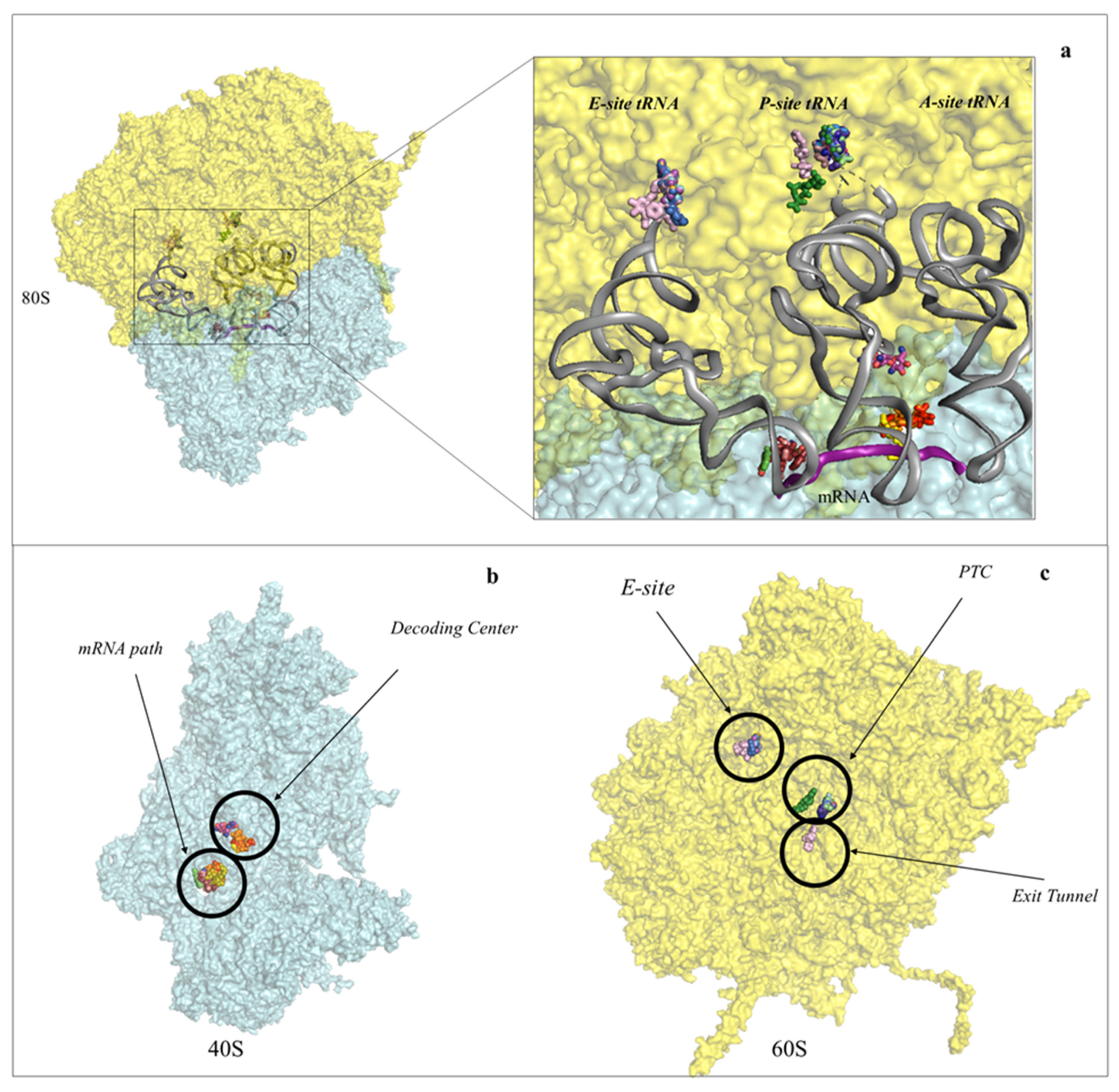
2. Inhibitors Targeting the Large Ribosomal Subunit
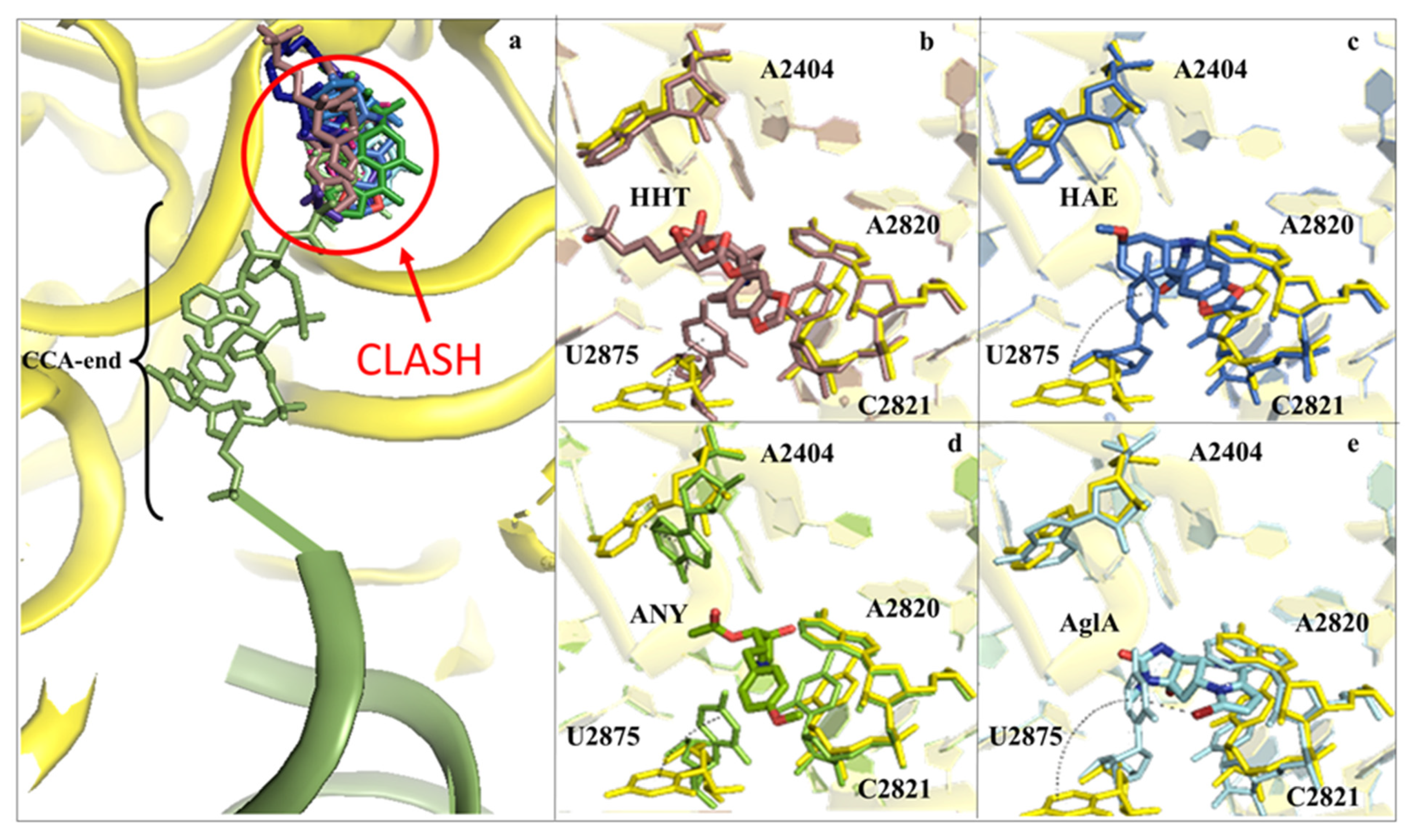
2.1. Drugs Binding to the 60S LSU Peptidyl Transferase Centre A-Site
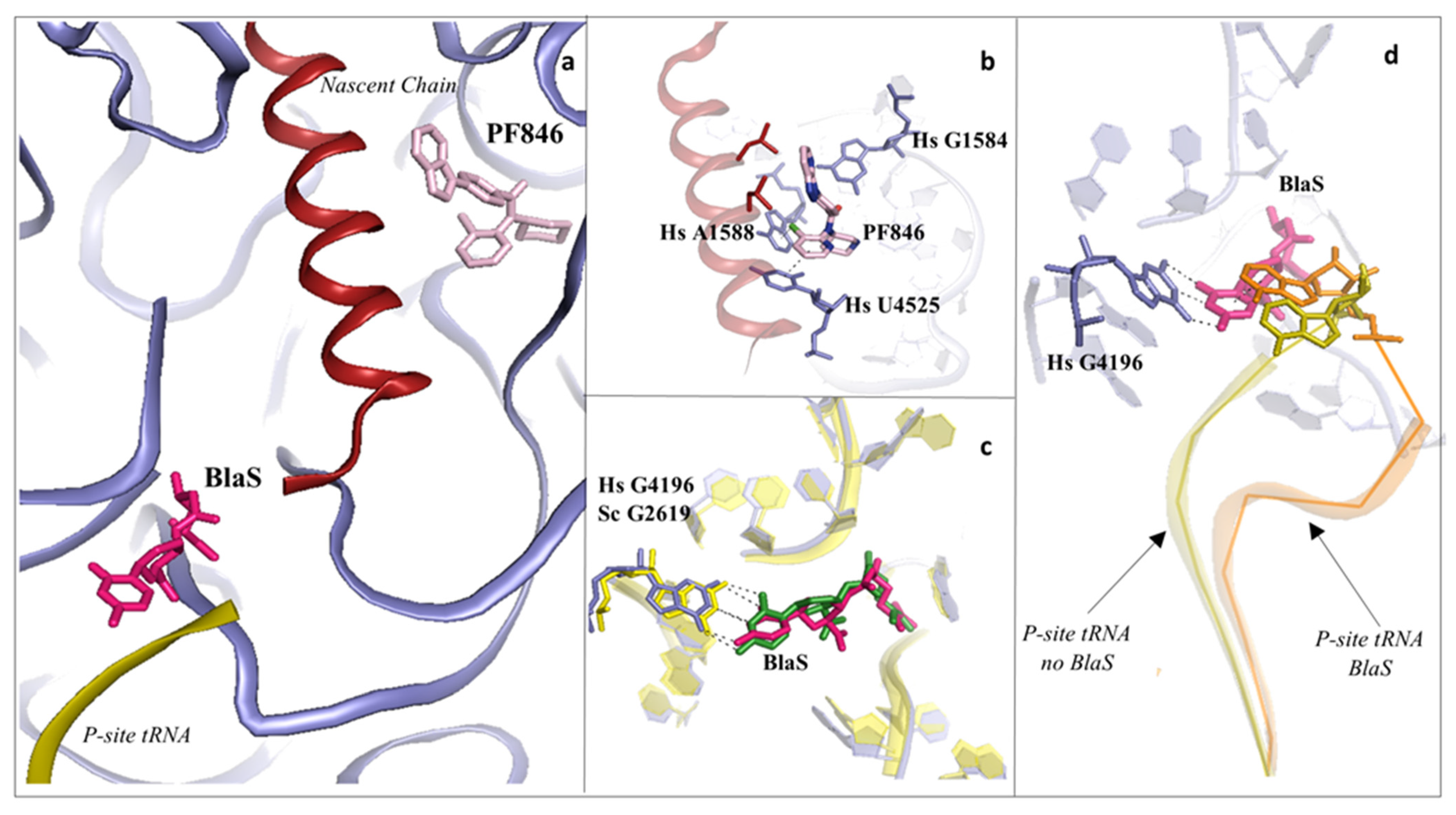
2.2. Drugs Binding to the Peptide Exit Tunnel
2.3. Drugs Binding to the 60S LSU Peptidyl Transferase Centre P-Site
2.4. Drugs Binding to the 60S LSU tRNA E-Site
3. Inhibitors Targeting the 40S Small Ribosomal Subunit
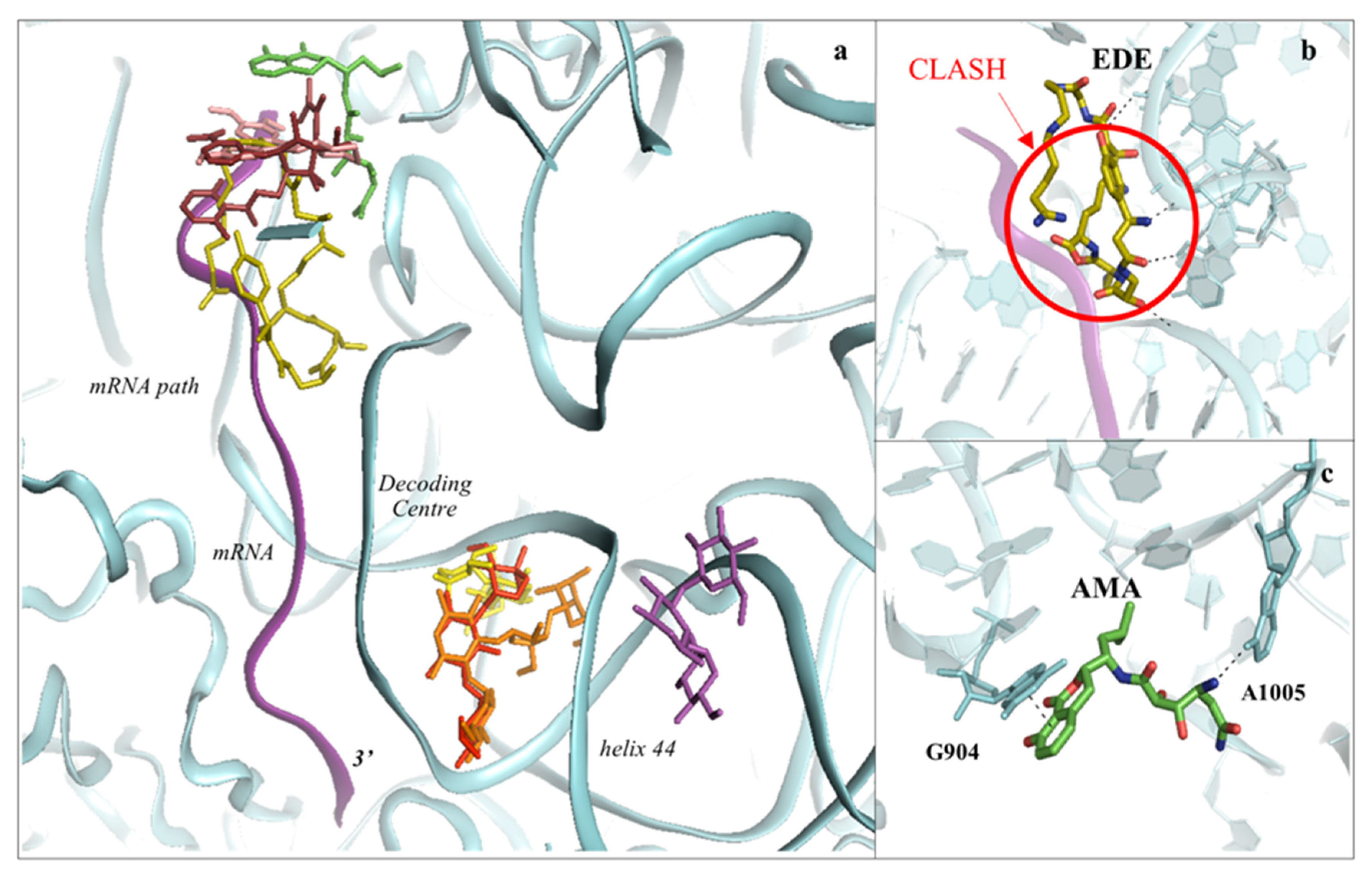
3.1. Drugs Binding to the mRNA Path
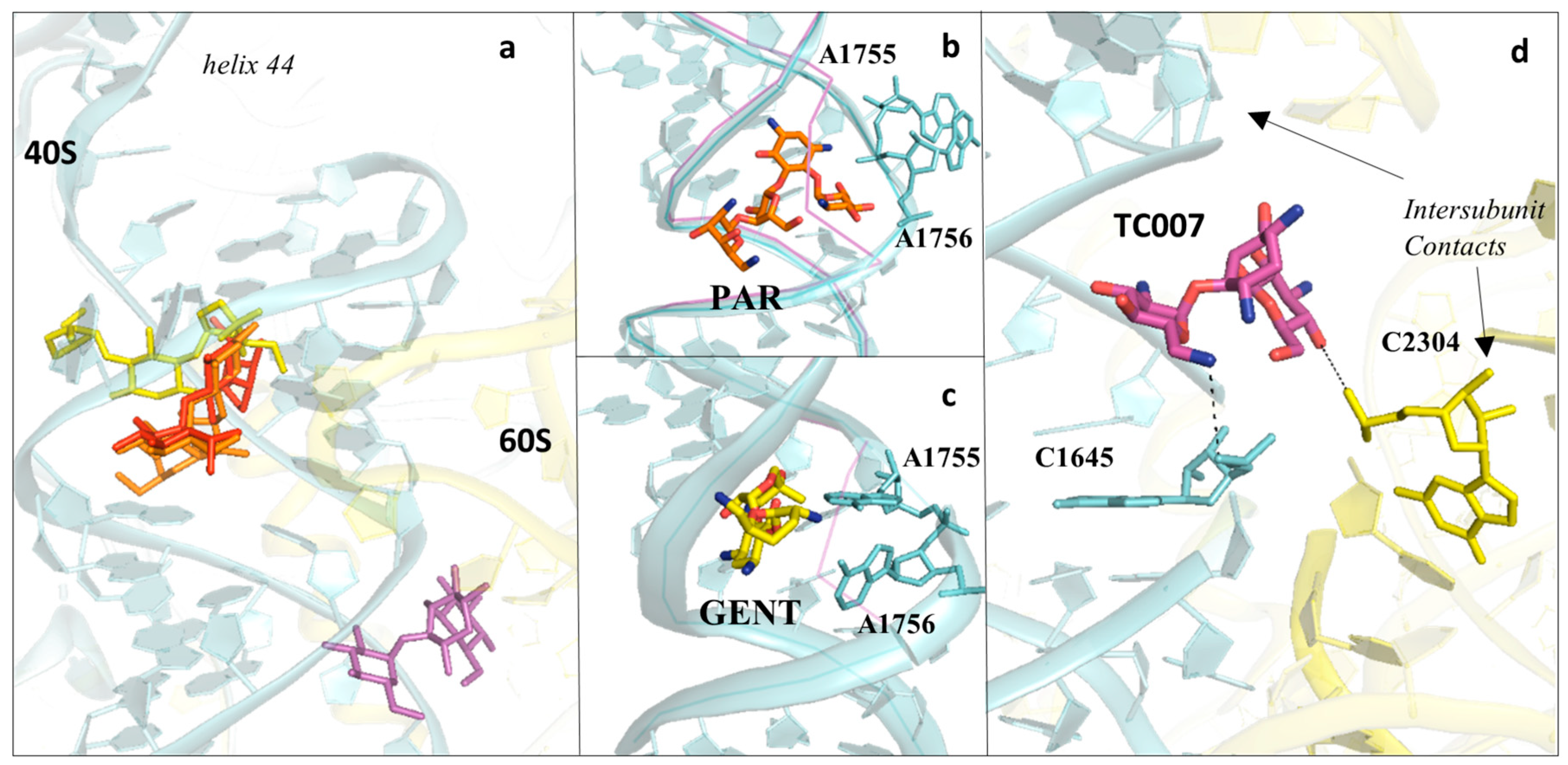
3.2. Drugs Binding to the Decoding Centre
3.3. Aminoglycosides Binding to the Eukaryotic Decoding Centre
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Melnikov, S.; Ben-Shem, A.; Garreau de Loubresse, N.; Jenner, L.; Yusupova, G.; Yusupov, M. One Core, Two Shells: Bacterial and Eukaryotic Ribosomes. Nat. Struct. Mol. Biol. 2012, 19, 560–567. [Google Scholar] [CrossRef]
- Yusupova, G.; Yusupov, M. Crystal Structure of Eukaryotic Ribosome and Its Complexes with Inhibitors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160184. [Google Scholar] [CrossRef]
- Yusupov, M.M.; Yusupova, G.Z.; Baucom, A.; Lieberman, K.; Earnest, T.N.; Cate, J.H.; Noller, H.F. Crystal Structure of the Ribosome at 5.5 A Resolution. Science 2001, 292, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Selmer, M.; Dunham, C.M.; Murphy, F.V., 4th; Weixlbaumer, A.; Petry, S.; Kelley, A.C.; Weir, J.R.; Ramakrishnan, V. Structure of the 70S Ribosome Complexed with MRNA and TRNA. Science 2006, 313, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Harms, J.; Schluenzen, F.; Zarivach, R.; Bashan, A.; Gat, S.; Agmon, I.; Bartels, H.; Franceschi, F.; Yonath, A. High Resolution Structure of the Large Ribosomal Subunit from a Mesophilic Eubacterium. Cell 2001, 107, 679–688. [Google Scholar] [CrossRef]
- Schmeing, T.M.; Ramakrishnan, V. What Recent Ribosome Structures Have Revealed about the Mechanism of Translation. Nature 2009, 461, 1234–1242. [Google Scholar] [CrossRef]
- Wimberly, B.T.; Brodersen, D.E.; Clemons, W.M.J.; Morgan-Warren, R.J.; Carter, A.P.; Vonrhein, C.; Hartsch, T.; Ramakrishnan, V. Structure of the 30S Ribosomal Subunit. Nature 2000, 407, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The Complete Atomic Structure of the Large Ribosomal Subunit at 2.4 A Resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Schuwirth, B.S.; Borovinskaya, M.A.; Hau, C.W.; Zhang, W.; Vila-Sanjurjo, A.; Holton, J.M.; Cate, J.H.D. Structures of the Bacterial Ribosome at 3.5 A Resolution. Science 2005, 310, 827–834. [Google Scholar] [CrossRef]
- Lake, J.A. Ribosome Structure Determined by Electron Microscopy of Escherichia Coli Small Subunits, Large Subunits and Monomeric Ribosomes. J. Mol. Biol. 1976, 105, 131–139. [Google Scholar] [CrossRef]
- Yusupova, G.; Jenner, L.; Rees, B.; Moras, D.; Yusupov, M. Structural Basis for Messenger RNA Movement on the Ribosome. Nature 2006, 444, 391–394. [Google Scholar] [CrossRef]
- Rozov, A.; Demeshkina, N.; Westhof, E.; Yusupov, M.; Yusupova, G. Structural Insights into the Translational Infidelity Mechanism. Nat. Commun. 2015, 6, 7251. [Google Scholar] [CrossRef]
- Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The Structure of the Eukaryotic Ribosome at 3.0 Å Resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef]
- Arenz, S.; Wilson, D.N. Bacterial Protein Synthesis as a Target for Antibiotic Inhibition. Cold Spring Harb. Perspect. Med. 2016, 6, a025361. [Google Scholar] [CrossRef]
- Polikanov, Y.S.; Aleksashin, N.A.; Beckert, B.; Wilson, D.N. The Mechanisms of Action of Ribosome-Targeting Peptide Antibiotics. Front. Mol. Biosci. 2018, 5, 48. [Google Scholar] [CrossRef]
- Vázquez-Laslop, N.; Mankin, A.S. How Macrolide Antibiotics Work. Trends Biochem. Sci. 2018, 43, 668–684. [Google Scholar] [CrossRef]
- Lin, J.; Zhou, D.; Steitz, T.A.; Polikanov, Y.S.; Gagnon, M.G. Ribosome-Targeting Antibiotics: Modes of Action, Mechanisms of Resistance, and Implications for Drug Design. Annu. Rev. Biochem. 2018, 87, 451–478. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-Targeting Antibiotics and Mechanisms of Bacterial Resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Osterman, I.A.; Wieland, M.; Maviza, T.P.; Lashkevich, K.A.; Lukianov, D.A.; Komarova, E.S.; Zakalyukina, Y.V.; Buschauer, R.; Shiriaev, D.I.; Leyn, S.A.; et al. Tetracenomycin X Inhibits Translation by Binding within the Ribosomal Exit Tunnel. Nat. Chem. Biol. 2020, 16, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Svetlov, M.S.; Koller, T.O.; Meydan, S.; Shankar, V.; Klepacki, D.; Polacek, N.; Guydosh, N.R.; Vázquez-Laslop, N.; Wilson, D.N.; Mankin, A.S. Context-Specific Action of Macrolide Antibiotics on the Eukaryotic Ribosome. Nat. Commun. 2021, 12, 2803. [Google Scholar] [CrossRef] [PubMed]
- Travin, D.Y.; Watson, Z.L.; Metelev, M.; Ward, F.R.; Osterman, I.A.; Khven, I.M.; Khabibullina, N.F.; Serebryakova, M.; Mergaert, P.; Polikanov, Y.S.; et al. Structure of Ribosome-Bound Azole-Modified Peptide Phazolicin Rationalizes Its Species-Specific Mode of Bacterial Translation Inhibition. Nat. Commun. 2019, 10, 4563. [Google Scholar] [CrossRef]
- Matzov, D.; Eyal, Z.; Benhamou, R.I.; Shalev-Benami, M.; Halfon, Y.; Krupkin, M.; Zimmerman, E.; Rozenberg, H.; Bashan, A.; Fridman, M.; et al. Structural Insights of Lincosamides Targeting the Ribosome of Staphylococcus Aureus. Nucleic Acids Res. 2017, 45, 10284–10292. [Google Scholar] [CrossRef] [PubMed]
- Matzov, D.; Bashan, A.; Yonath, A. A Bright Future for Antibiotics? Annu. Rev. Biochem. 2017, 86, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Khan, S.N.; Harvey, I.; Merrick, W.; Pelletier, J. Eukaryotic Protein Synthesis Inhibitors Identified by Comparison of Cytotoxicity Profiles. RNA 2004, 10, 528–543. [Google Scholar] [CrossRef] [PubMed]
- Fresno, M.; Jiménez, A.; Vázquez, D. Inhibition of Translation in Eukaryotic Systems by Harringtonine. Eur. J. Biochem. 1977, 72, 323–330. [Google Scholar] [CrossRef]
- Barbacid, M.; Fresno, M.; Vazquez, D. Inhibitors of Polypeptide Elongation on Yeast Polysomes. J. Antibiot. 1975, 28, 453–462. [Google Scholar] [CrossRef][Green Version]
- Cuendet, M.; Pezzuto, J.M. Antitumor Activity of Bruceantin: An Old Drug with New Promise. J. Nat. Prod. 2004, 67, 269–272. [Google Scholar] [CrossRef]
- Schneider-Poetsch, T.; Ju, J.; Eyler, D.E.; Dang, Y.; Bhat, S.; Merrick, W.C.; Green, R.; Shen, B.; Liu, J.O. Inhibition of Eukaryotic Translation Elongation by Cycloheximide and Lactimidomycin. Nat. Chem. Biol. 2010, 6, 209–217. [Google Scholar] [CrossRef]
- Evidente, A.; Kireev, A.S.; Jenkins, A.R.; Romero, A.E.; Steelant, W.F.A.; Van Slambrouck, S.; Kornienko, A. Biological Evaluation of Structurally Diverse Amaryllidaceae Alkaloids and Their Synthetic Derivatives: Discovery of Novel Leads for Anticancer Drug Design. Planta Med. 2009, 75, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Dölz, H.; Vázquez, D.; Jiménez, A. Quantitation of the Specific Interaction of [14a-3H] Cryptopleurine with 80S and 40S Ribosomal Species from the Yeast Saccharomyces Cerevisiae. Biochemistry 1982, 21, 3181–3187. [Google Scholar] [CrossRef]
- Dmitriev, S.E.; Vladimirov, D.O.; Lashkevich, K.A. A Quick Guide to Small-Molecule Inhibitors of Eukaryotic Protein Synthesis. Biochemistry 2020, 85, 1389–1421. [Google Scholar] [CrossRef] [PubMed]
- Burgers, L.D.; Fürst, R. Natural Products as Drugs and Tools for Influencing Core Processes of Eukaryotic MRNA Translation. Pharmacol. Res. 2021, 170, 105535. [Google Scholar] [CrossRef]
- Brönstrup, M.; Sasse, F. Natural Products Targeting the Elongation Phase of Eukaryotic Protein Biosynthesis. Nat. Prod. Rep. 2020, 37, 752–762. [Google Scholar] [CrossRef]
- Garreau de Loubresse, N.; Prokhorova, I.; Holtkamp, W.; Rodnina, M.V.; Yusupova, G.; Yusupov, M. Structural Basis for the Inhibition of the Eukaryotic Ribosome. Nature 2014, 513, 517–522. [Google Scholar] [CrossRef]
- Gilles, A.; Frechin, L.; Natchiar, K.; Biondani, G.; von Loeffelholz, O.; Holvec, S.; Malaval, J.-L.; Winum, J.-Y.; Klaholz, B.P.; Peyron, J.-F. Targeting the Human 80S Ribosome in Cancer: From Structure to Function and Drug Design for Innovative Adjuvant Therapeutic Strategies. Cells 2020, 9, 629. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; O’Brien, S.; Cortes, J. Homoharringtonine/Omacetaxine Mepesuccinate: The Long and Winding Road to Food and Drug Administration Approval. Clin. Lymphoma Myeloma Leuk. 2013, 13, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Yakhni, M.; Briat, A.; El Guerrab, A.; Furtado, L.; Kwiatkowski, F.; Miot-Noirault, E.; Cachin, F.; Penault-Llorca, F.; Radosevic-Robin, N. Homoharringtonine, an Approved Anti-Leukemia Drug, Suppresses Triple Negative Breast Cancer Growth through a Rapid Reduction of Anti-Apoptotic Protein Abundance. Am. J. Cancer Res. 2019, 9, 1043–1060. [Google Scholar]
- Chowdhury, H.M.; Siddiqui, M.A.; Kanneganti, S.; Sharmin, N.; Chowdhury, M.W.; Nasim, M.T. Aminoglycoside-Mediated Promotion of Translation Readthrough Occurs through a Non-Stochastic Mechanism That Competes with Translation Termination. Hum. Mol. Genet. 2018, 27, 373–384. [Google Scholar] [CrossRef]
- Bidou, L.; Allamand, V.; Rousset, J.-P.; Namy, O. Sense from Nonsense: Therapies for Premature Stop Codon Diseases. Trends Mol. Med. 2012, 18, 679–688. [Google Scholar] [CrossRef]
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A Meta-Analysis of Nonsense Mutations Causing Human Genetic Disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Genuth, N.R.; Barna, M. The Discovery of Ribosome Heterogeneity and Its Implications for Gene Regulation and Organismal Life. Mol. Cell 2018, 71, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.M.; Kurylo, C.M.; Batchelder, J.E.; Theresa Vincent, C.; Blanchard, S.C. Implications of Sequence Variation on the Evolution of RRNA. Chromosome Res. 2019, 27, 89–93. [Google Scholar] [CrossRef]
- Norris, K.; Hopes, T.; Aspden, J.L. Ribosome Heterogeneity and Specialization in Development. Wiley Interdiscip. Rev. RNA 2021, 12, e1644. [Google Scholar] [CrossRef] [PubMed]
- Bastide, A.; David, A. The Ribosome, (Slow) Beating Heart of Cancer (Stem) Cell. Oncogenesis 2018, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Polikanov, Y.S.; Steitz, T.A.; Innis, C.A. A Proton Wire to Couple Aminoacyl-TRNA Accommodation and Peptide-Bond Formation on the Ribosome. Nat. Struct. Mol. Biol. 2014, 21, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Świderek, K.; Marti, S.; Tuñón, I.; Moliner, V.; Bertran, J. Peptide Bond Formation Mechanism Catalyzed by Ribosome. J. Am. Chem. Soc. 2015, 137, 12024–12034. [Google Scholar] [CrossRef]
- Dao Duc, K.; Batra, S.S.; Bhattacharya, N.; Cate, J.H.D.; Song, Y.S. Differences in the Path to Exit the Ribosome across the Three Domains of Life. Nucleic Acids Res. 2019, 47, 4198–4210. [Google Scholar] [CrossRef]
- McClary, B.; Zinshteyn, B.; Meyer, M.; Jouanneau, M.; Pellegrino, S.; Yusupova, G.; Schuller, A.; Reyes, J.C.P.; Lu, J.; Guo, Z.; et al. Inhibition of Eukaryotic Translation by the Antitumor Natural Product Agelastatin A. Cell Chem. Biol. 2017, 24, 605–613.e5. [Google Scholar] [CrossRef]
- Pellegrino, S.; Meyer, M.; Zorbas, C.; Bouchta, S.A.; Saraf, K.; Pelly, S.C.; Yusupova, G.; Evidente, A.; Mathieu, V.; Kornienko, A.; et al. The Amaryllidaceae Alkaloid Haemanthamine Binds the Eukaryotic Ribosome to Repress Cancer Cell Growth. Structure 2018, 26, 416–425.e4. [Google Scholar] [CrossRef]
- Li, W.; Chang, S.T.-L.; Ward, F.R.; Cate, J.H.D. Selective Inhibition of Human Translation Termination by a Drug-like Compound. Nat. Commun. 2020, 11, 4941. [Google Scholar] [CrossRef]
- Pellegrino, S.; Meyer, M.; Könst, Z.A.; Holm, M.; Voora, V.K.; Kashinskaya, D.; Zanette, C.; Mobley, D.L.; Yusupova, G.; Vanderwal, C.D.; et al. Understanding the Role of Intermolecular Interactions between Lissoclimides and the Eukaryotic Ribosome. Nucleic Acids Res. 2019, 47, 3223–3232. [Google Scholar] [CrossRef]
- Könst, Z.A.; Szklarski, A.R.; Pellegrino, S.; Michalak, S.E.; Meyer, M.; Zanette, C.; Cencic, R.; Nam, S.; Voora, V.K.; Horne, D.A.; et al. Synthesis Facilitates an Understanding of the Structural Basis for Translation Inhibition by the Lissoclimides. Nat. Chem. 2017, 9, 1140–1149. [Google Scholar] [CrossRef]
- Wu, C.C.-C.; Peterson, A.; Zinshteyn, B.; Regot, S.; Green, R. Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 2020, 182, 404–416.e14. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.J.; van Staden, J. Cytotoxic Agents in the Minor Alkaloid Groups of the Amaryllidaceae. Planta Med. 2021. [Google Scholar] [CrossRef]
- Liaud, N.; Horlbeck, M.A.; Gilbert, L.A.; Gjoni, K.; Weissman, J.S.; Cate, J.H.D. Cellular Response to Small Molecules That Selectively Stall Protein Synthesis by the Ribosome. PLoS Genet. 2019, 15, e1008057. [Google Scholar] [CrossRef] [PubMed]
- Petersen, D.N.; Hawkins, J.; Ruangsiriluk, W.; Stevens, K.A.; Maguire, B.A.; O’Connell, T.N.; Rocke, B.N.; Boehm, M.; Ruggeri, R.B.; Rolph, T.; et al. A Small-Molecule Anti-Secretagogue of PCSK9 Targets the 80S Ribosome to Inhibit PCSK9 Protein Translation. Cell Chem. Biol. 2016, 23, 1362–1371. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, W.; Ward, F.R.; McClure, K.F.; Chang, S.T.-L.; Montabana, E.; Liras, S.; Dullea, R.G.; Cate, J.H.D. Structural Basis for Selective Stalling of Human Ribosome Nascent Chain Complexes by a Drug-like Molecule. Nat. Struct. Mol. Biol. 2019, 26, 501–509. [Google Scholar] [CrossRef]
- Lintner, N.G.; McClure, K.F.; Petersen, D.; Londregan, A.T.; Piotrowski, D.W.; Wei, L.; Xiao, J.; Bolt, M.; Loria, P.M.; Maguire, B.; et al. Selective Stalling of Human Translation through Small-Molecule Engagement of the Ribosome Nascent Chain. PLoS Biol. 2017, 15, e2001882. [Google Scholar] [CrossRef]
- Powers, K.T.; Stevenson-Jones, F.; Yadav, S.K.N.; Amthor, B.; Bufton, J.C.; Borucu, U.; Shen, D.; Becker, J.P.; Lavysh, D.; Hentze, M.W.; et al. Blasticidin S Inhibits Mammalian Translation and Enhances Production of Protein Encoded by Nonsense MRNA. Nucleic Acids Res. 2021, 49, 7665–7679. [Google Scholar] [CrossRef]
- Svidritskiy, E.; Ling, C.; Ermolenko, D.N.; Korostelev, A.A. Blasticidin S Inhibits Translation by Trapping Deformed TRNA on the Ribosome. Proc. Natl. Acad. Sci. USA 2013, 110, 12283–12288. [Google Scholar] [CrossRef]
- Klinge, S.; Voigts-Hoffmann, F.; Leibundgut, M.; Arpagaus, S.; Ban, N. Crystal Structure of the Eukaryotic 60S Ribosomal Subunit in Complex with Initiation Factor 6. Science 2011, 334, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Myasnikov, A.G.; Kundhavai Natchiar, S.; Nebout, M.; Hazemann, I.; Imbert, V.; Khatter, H.; Peyron, J.-F.; Klaholz, B.P. Structure-Function Insights Reveal the Human Ribosome as a Cancer Target for Antibiotics. Nat. Commun. 2016, 7, 12856. [Google Scholar] [CrossRef] [PubMed]
- Gürel, G.; Blaha, G.; Steitz, T.A.; Moore, P.B. Structures of Triacetyloleandomycin and Mycalamide A Bind to the Large Ribosomal Subunit of Haloarcula Marismortui. Antimicrob. Agents Chemother. 2009, 53, 5010–5014. [Google Scholar] [CrossRef] [PubMed]
- Robert, F.; Gao, H.Q.; Donia, M.; Merrick, W.C.; Hamann, M.T.; Pelletier, J. Chlorolissoclimides: New Inhibitors of Eukaryotic Protein Synthesis. RNA 2006, 12, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Llano-Sotelo, B.; Dunkle, J.; Klepacki, D.; Zhang, W.; Fernandes, P.; Cate, J.H.D.; Mankin, A.S. Binding and Action of CEM-101, a New Fluoroketolide Antibiotic That Inhibits Protein Synthesis. Antimicrob. Agents Chemother. 2010, 54, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.; Wilson, D.N.; Teraoka, Y.; Szaflarski, W.; Fucini, P.; Kalpaxis, D.; Nierhaus, K.H. Dissecting the Ribosomal Inhibition Mechanisms of Edeine and Pactamycin: The Universally Conserved Residues G693 and C795 Regulate P-Site RNA Binding. Mol. Cell 2004, 13, 113–124. [Google Scholar] [CrossRef]
- Bucher, K.; Skogerson, L. Cryptopleurine—An Inhibitor of Translocation. Biochemistry 1976, 15, 4755–4759. [Google Scholar] [CrossRef]
- Grant, P.; Sánchez, L.; Jiménez, A. Cryptopleurine Resistance: Genetic Locus for a 40S Ribosomal Component in Saccharomyces Cerevisiae. J. Bacteriol. 1974, 120, 1308–1314. [Google Scholar] [CrossRef]
- Pioletti, M.; Schlünzen, F.; Harms, J.; Zarivach, R.; Glühmann, M.; Avila, H.; Bashan, A.; Bartels, H.; Auerbach, T.; Jacobi, C.; et al. Crystal Structures of Complexes of the Small Ribosomal Subunit with Tetracycline, Edeine and IF3. EMBO J. 2001, 20, 1829–1839. [Google Scholar] [CrossRef]
- Kozak, M.; Shatkin, A.J. Migration of 40 S Ribosomal Subunits on Messenger RNA in the Presence of Edeine. J. Biol. Chem. 1978, 253, 6568–6577. [Google Scholar] [CrossRef]
- Prokhorova, I.V.; Akulich, K.A.; Makeeva, D.S.; Osterman, I.A.; Skvortsov, D.A.; Sergiev, P.V.; Dontsova, O.A.; Yusupova, G.; Yusupov, M.M.; Dmitriev, S.E. Amicoumacin A Induces Cancer Cell Death by Targeting the Eukaryotic Ribosome. Sci. Rep. 2016, 6, 27720. [Google Scholar] [CrossRef]
- Polikanov, Y.S.; Osterman, I.A.; Szal, T.; Tashlitsky, V.N.; Serebryakova, M.V.; Kusochek, P.; Bulkley, D.; Malanicheva, I.A.; Efimenko, T.A.; Efremenkova, O.V.; et al. Amicoumacin a Inhibits Translation by Stabilizing MRNA Interaction with the Ribosome. Mol. Cell 2014, 56, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef]
- Prokhorova, I.; Altman, R.B.; Djumagulov, M.; Shrestha, J.P.; Urzhumtsev, A.; Ferguson, A.; Chang, C.-W.T.; Yusupov, M.; Blanchard, S.C.; Yusupova, G. Aminoglycoside Interactions and Impacts on the Eukaryotic Ribosome. Proc. Natl. Acad. Sci. USA 2017, 114, E10899–E10908. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.; Uemura, S.; Johansson, M.; Puglisi, E.V.; Marshall, R.A.; Aitken, C.E.; Korlach, J.; Ehrenberg, M.; Puglisi, J.D. The Impact of Aminoglycosides on the Dynamics of Translation Elongation. Cell Rep. 2013, 3, 497–508. [Google Scholar] [CrossRef]
- Feldman, M.B.; Terry, D.S.; Altman, R.B.; Blanchard, S.C. Aminoglycoside Activity Observed on Single Pre-Translocation Ribosome Complexes. Nat. Chem. Biol. 2010, 6, 244. [Google Scholar] [CrossRef] [PubMed]
- Borovinskaya, M.A.; Pai, R.D.; Zhang, W.; Schuwirth, B.S.; Holton, J.M.; Hirokawa, G.; Kaji, H.; Kaji, A.; Cate, J.H.D. Structural Basis for Aminoglycoside Inhibition of Bacterial Ribosome Recycling. Nat. Struct. Mol. Biol. 2007, 14, 727–732. [Google Scholar] [CrossRef]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in Therapeutic Use of a Drug-Stimulated Translational Readthrough of Premature Termination Codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef]
- Keeling, K.M.; Xue, X.; Gunn, G.; Bedwell, D.M. Therapeutics Based on Stop Codon Readthrough. Annu. Rev. Genom. Hum. Genet. 2014, 15, 371–394. [Google Scholar] [CrossRef]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Mendell, J.R. Aminoglycoside-Induced Mutation Suppression (Stop Codon Readthrough) as a Therapeutic Strategy for Duchenne Muscular Dystrophy. Ther. Adv. Neurol. Disord. 2010, 3, 379–389. [Google Scholar] [CrossRef]
- Ng, M.Y.; Li, H.; Ghelfi, M.D.; Goldman, Y.E.; Cooperman, B.S. Ataluren and Aminoglycosides Stimulate Read-through of Nonsense Codons by Orthogonal Mechanisms. Proc. Natl. Acad. Sci. USA 2021, 118, e2020599118. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-L.R.; Dougherty, J.P. Pharmaceutical Therapies to Recode Nonsense Mutations in Inherited Diseases. Pharmacol. Ther. 2012, 136, 227–266. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Wang, D.; Conard, S.E.; Bedwell, D.M. Suppression of Premature Termination Codons as a Therapeutic Approach. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 444–463. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Hamed, S.; Hadley, D.W.; Gropman, A.L.; Burstein, A.H.; Escolar, D.M.; Hoffman, E.P.; Fischbeck, K.H. Gentamicin Treatment of Duchenne and Becker Muscular Dystrophy Due to Nonsense Mutations. Ann. Neurol. 2001, 49, 706–711. [Google Scholar] [CrossRef]
- Mattis, V.B.; Tom Chang, C.-W.; Lorson, C.L. Analysis of a Read-through Promoting Compound in a Severe Mouse Model of Spinal Muscular Atrophy. Neurosci. Lett. 2012, 525, 72–75. [Google Scholar] [CrossRef]
- Mattis, V.B.; Rai, R.; Wang, J.; Chang, C.-W.T.; Coady, T.; Lorson, C.L. Novel Aminoglycosides Increase SMN Levels in Spinal Muscular Atrophy Fibroblasts. Hum. Genet. 2006, 120, 589–601. [Google Scholar] [CrossRef]
- Wasserman, M.R.; Pulk, A.; Zhou, Z.; Altman, R.B.; Zinder, J.C.; Green, K.D.; Garneau-Tsodikova, S.; Cate, J.H.D.; Blanchard, S.C. Chemically Related 4,5-Linked Aminoglycoside Antibiotics Drive Subunit Rotation in Opposite Directions. Nat. Commun. 2015, 6, 7896. [Google Scholar] [CrossRef]
- Borovinskaya, M.A.; Shoji, S.; Fredrick, K.; Cate, J.H.D. Structural Basis for Hygromycin B Inhibition of Protein Biosynthesis. RNA 2008, 14, 1590–1599. [Google Scholar] [CrossRef]
- Brodersen, D.E.; Clemons, W.M.J.; Carter, A.P.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. The Structural Basis for the Action of the Antibiotics Tetracycline, Pactamycin, and Hygromycin B on the 30S Ribosomal Subunit. Cell 2000, 103, 1143–1154. [Google Scholar] [CrossRef]
- Natchiar, S.K.; Myasnikov, A.G.; Kratzat, H.; Hazemann, I.; Klaholz, B.P. Visualization of Chemical Modifications in the Human 80S Ribosome Structure. Nature 2017, 551, 472–477. [Google Scholar] [CrossRef]
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-Resolution Protein Structure Determination by Cryo-EM. Nature 2020, 587, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Nakane, T.; Kotecha, A.; Sente, A.; McMullan, G.; Masiulis, S.; Brown, P.M.G.E.; Grigoras, I.T.; Malinauskaite, L.; Malinauskas, T.; Miehling, J.; et al. Single-Particle Cryo-EM at Atomic Resolution. Nature 2020, 587, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Watson, Z.L.; Ward, F.R.; Méheust, R.; Ad, O.; Schepartz, A.; Banfield, J.F.; Cate, J.H. Structure of the Bacterial Ribosome at 2 Å Resolution. Elife 2020, 9, e60482. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrino, S.; Terrosu, S.; Yusupova, G.; Yusupov, M. Inhibition of the Eukaryotic 80S Ribosome as a Potential Anticancer Therapy: A Structural Perspective. Cancers 2021, 13, 4392. https://doi.org/10.3390/cancers13174392
Pellegrino S, Terrosu S, Yusupova G, Yusupov M. Inhibition of the Eukaryotic 80S Ribosome as a Potential Anticancer Therapy: A Structural Perspective. Cancers. 2021; 13(17):4392. https://doi.org/10.3390/cancers13174392
Chicago/Turabian StylePellegrino, Simone, Salvatore Terrosu, Gulnara Yusupova, and Marat Yusupov. 2021. "Inhibition of the Eukaryotic 80S Ribosome as a Potential Anticancer Therapy: A Structural Perspective" Cancers 13, no. 17: 4392. https://doi.org/10.3390/cancers13174392
APA StylePellegrino, S., Terrosu, S., Yusupova, G., & Yusupov, M. (2021). Inhibition of the Eukaryotic 80S Ribosome as a Potential Anticancer Therapy: A Structural Perspective. Cancers, 13(17), 4392. https://doi.org/10.3390/cancers13174392