
Effective Oncoleaking Treatment of Pancreatic Cancer by Claudin-Targeted Suicide Gene Therapy with Clostridium perfringens Enterotoxin (CPE)

 ,
,  ,
,  and
and 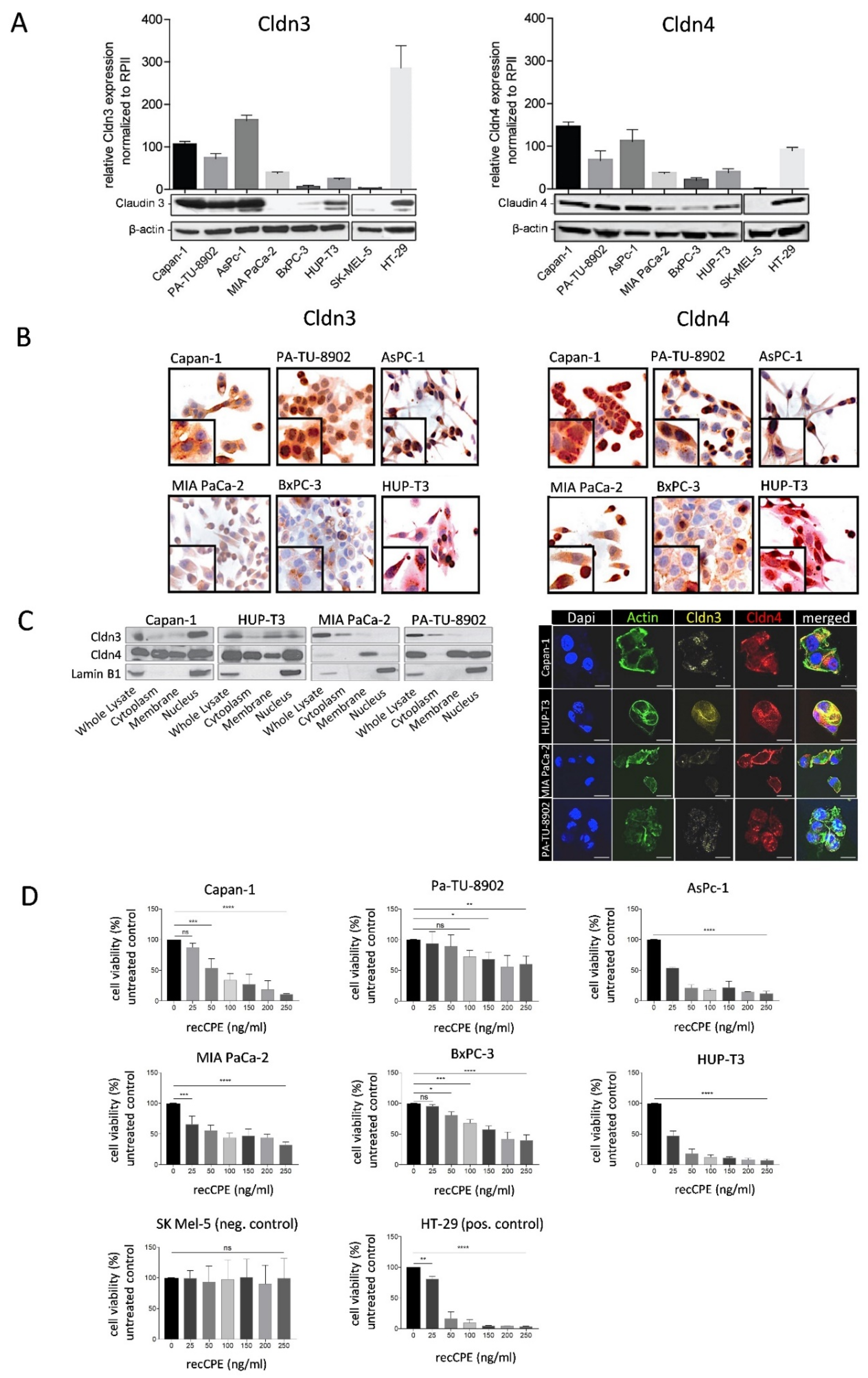{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Pancreatic Cancer Patient Derived Xenograft (PDX) Models
2.3. Quantitative Real-Time RT-PCR
2.4. Western Blot
2.5. Clostridium Perfringens Enterotoxin (CPE) Expressing Plasmids
2.6. Transfection of Cells with Plasmid DNA and siRNA
2.7. MTT Cytotoxicity Assay
2.8. CPE ELISA
2.9. Immunocytochemistry, Immunohistochemistry and Immunofluorescence
2.10. In Vivo optCPE Gene Transfer
2.11. Statistical Analysis
3. Results
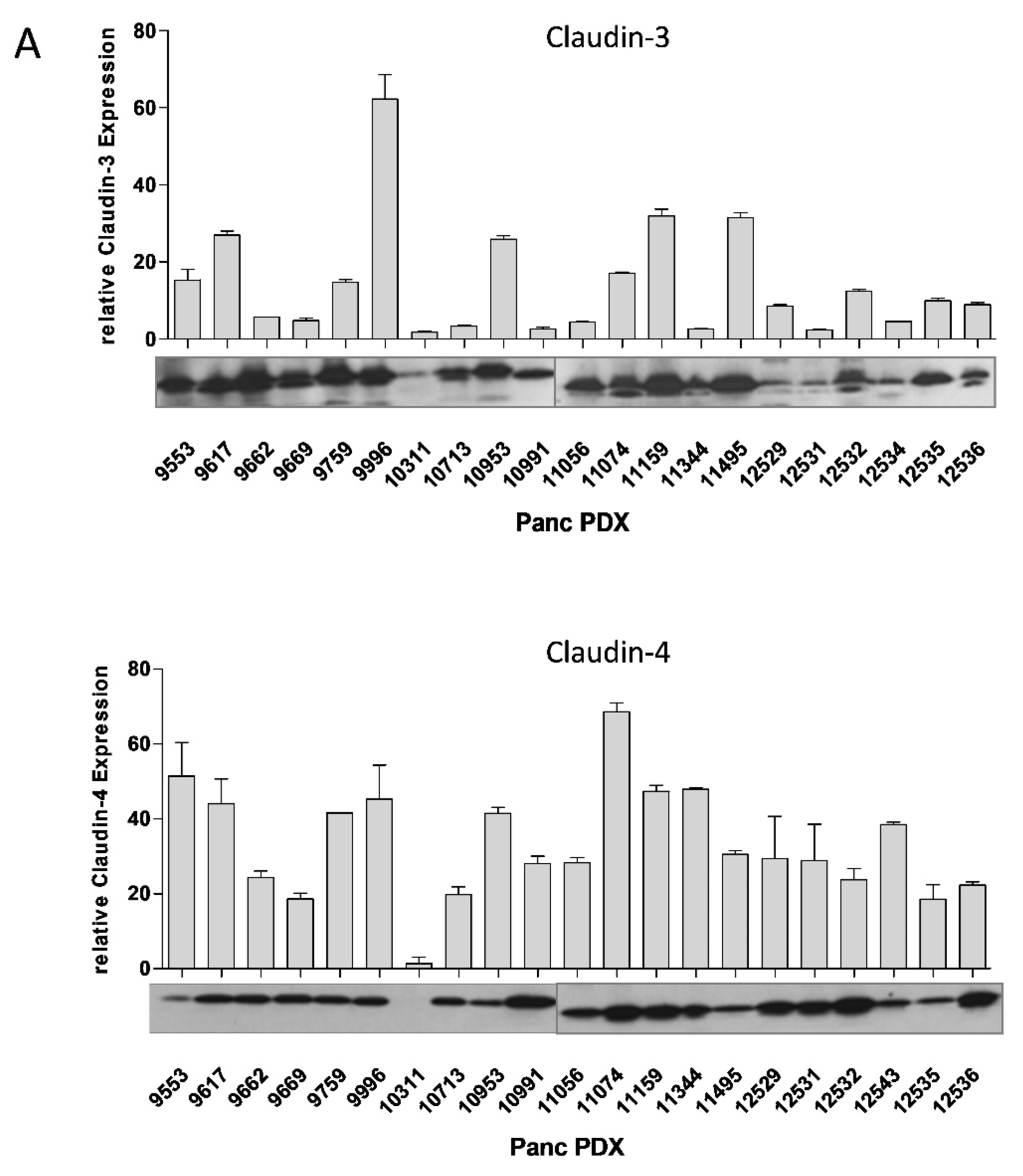
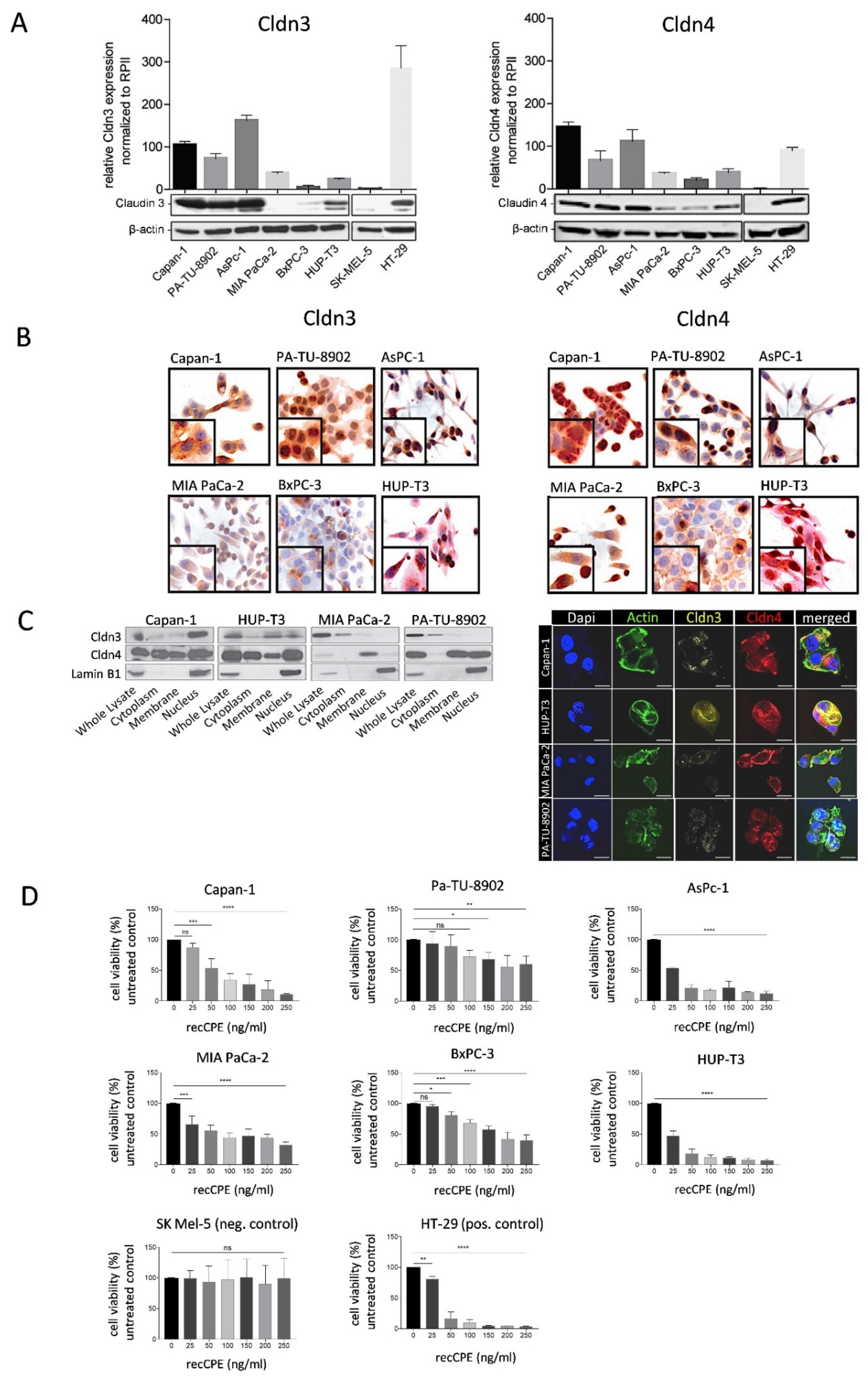
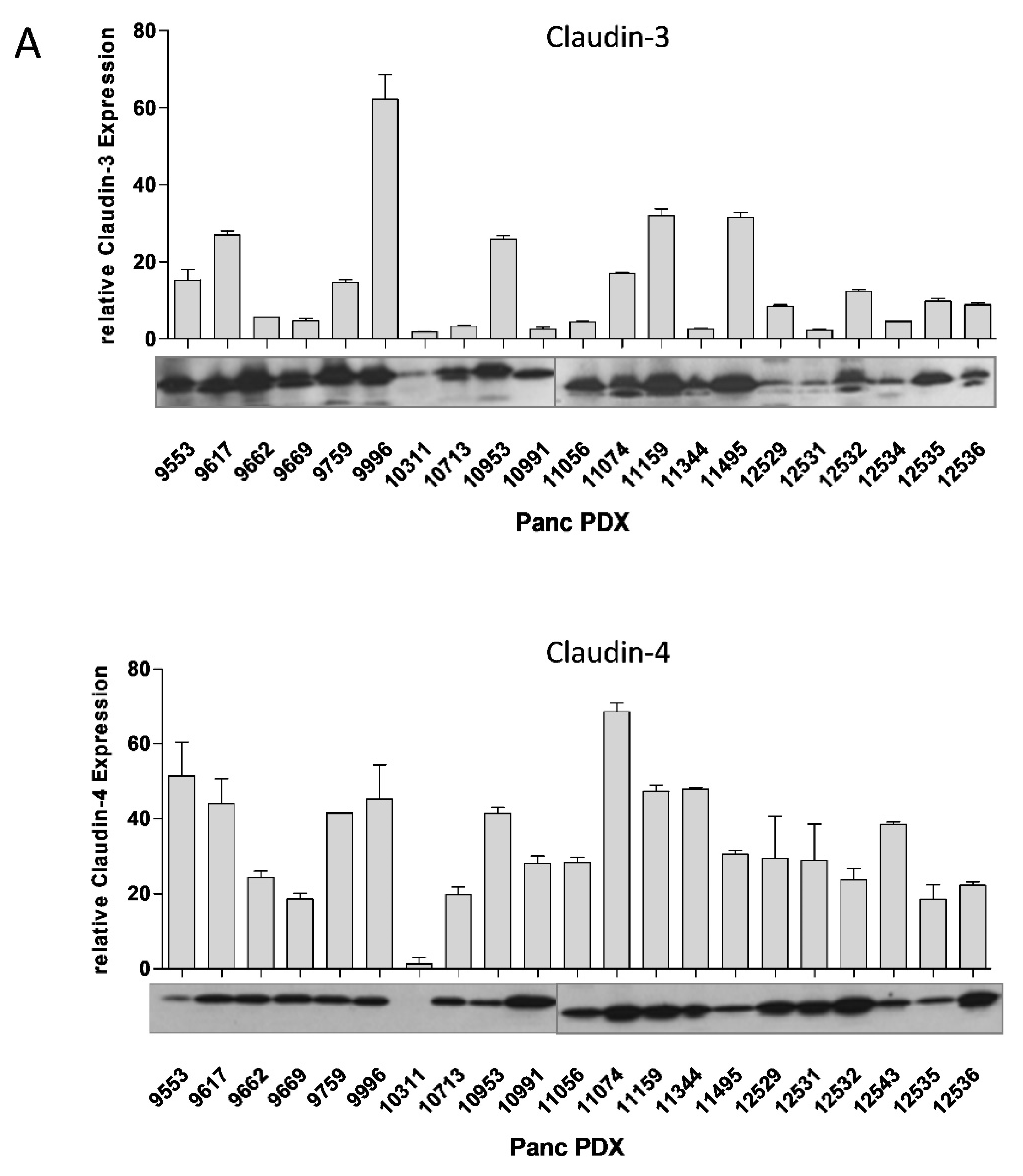
3.1. Cldn3/4 Expression in Human Pancreas Carcinoma Cell Lines
3.2. Sensitivity of Pancreas Carcinoma Cells toward Recombinant CPE
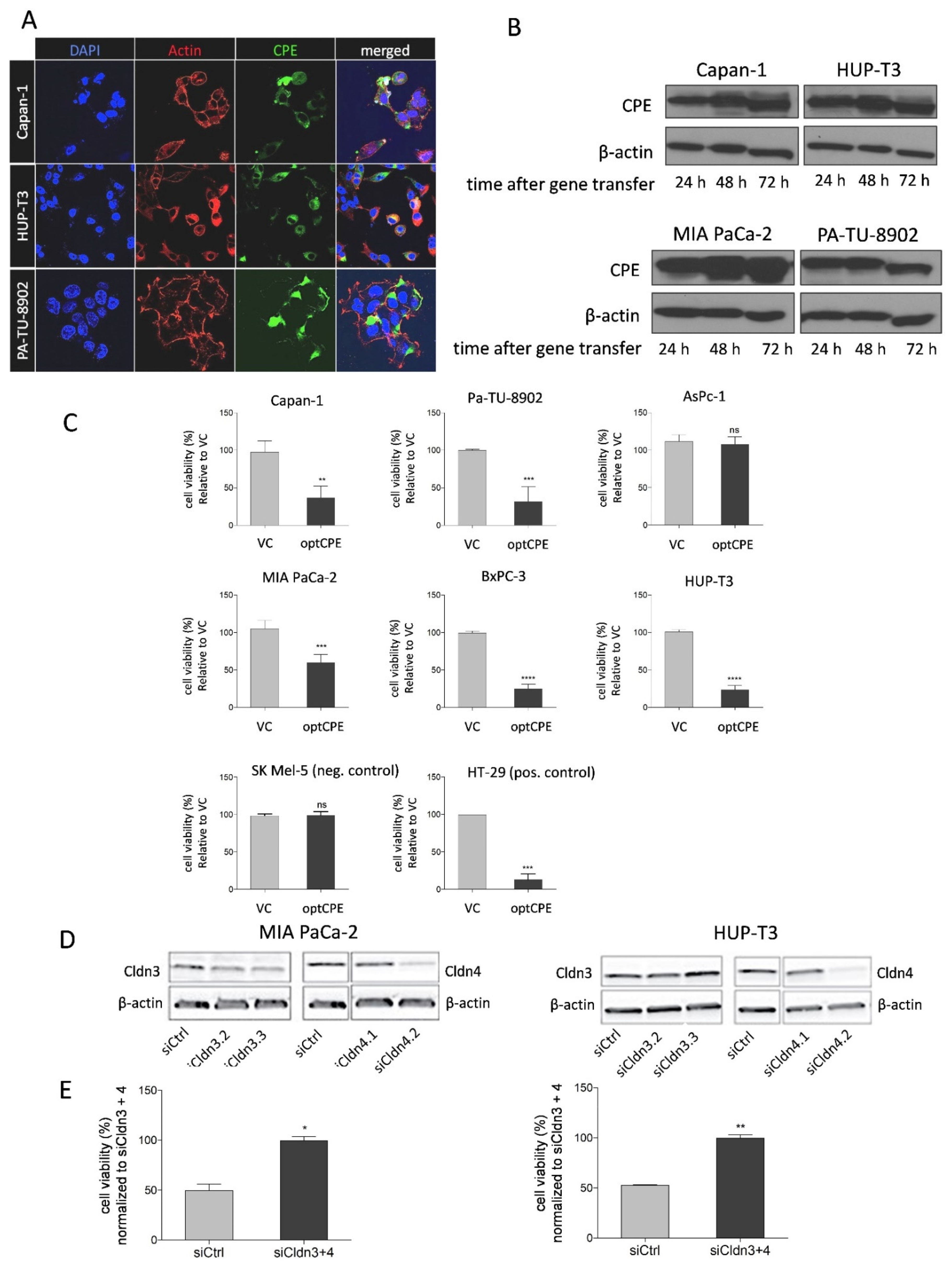
3.3. CPE Gene Transfer Permits Effective and Selective Cell Killing
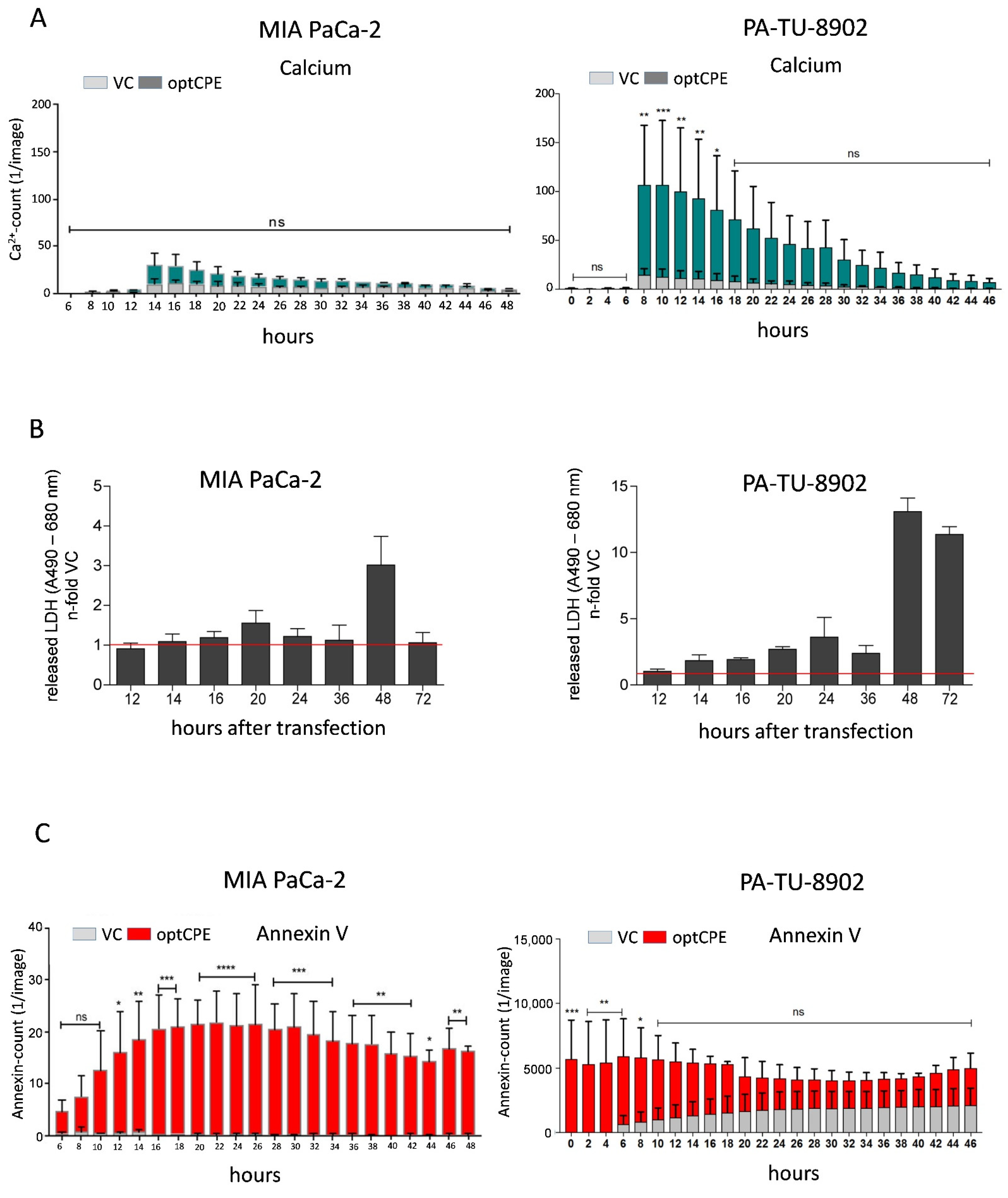
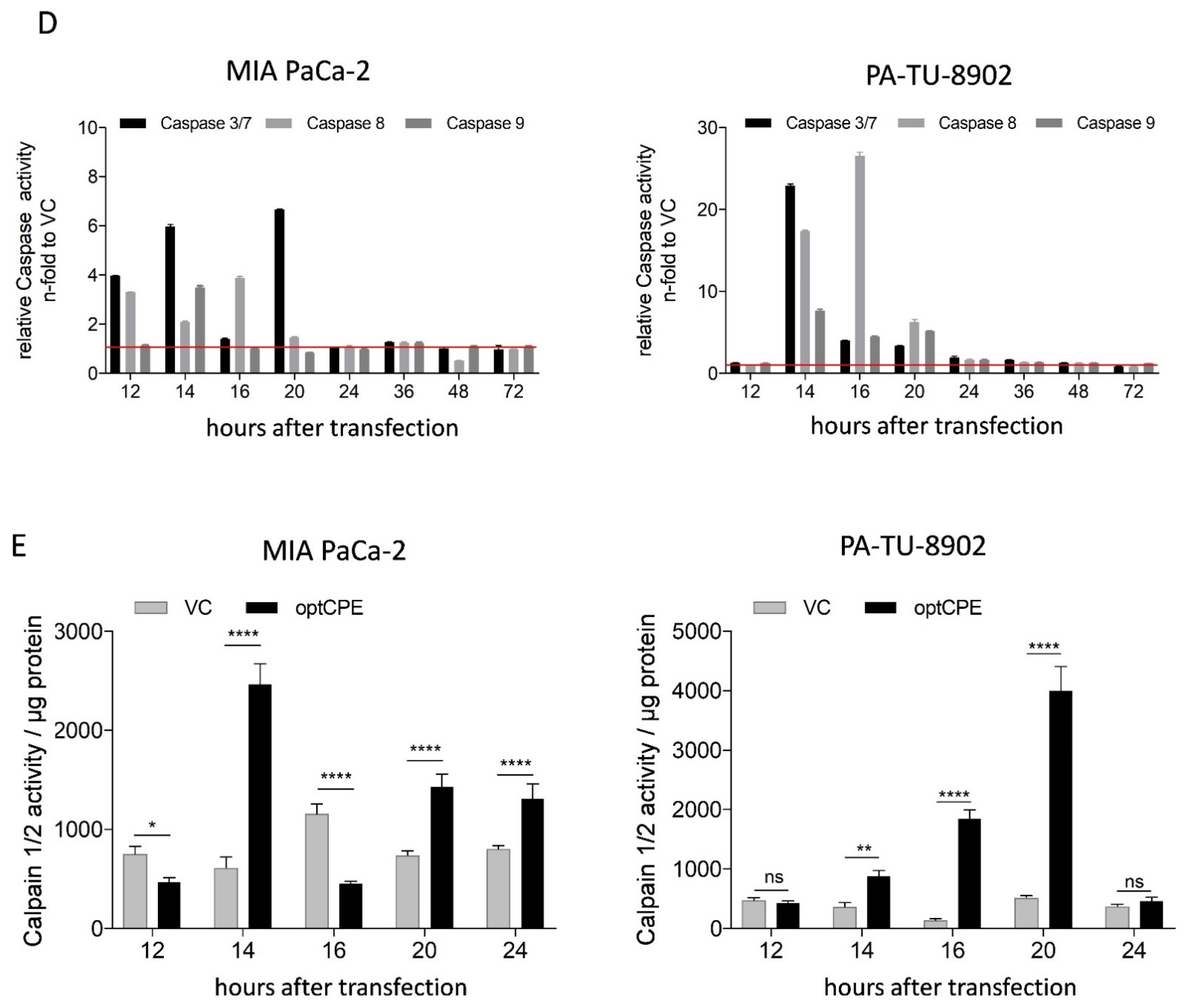
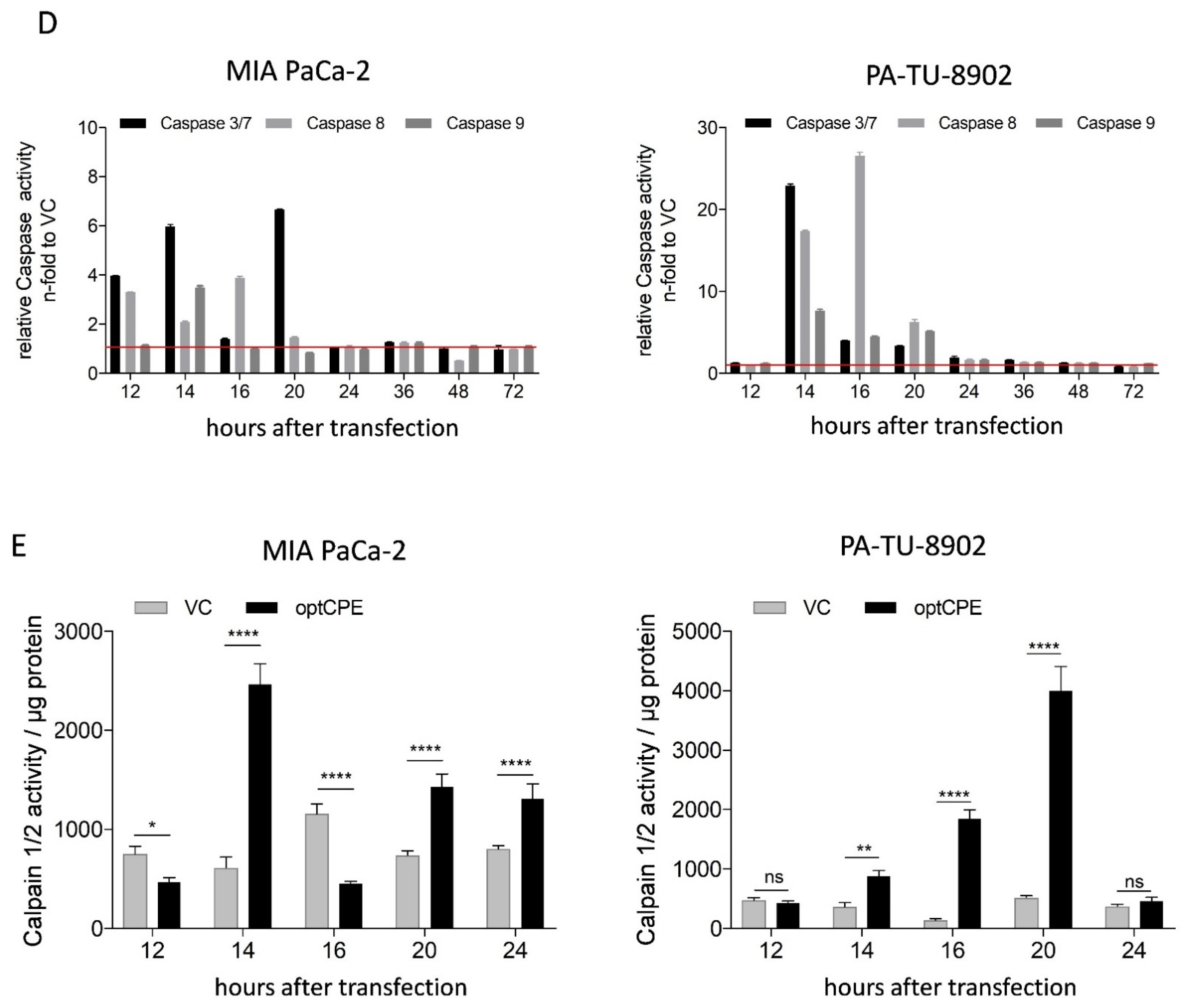
3.4. Analysis of Rapid Oncoleaking Cell Death Mechanism by CPE Gene Transfer
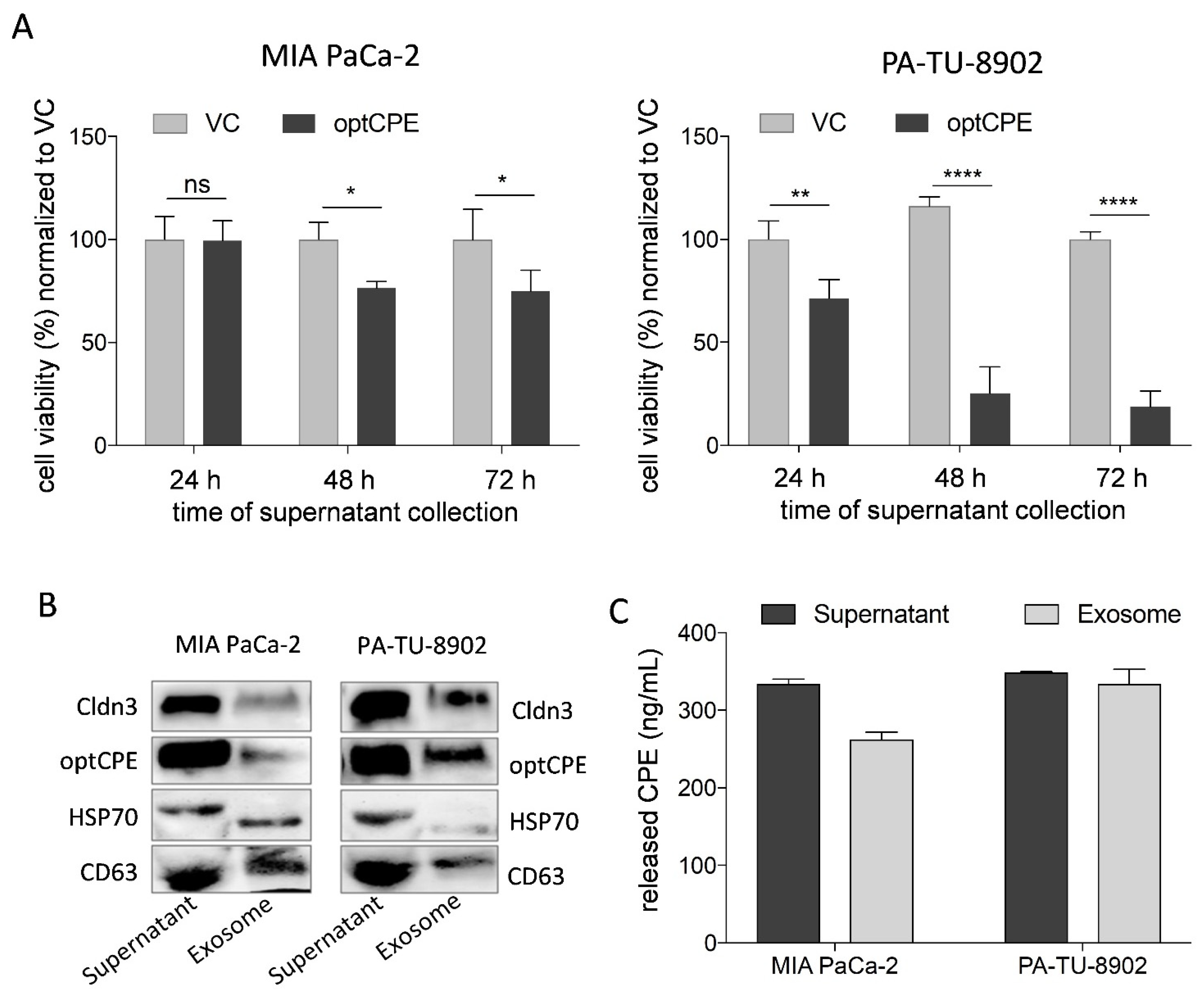
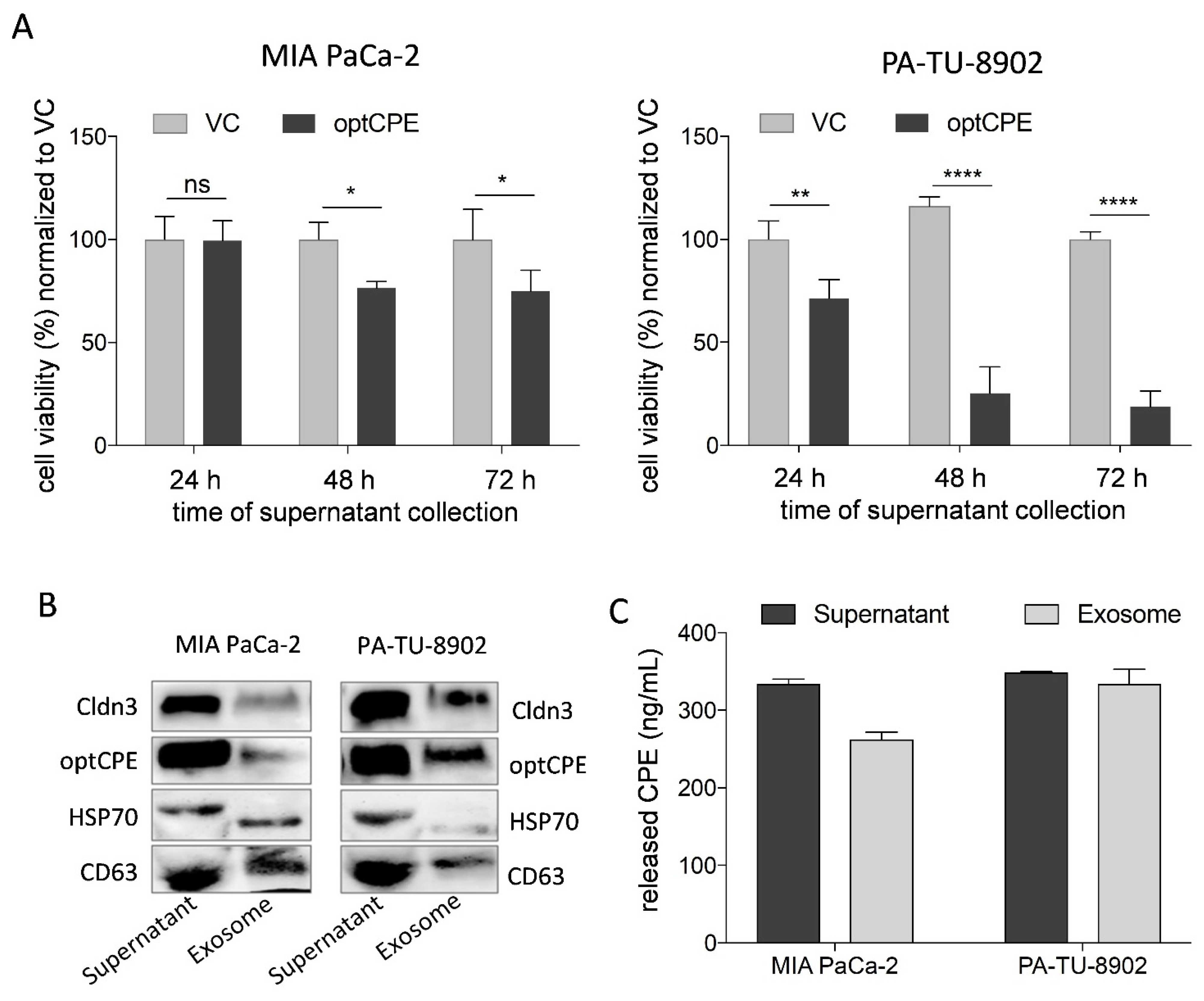
3.5. Mechanism of Bystander Effect in CPE Gene Therapy
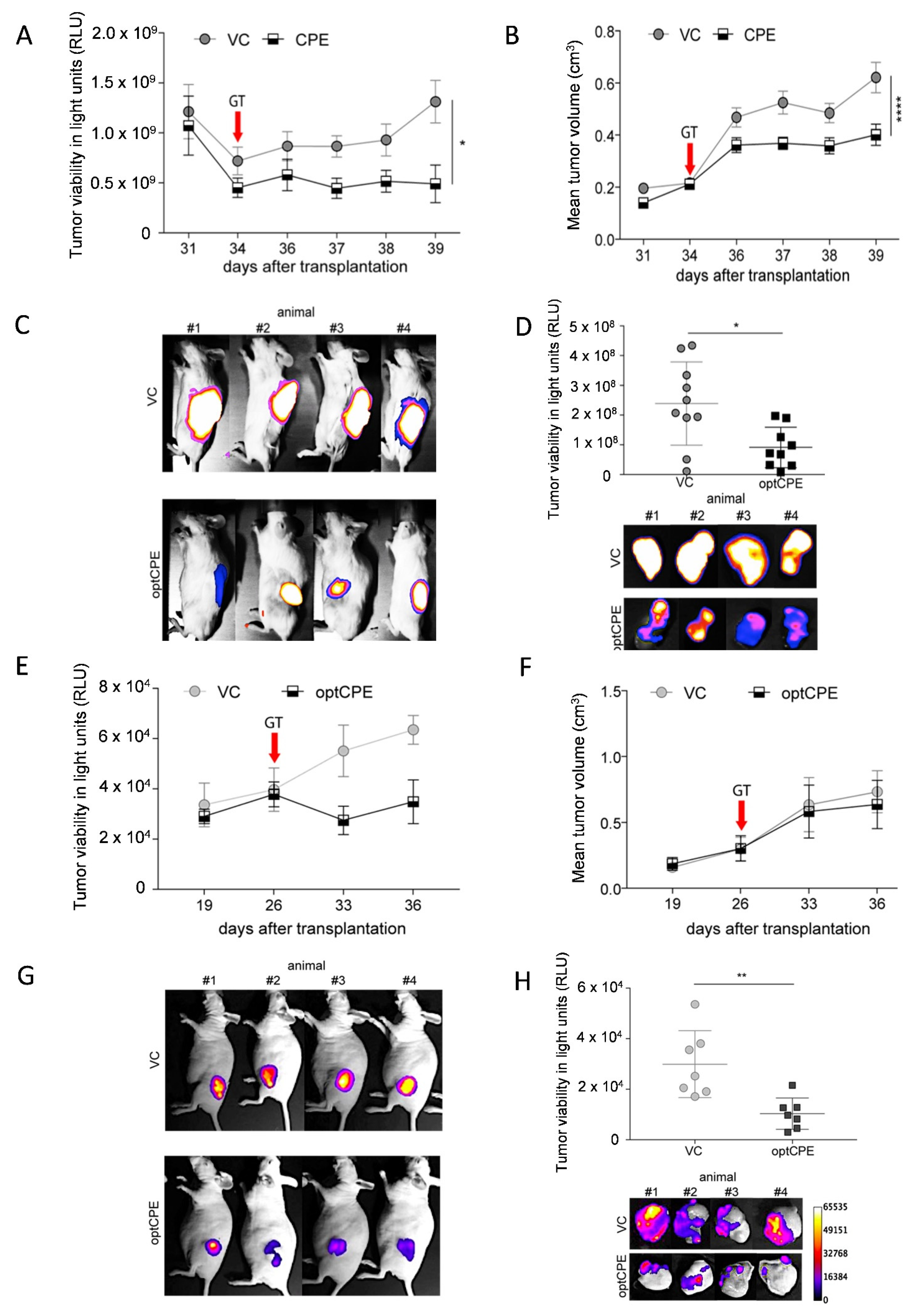
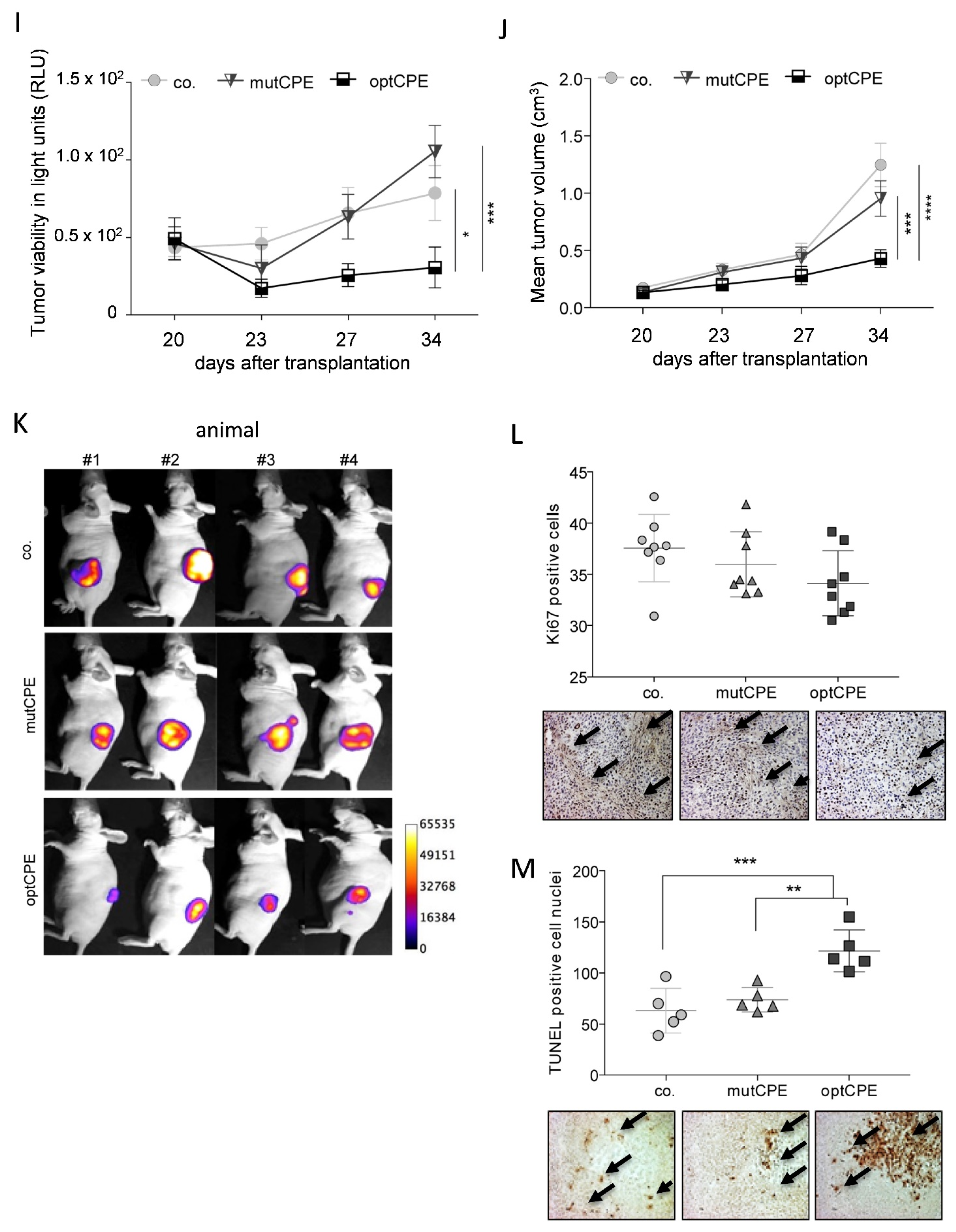
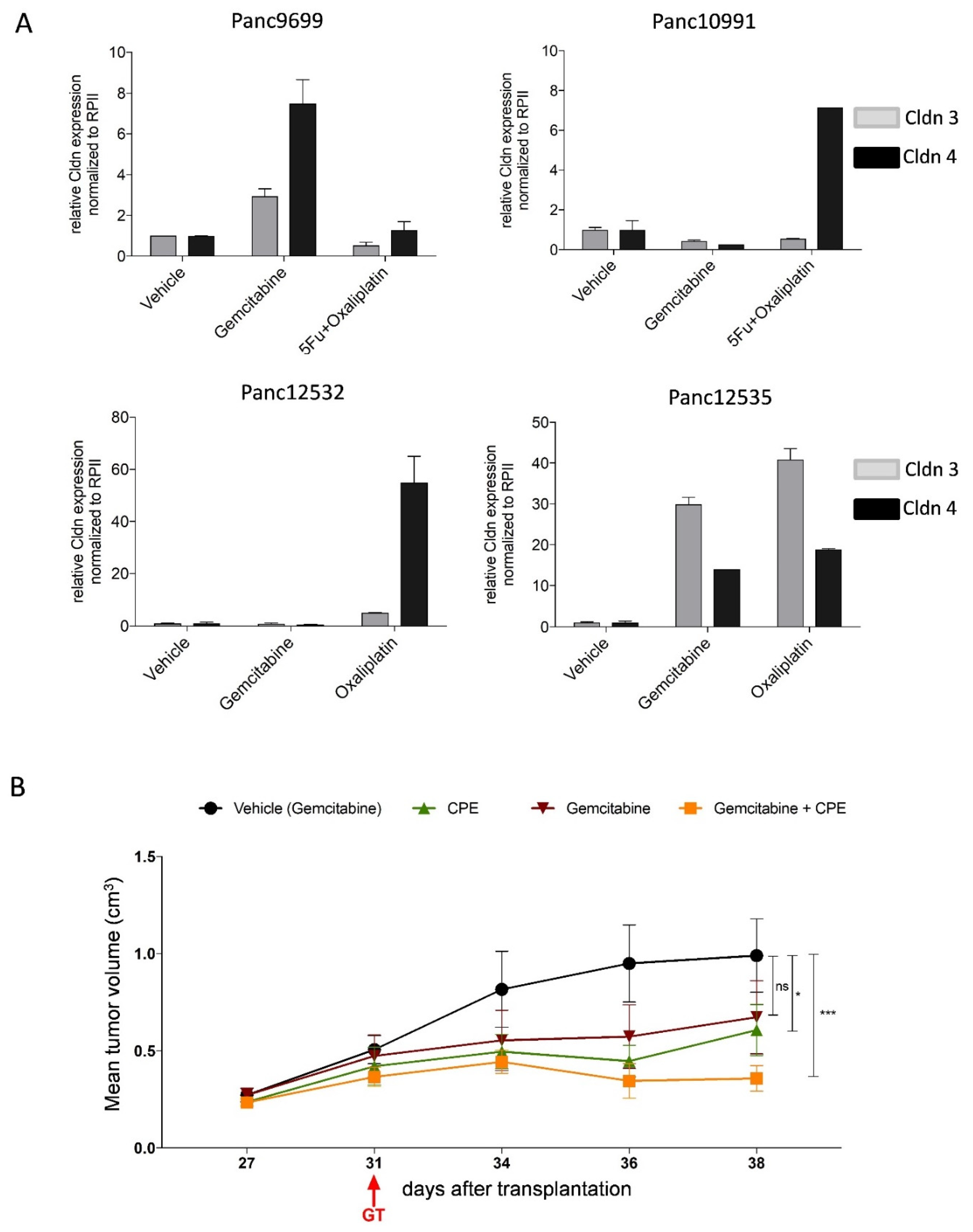
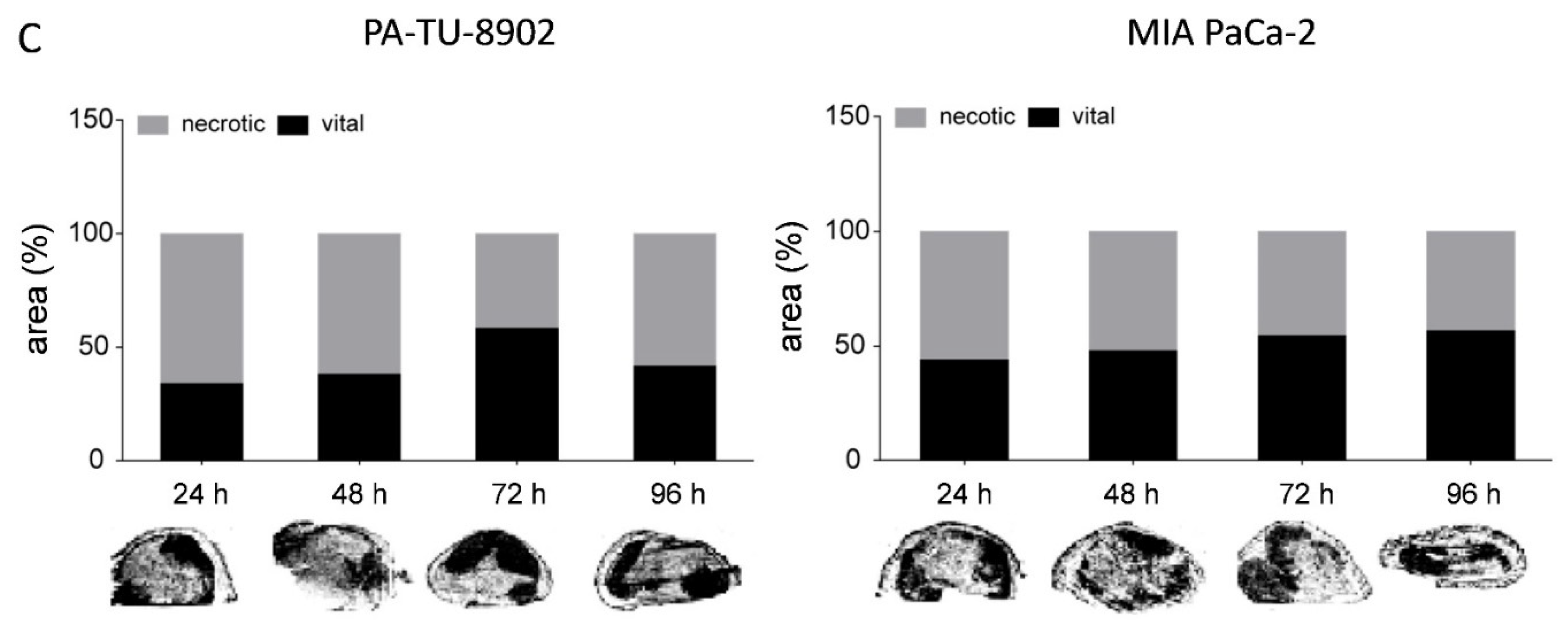
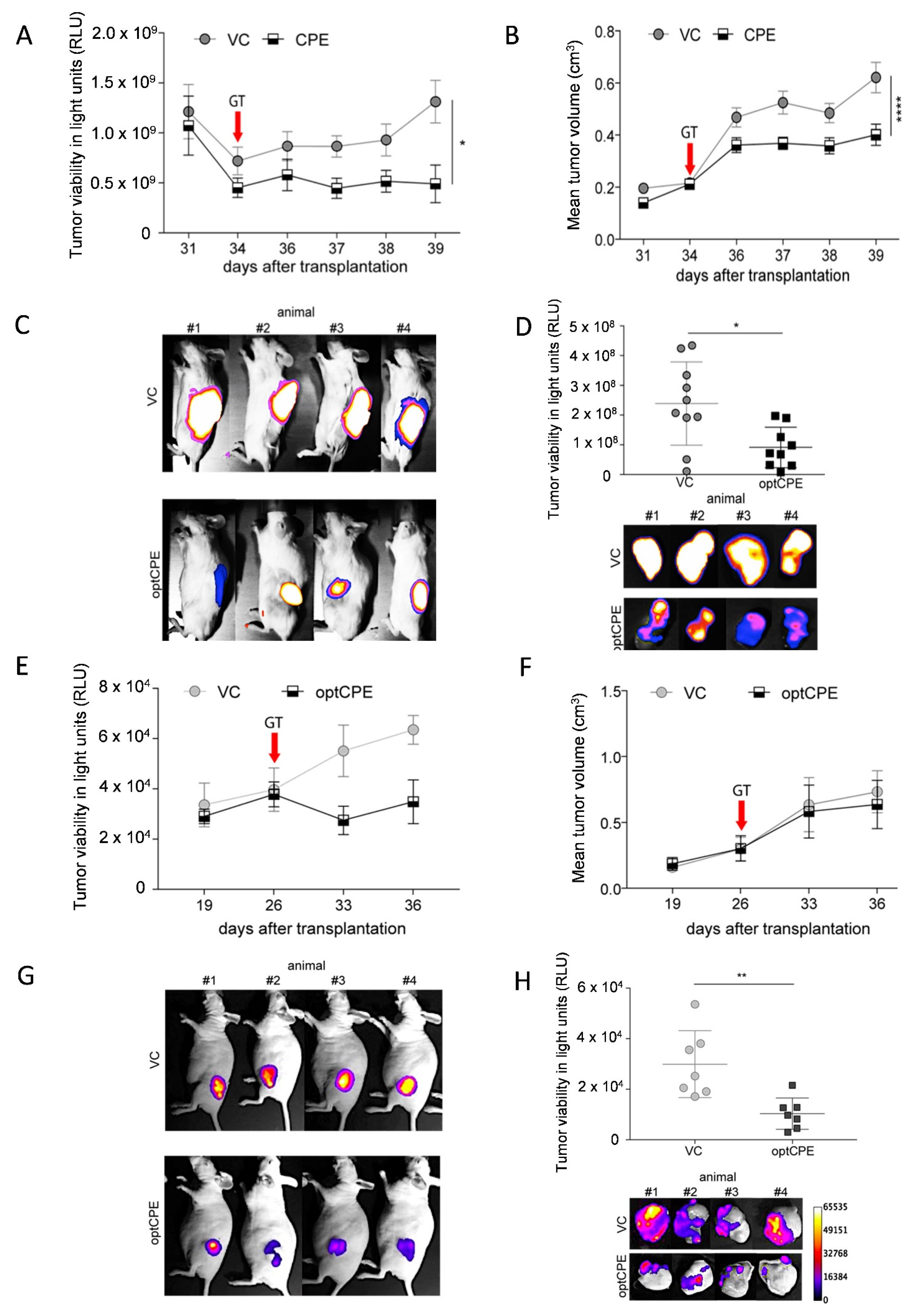
3.6. In Vivo Kinetic and Antitumoral Effects of CPE In Vivo Gene Transfer
3.7. Oncoleaking Efficacy of In Vivo CPE Gene Transfer
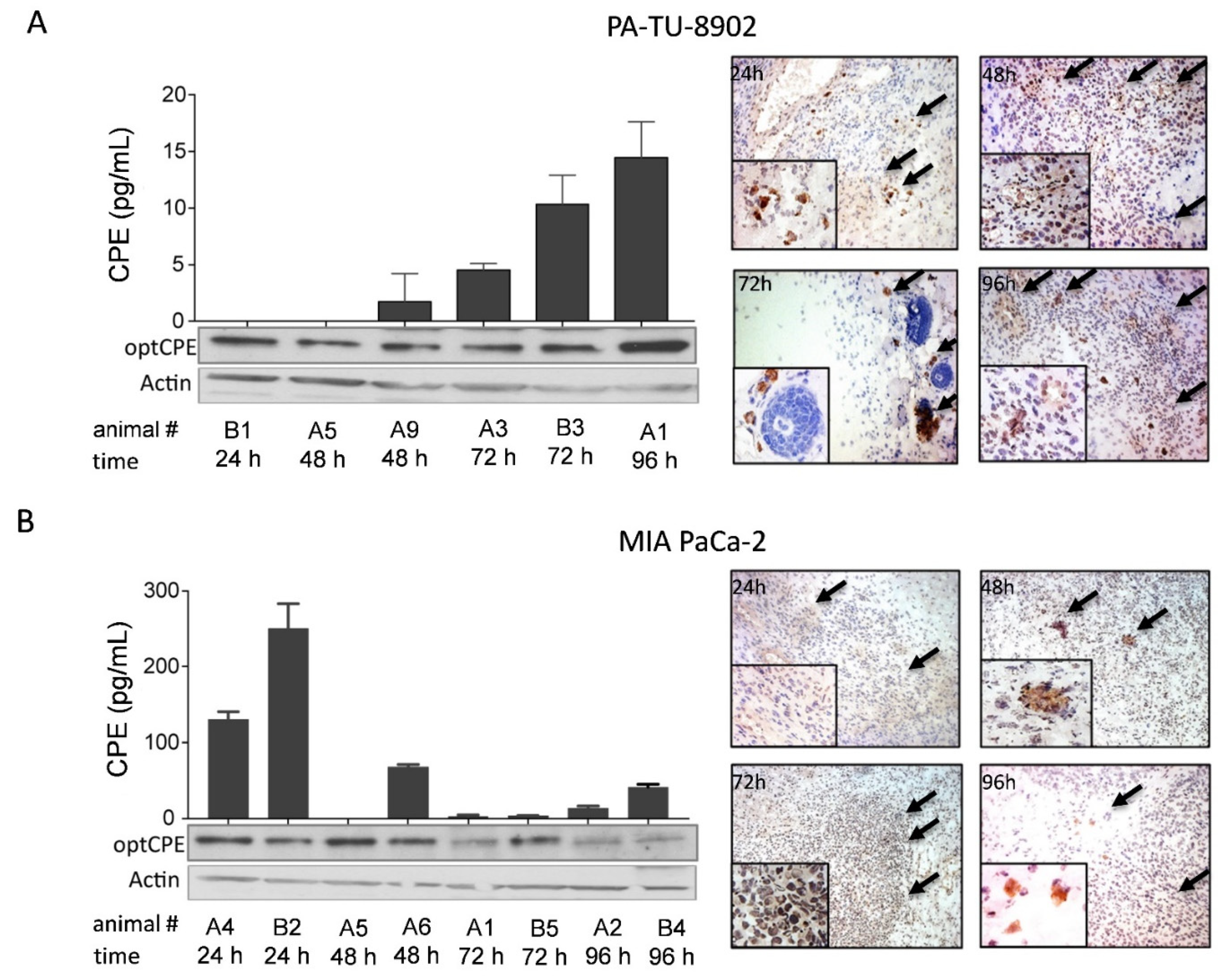
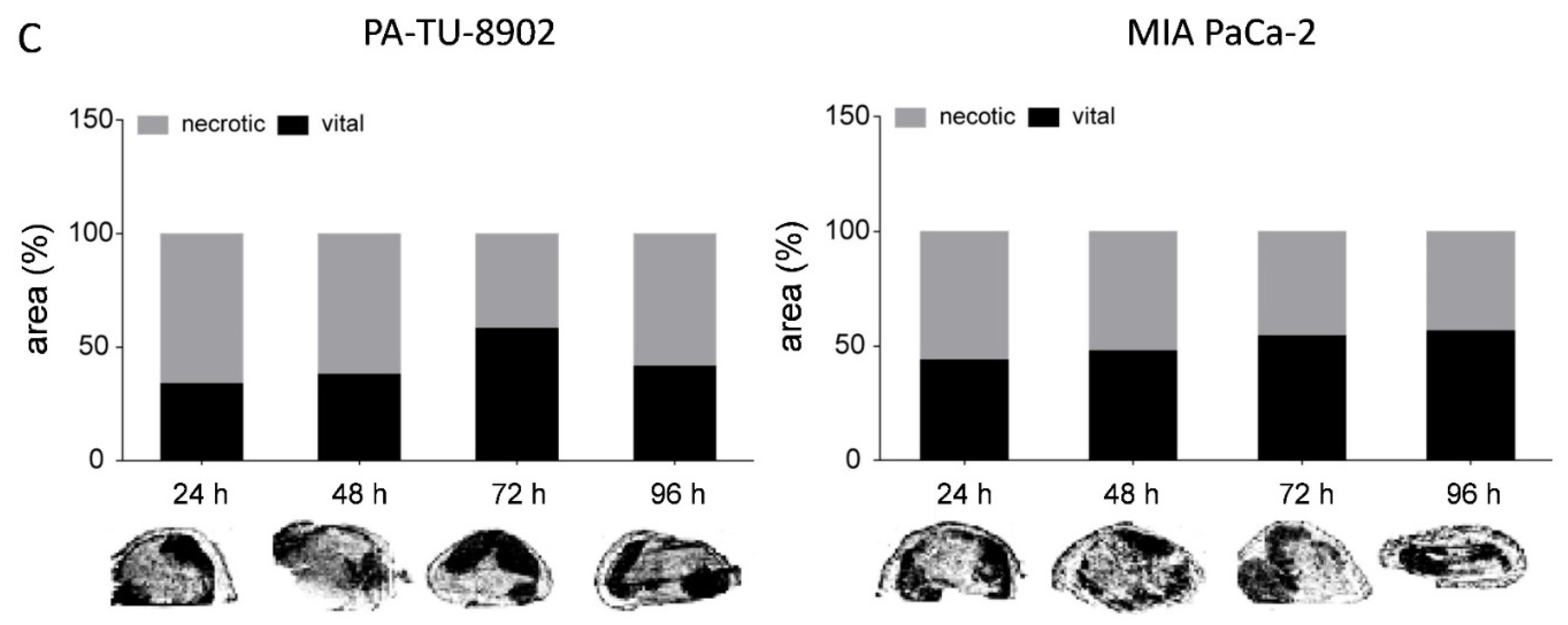
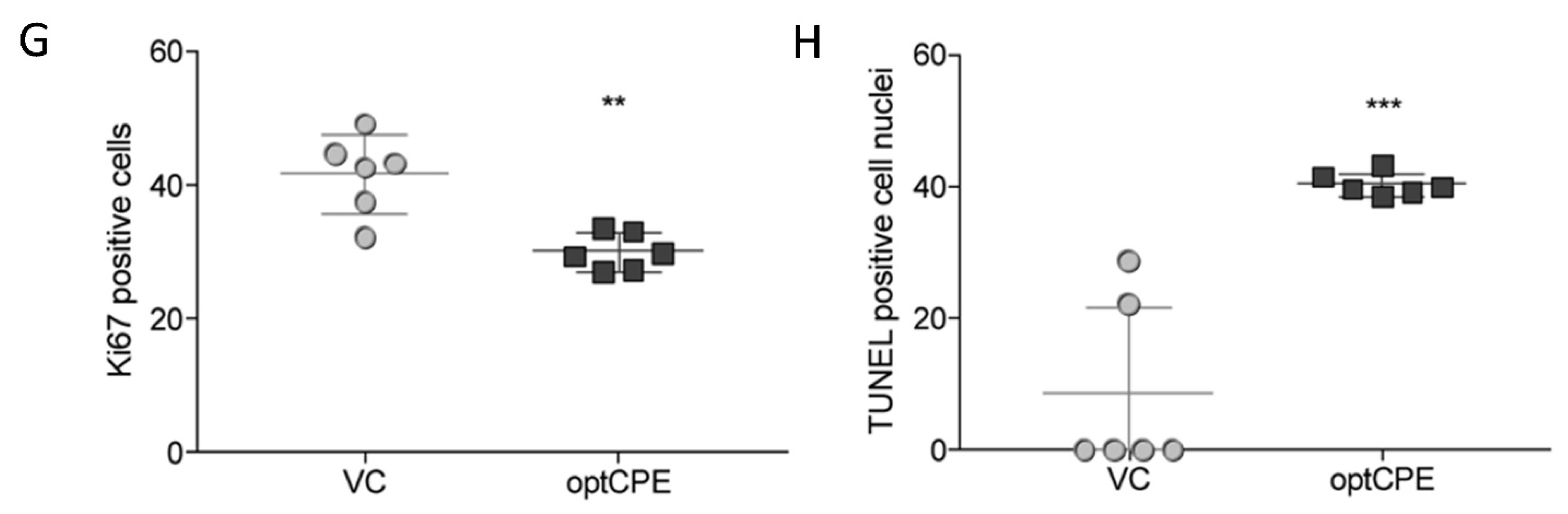
3.8. In Vivo Selectivity of CPE Gene Therapy and Mechanism of In Vivo Action
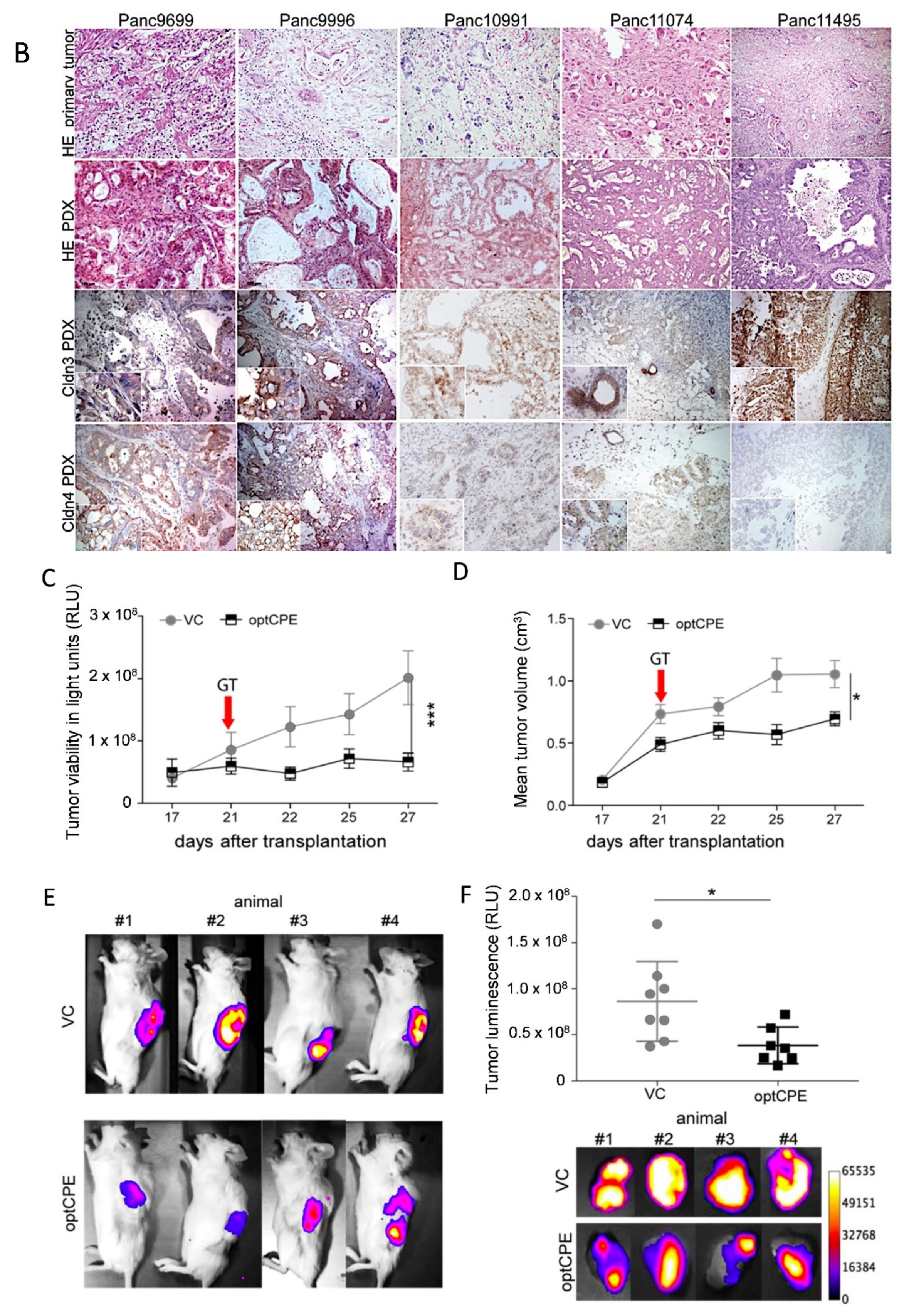
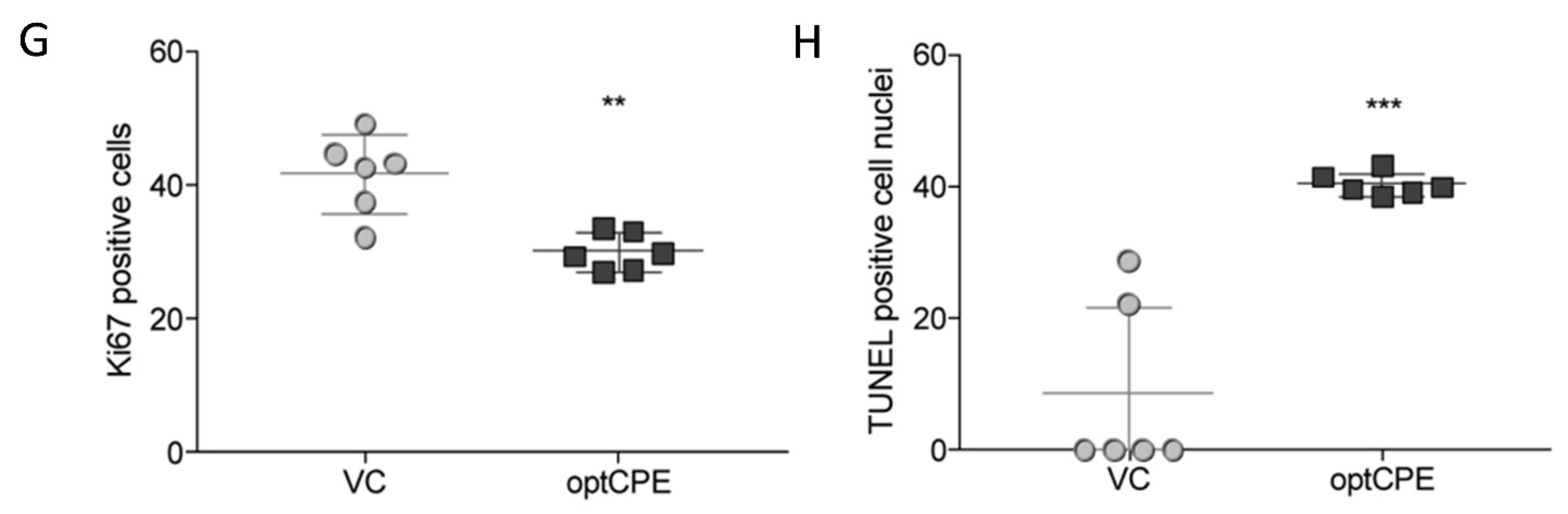
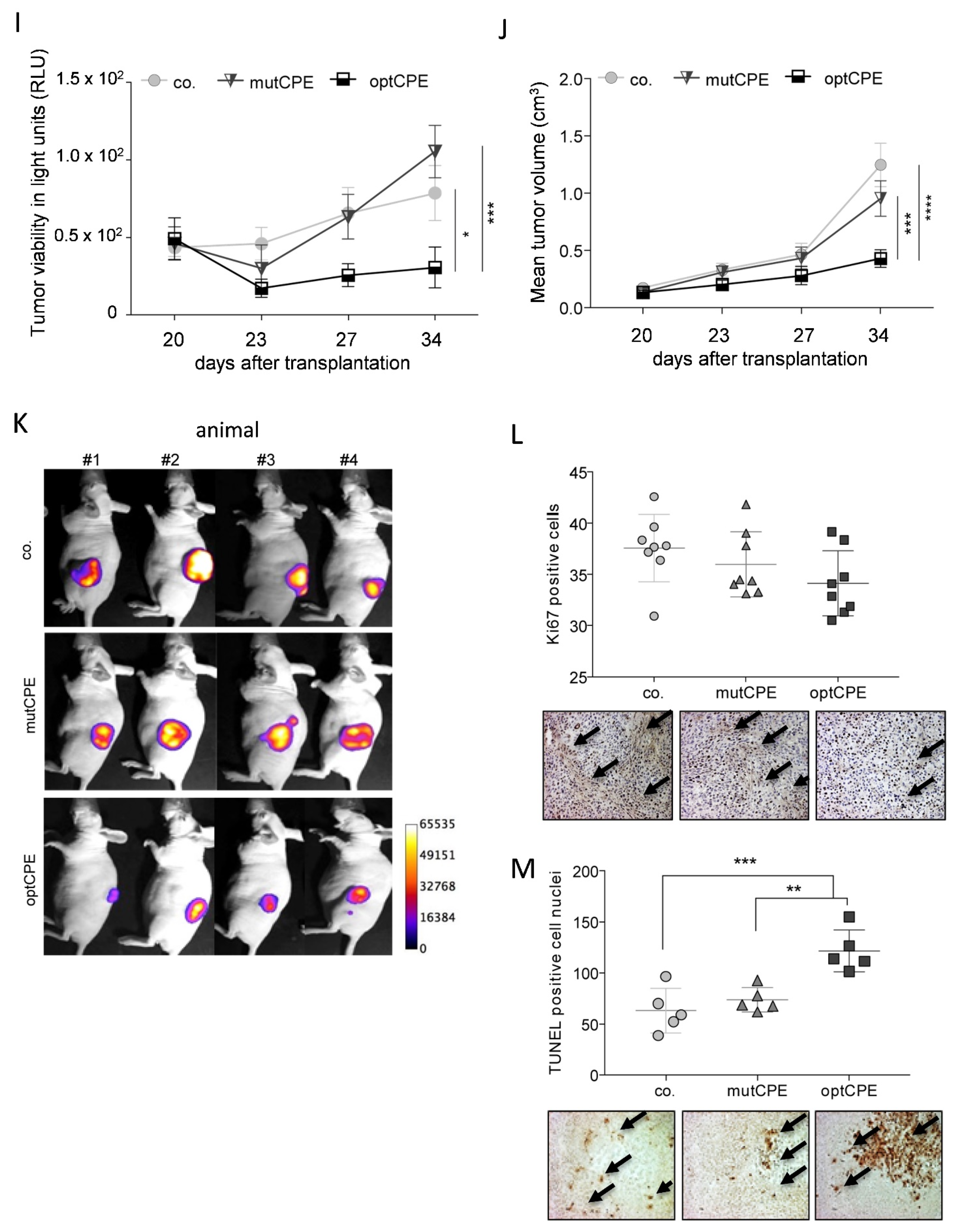
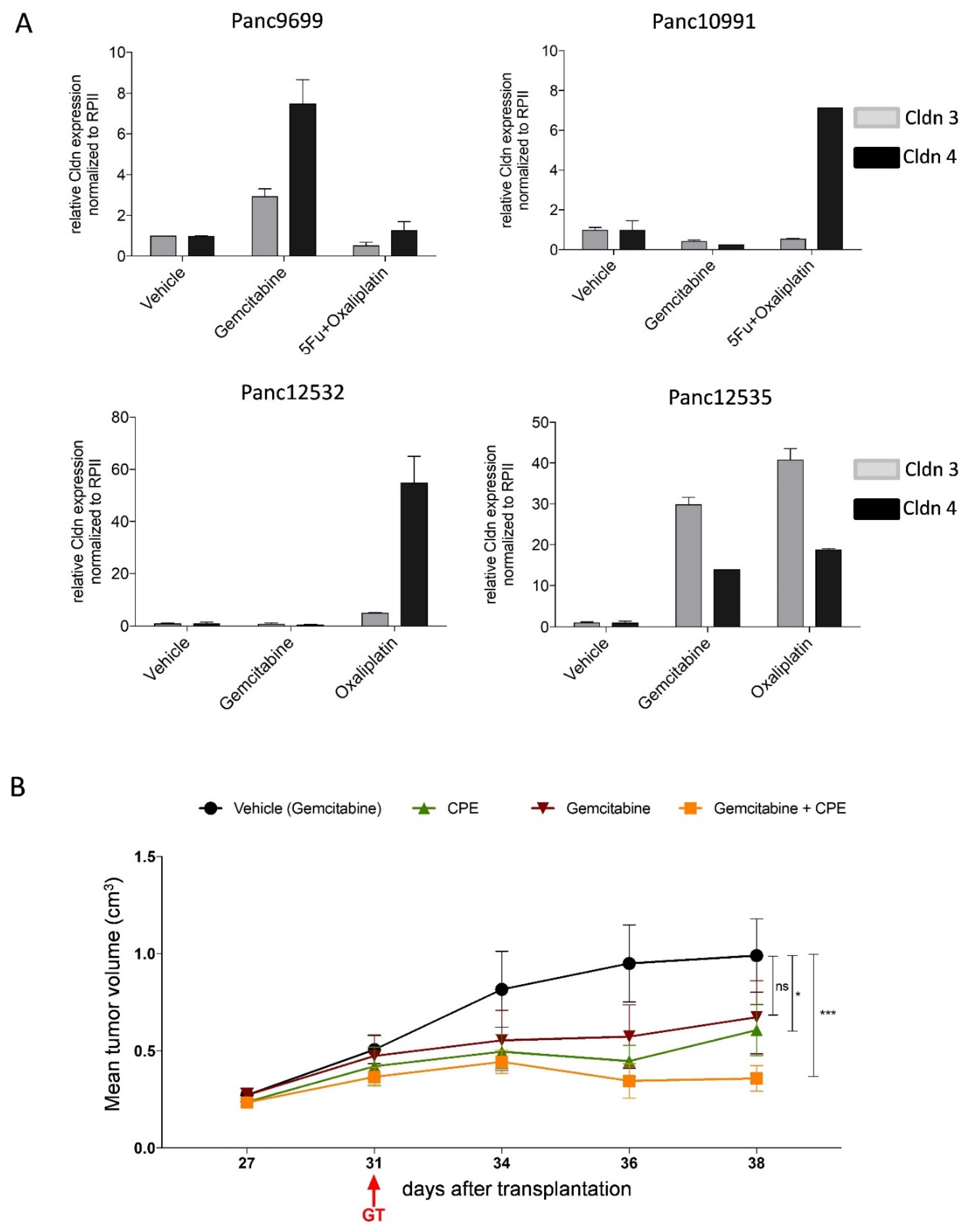
3.9. Oncoleaking CPE Gene Therapy in Pancreas Carcinoma PDX
3.10. Combined CPE Oncoleaking Gene Therapy and Drug Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Lohr, J.M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Raimondi, S.; Lowenfels, A.B.; Morselli-Labate, A.M.; Maisonneuve, P.; Pezzilli, R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Renouf, D.; Moore, M. Evolution of systemic therapy for advanced pancreatic cancer. Expert Rev. Anticancer Ther. 2010, 10, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Louvet, C.; Labianca, R.; Hammel, P.; Lledo, G.; Zampino, M.G.; Andre, T.; Zaniboni, A.; Ducreux, M.; Aitini, E.; Taieb, J.; et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: Results of a GERCOR and GISCAD phase III trial. J. Clin. Oncol. 2005, 23, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Quietzsch, D.; Gieseler, F.; Gonnermann, M.; Schonekas, H.; Rost, A.; Neuhaus, H.; Haag, C.; Clemens, M.; Heinrich, B.; et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J. Clin. Oncol. 2006, 24, 3946–3952. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Winter, J.M.; Brennan, M.F.; Tang, L.H.; D’Angelica, M.I.; Dematteo, R.P.; Fong, Y.; Klimstra, D.S.; Jarnagin, W.R.; Allen, P.J. Survival after resection of pancreatic adenocarcinoma: Results from a single institution over three decades. Ann. Surg. Oncol. 2012, 19, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Felgner, S.; Kocijancic, D.; Frahm, M.; Weiss, S. Bacteria in Cancer Therapy: Renaissance of an Old Concept. Int. J. Microbiol. 2016, 2016, 8451728. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.Y.; Chen, D.; Chan, J.; Yu, D.; Ko, E.; Pang, S. Regression of prostate cancer xenografts by a lentiviral vector specifically expressing diphtheria toxin A. Cancer Gene Ther. 2003, 10, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michl, P.; Gress, T.M. Bacteria and bacterial toxins as therapeutic agents for solid tumors. Curr. Cancer Drug Targets 2004, 4, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Shapira, A.; Benhar, I. Toxin-based therapeutic approaches. Toxins 2010, 2, 2519–2583. [Google Scholar] [CrossRef] [Green Version]
- Pahle, J.; Walther, W. Bacterial Toxins for Oncoleaking Suicidal Cancer Gene Therapy. Recent Results Cancer Res. 2016, 209, 95–110. [Google Scholar] [CrossRef]
- Rood, J.I. Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol. 1998, 52, 333–360. [Google Scholar] [CrossRef]
- Krause, G.; Winkler, L.; Mueller, S.L.; Haseloff, R.F.; Piontek, J.; Blasig, I.E. Structure and function of claudins. Biochim. Biophys. Acta 2008, 1778, 631–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunzel, D. Claudins: Vital partners in transcellular and paracellular transport coupling. Pflugers Arch. 2017, 469, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Smedley, J.G., 3rd; McClane, B.A. Fine mapping of the N-terminal cytotoxicity region of Clostridium perfringens enterotoxin by site-directed mutagenesis. Infect. Immun. 2004, 72, 6914–6923. [Google Scholar] [CrossRef] [Green Version]
- Robertson, S.L.; Smedley, J.G., 3rd; Singh, U.; Chakrabarti, G.; Van Itallie, C.M.; Anderson, J.M.; McClane, B.A. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell Microbiol. 2007, 9, 2734–2755. [Google Scholar] [CrossRef]
- Kitadokoro, K.; Nishimura, K.; Kamitani, S.; Fukui-Miyazaki, A.; Toshima, H.; Abe, H.; Kamata, Y.; Sugita-Konishi, Y.; Yamamoto, S.; Karatani, H.; et al. Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J. Biol. Chem. 2011, 286, 19549–19555. [Google Scholar] [CrossRef] [Green Version]
- Freedman, J.C.; Shrestha, A.; McClane, B.A. Clostridium perfringens Enterotoxin: Action, Genetics, and Translational Applications. Toxins 2016, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, G.; Zhou, X.; McClane, B.A. Death pathways activated in CaCo-2 cells by Clostridium perfringens enterotoxin. Infect. Immun. 2003, 71, 4260–4270. [Google Scholar] [CrossRef] [Green Version]
- Kominsky, S.L.; Tyler, B.; Sosnowski, J.; Brady, K.; Doucet, M.; Nell, D.; Smedley, J.G., 3rd; McClane, B.; Brem, H.; Sukumar, S. Clostridium perfringens enterotoxin as a novel-targeted therapeutic for brain metastasis. Cancer Res. 2007, 67, 7977–7982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, K.J.; Agarwal, R.; Morin, P.J. The claudin gene family: Expression in normal and neoplastic tissues. BMC Cancer 2006, 6, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Ding, L.; Lu, Q.; Chen, Y.H. Claudins in intestines: Distribution and functional significance in health and diseases. Tissue Barriers 2013, 1, e24978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kominsky, S.L.; Vali, M.; Korz, D.; Gabig, T.G.; Weitzman, S.A.; Argani, P.; Sukumar, S. Clostridium perfringens enterotoxin elicits rapid and specific cytolysis of breast carcinoma cells mediated through tight junction proteins claudin 3 and 4. Am. J. Pathol. 2004, 164, 1627–1633. [Google Scholar] [CrossRef] [Green Version]
- Black, J.D.; Lopez, S.; Cocco, E.; Schwab, C.L.; English, D.P.; Santin, A.D. Clostridium perfringens enterotoxin (CPE) and CPE-binding domain (c-CPE) for the detection and treatment of gynecologic cancers. Toxins 2015, 7, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Michl, P.; Buchholz, M.; Rolke, M.; Kunsch, S.; Lohr, M.; McClane, B.; Tsukita, S.; Leder, G.; Adler, G.; Gress, T.M. Claudin-4: A new target for pancreatic cancer treatment using Clostridium perfringens enterotoxin. Gastroenterology 2001, 121, 678–684. [Google Scholar] [CrossRef]
- Walther, W.; Petkov, S.; Kuvardina, O.N.; Aumann, J.; Kobelt, D.; Fichtner, I.; Lemm, M.; Piontek, J.; Blasig, I.E.; Stein, U.; et al. Novel Clostridium perfringens enterotoxin suicide gene therapy for selective treatment of claudin-3- and -4-overexpressing tumors. Gene Ther. 2012, 19, 494–503. [Google Scholar] [CrossRef]
- Pahle, J.; Menzel, L.; Niesler, N.; Kobelt, D.; Aumann, J.; Rivera, M.; Walther, W. Rapid eradication of colon carcinoma by Clostridium perfringens Enterotoxin suicidal gene therapy. BMC Cancer 2017, 17, 129. [Google Scholar] [CrossRef] [Green Version]
- Behrens, D.; Walther, W.; Fichtner, I. Pancreatic cancer models for translational research. Pharmacol. Ther. 2017, 173, 146–158. [Google Scholar] [CrossRef]
- Eichner, M.; Augustin, C.; Fromm, A.; Piontek, A.; Walther, W.; Bucker, R.; Fromm, M.; Krause, G.; Schulzke, J.D.; Gunzel, D.; et al. In Colon Epithelia, Clostridium perfringens Enterotoxin Causes Focal Leaks by Targeting Claudins Which are Apically Accessible Due to Tight Junction Derangement. J. Infect. Dis. 2017, 217, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piontek, A.; Eichner, M.; Zwanziger, D.; Beier, L.S.; Protze, J.; Walther, W.; Theurer, S.; Schmid, K.W.; Fuhrer-Sakel, D.; Piontek, J.; et al. Targeting claudin-overexpressing thyroid and lung cancer by modified Clostridium perfringens enterotoxin. Mol. Oncol. 2020, 14, 261–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinoda, T.; Shinya, N.; Ito, K.; Ohsawa, N.; Terada, T.; Hirata, K.; Kawano, Y.; Yamamoto, M.; Kimura-Someya, T.; Yokoyama, S.; et al. Structural basis for disruption of claudin assembly in tight junctions by an enterotoxin. Sci. Rep. 2016, 6, 33632. [Google Scholar] [CrossRef] [PubMed]
- Walther, W.; Siegel, R.; Kobelt, D.; Knosel, T.; Dietel, M.; Bembenek, A.; Aumann, J.; Schleef, M.; Baier, R.; Stein, U.; et al. Novel jet-injection technology for nonviral intratumoral gene transfer in patients with melanoma and breast cancer. Clin. Cancer Res. 2008, 14, 7545–7553. [Google Scholar] [CrossRef] [Green Version]
- Kosmidis, C.; Sapalidis, K.; Kotidis, E.; Mixalopoulos, N.; Zarogoulidis, P.; Tsavlis, D.; Baka, S.; Man, Y.G.; Kanellos, J. Pancreatic cancer from bench to bedside: Molecular pathways and treatment options. Ann. Transl. Med. 2016, 4, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teague, A.; Lim, K.H.; Wang-Gillam, A. Advanced pancreatic adenocarcinoma: A review of current treatment strategies and developing therapies. Ther. Adv. Med. Oncol. 2015, 7, 68–84. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Kojima, T.; Ito, T.; Kyuno, D.; Kimura, Y.; Imamura, M.; Hirata, K.; Sawada, N. Effects of Clostridium perfringens enterotoxin via claudin-4 on normal human pancreatic duct epithelial cells and cancer cells. Cell Mol. Biol. Lett. 2011, 16, 385–397. [Google Scholar] [CrossRef]
- Maeda, T.; Murata, M.; Chiba, H.; Takasawa, A.; Tanaka, S.; Kojima, T.; Masumori, N.; Tsukamoto, T.; Sawada, N. Claudin-4-targeted therapy using Clostridium perfringens enterotoxin for prostate cancer. Prostate 2012, 72, 351–360. [Google Scholar] [CrossRef]
- Kwon, M.J. Emerging roles of claudins in human cancer. Int. J. Mol. Sci. 2013, 14, 18148–18180. [Google Scholar] [CrossRef] [Green Version]
- Rangel, L.B.; Agarwal, R.; D’Souza, T.; Pizer, E.S.; Alo, P.L.; Lancaster, W.D.; Gregoire, L.; Schwartz, D.R.; Cho, K.R.; Morin, P.J. Tight junction proteins claudin-3 and claudin-4 are frequently overexpressed in ovarian cancer but not in ovarian cystadenomas. Clin. Cancer Res. 2003, 9, 2567–2575. [Google Scholar] [PubMed]
- Blanchard, A.A.; Skliris, G.P.; Watson, P.H.; Murphy, L.C.; Penner, C.; Tomes, L.; Young, T.L.; Leygue, E.; Myal, Y. Claudins 1, 3, and 4 protein expression in ER negative breast cancer correlates with markers of the basal phenotype. Virchows Arch. 2009, 454, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Lu, Z.; Lu, Q.; Chen, Y.H. The claudin family of proteins in human malignancy: A clinical perspective. Cancer Manag. Res. 2013, 5, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Nichols, L.S.; Ashfaq, R.; Iacobuzio-Donahue, C.A. Claudin 4 protein expression in primary and metastatic pancreatic cancer: Support for use as a therapeutic target. Am. J. Clin. Pathol. 2004, 121, 226–230. [Google Scholar] [CrossRef]
- Landers, K.A.; Samaratunga, H.; Teng, L.; Buck, M.; Burger, M.J.; Scells, B.; Lavin, M.F.; Gardiner, R.A. Identification of claudin-4 as a marker highly overexpressed in both primary and metastatic prostate cancer. Br. J. Cancer 2008, 99, 491–501. [Google Scholar] [CrossRef]
- Neesse, A.; Griesmann, H.; Gress, T.M.; Michl, P. Claudin-4 as therapeutic target in cancer. Arch. Biochem. Biophys. 2012, 524, 64–70. [Google Scholar] [CrossRef]
- Mitchell, L.A.; Koval, M. Specificity of interaction between clostridium perfringens enterotoxin and claudin-family tight junction proteins. Toxins 2010, 2, 1595–1611. [Google Scholar] [CrossRef] [Green Version]
- Robertson, S.L.; McClane, B.A. Interactions between Clostridium perfringens enterotoxin and claudins. Methods Mol. Biol. 2011, 762, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Veshnyakova, A.; Protze, J.; Rossa, J.; Blasig, I.E.; Krause, G.; Piontek, J. On the interaction of Clostridium perfringens enterotoxin with claudins. Toxins 2010, 2, 1336–1356. [Google Scholar] [CrossRef] [Green Version]
- Zeiss, C.J. The apoptosis-necrosis continuum: Insights from genetically altered mice. Vet. Pathol. 2003, 40, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, M.; Sugimoto, N. Calcium-independent and dependent steps in action of Clostridium perfringens enterotoxin on HeLa and Vero cells. Biochem. Biophys. Res. Commun. 1979, 91, 629–636. [Google Scholar] [CrossRef]
- Garcia-Belinchon, M.; Sanchez-Osuna, M.; Martinez-Escardo, L.; Granados-Colomina, C.; Pascual-Guiral, S.; Iglesias-Guimarais, V.; Casanelles, E.; Ribas, J.; Yuste, V.J. An Early and Robust Activation of Caspases Heads Cells for a Regulated Form of Necrotic-like Cell Death. J. Biol. Chem. 2015, 290, 20841–20855. [Google Scholar] [CrossRef] [Green Version]
- Denecker, G.; Vercammen, D.; Declercq, W.; Vandenabeele, P. Apoptotic and necrotic cell death induced by death domain receptors. Cell Mol. Life Sci. 2001, 58, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Hendricks, M.R.; Bomberger, J.M.; McClane, B.A. Bystander Host Cell Killing Effects of Clostridium perfringens Enterotoxin. mBio 2016, 7, e02015-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goepfert, C.; Gazdhar, A.; Frey, F.J.; Frey, B.M. Effect of electroporation-mediated diphtheria toxin A expression on PSA positive human prostate xenograft tumors in SCID mice. Prostate 2011, 71, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Lal-Nag, M.; Battis, M.; Santin, A.D.; Morin, P.J. Claudin-6: A novel receptor for CPE-mediated cytotoxicity in ovarian cancer. Oncogenesis 2012, 1, e33. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Cane, S.; Bellone, S.; Palmieri, M.; Siegel, E.R.; Thomas, M.; Roman, J.J.; Burnett, A.; Cannon, M.J.; Pecorelli, S. Treatment of chemotherapy-resistant human ovarian cancer xenografts in C.B-17/SCID mice by intraperitoneal administration of Clostridium perfringens enterotoxin. Cancer Res. 2005, 65, 4334–4342. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pahle, J.; Kobelt, D.; Aumann, J.; Behrens, D.; Daberkow, O.; Mokritzkij, M.; Piontek, J.; Stein, U.; Walther, W. Effective Oncoleaking Treatment of Pancreatic Cancer by Claudin-Targeted Suicide Gene Therapy with Clostridium perfringens Enterotoxin (CPE). Cancers 2021, 13, 4393. https://doi.org/10.3390/cancers13174393
Pahle J, Kobelt D, Aumann J, Behrens D, Daberkow O, Mokritzkij M, Piontek J, Stein U, Walther W. Effective Oncoleaking Treatment of Pancreatic Cancer by Claudin-Targeted Suicide Gene Therapy with Clostridium perfringens Enterotoxin (CPE). Cancers. 2021; 13(17):4393. https://doi.org/10.3390/cancers13174393
Chicago/Turabian StylePahle, Jessica, Dennis Kobelt, Jutta Aumann, Diana Behrens, Ole Daberkow, Margarita Mokritzkij, Jörg Piontek, Ulrike Stein, and Wolfgang Walther. 2021. "Effective Oncoleaking Treatment of Pancreatic Cancer by Claudin-Targeted Suicide Gene Therapy with Clostridium perfringens Enterotoxin (CPE)" Cancers 13, no. 17: 4393. https://doi.org/10.3390/cancers13174393
APA StylePahle, J., Kobelt, D., Aumann, J., Behrens, D., Daberkow, O., Mokritzkij, M., Piontek, J., Stein, U., & Walther, W. (2021). Effective Oncoleaking Treatment of Pancreatic Cancer by Claudin-Targeted Suicide Gene Therapy with Clostridium perfringens Enterotoxin (CPE). Cancers, 13(17), 4393. https://doi.org/10.3390/cancers13174393