
Identification of Androgen Receptor Metabolic Correlome Reveals the Repression of Ceramide Kinase by Androgens

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
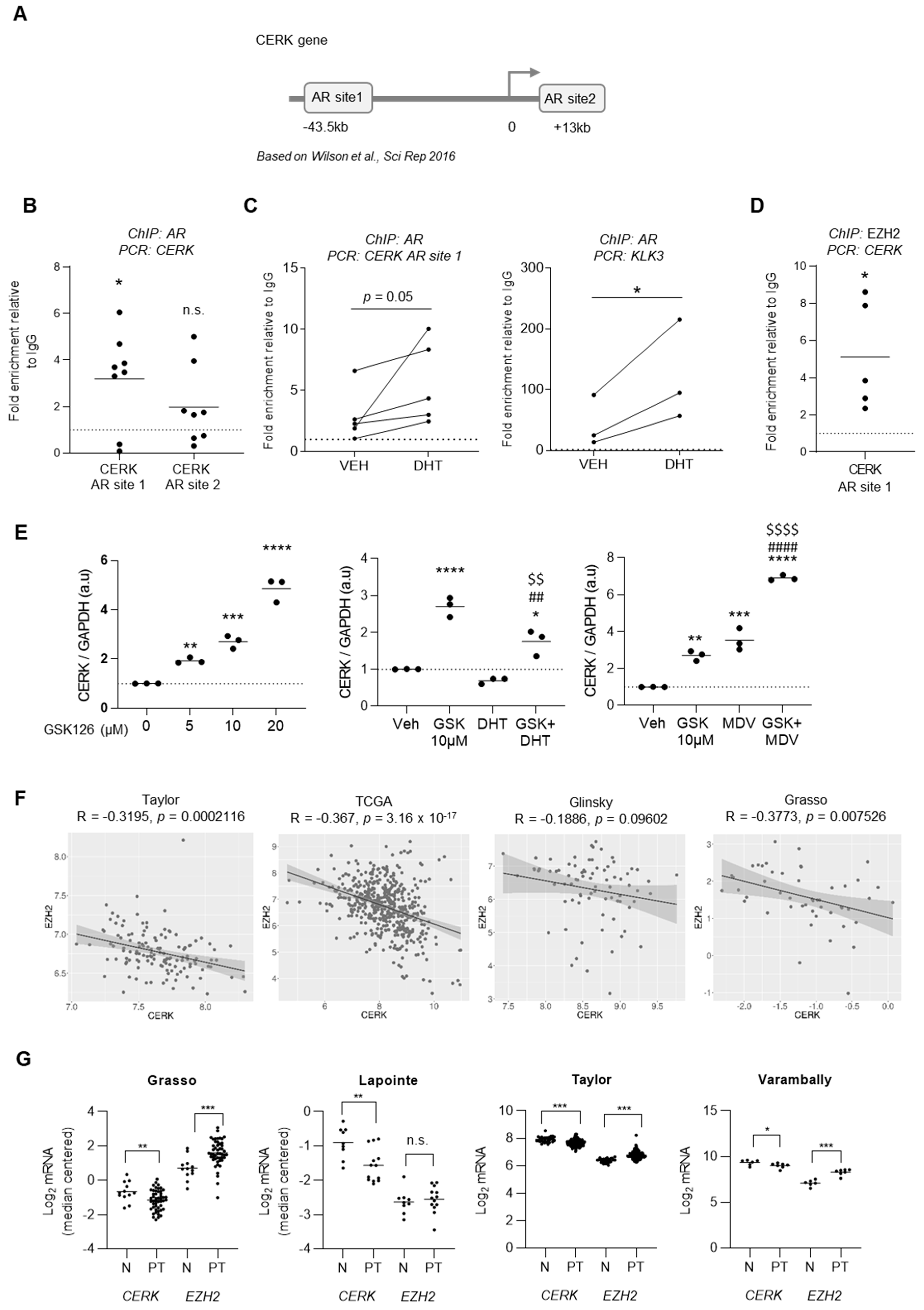
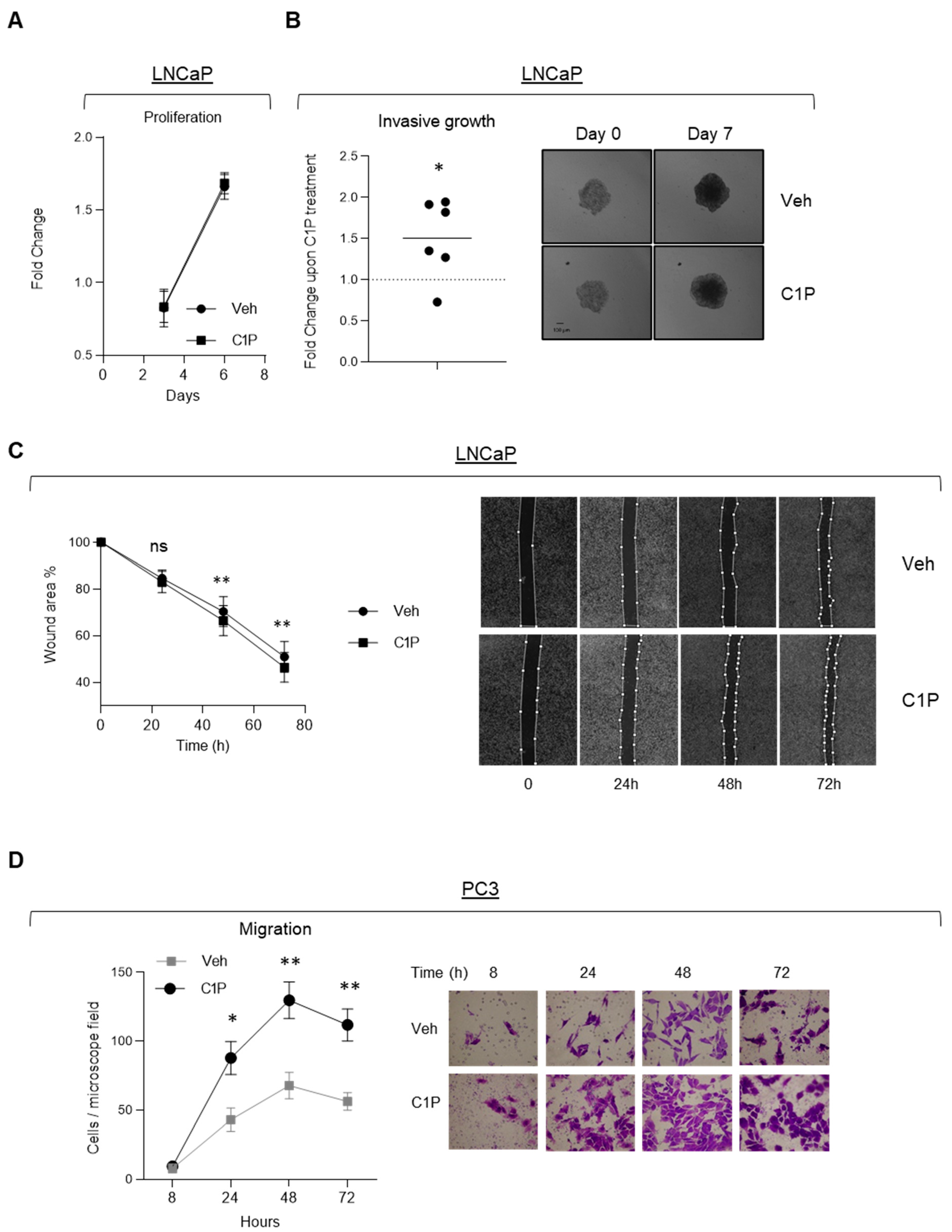
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of Stable Cell Lines
4.3. Reagents
4.4. Animals
4.5. qRTPCR
4.6. Sphingolipid Metabolic Analysis
4.7. ChIP
4.8. Cellular Assays
4.8.1. Cell Growth Analysis
4.8.2. Invasive Growth
4.8.3. Wound Healing
4.8.4. Migration
4.8.5. Anchorage-Independent Growth
4.9. Bioinformatics Analysis and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Litwack, G. Steroid Hormones Chapter 16. In Human Biochemistry; Litwack, G., Ed.; Academic Press: Boston, MA, USA, 2018; pp. 467–506. [Google Scholar] [CrossRef]
- Whirledge, S.; Cidlowski, J.A. Chapter 5—Steroid Hormone Action. In Yen and Jaffe’s Reproductive Endocrinology, 8th ed.; Strauss, J.F., Barbieri, R.L., Eds.; Content Repository Only: Philadelphia, PA, USA, 2019; pp. 115–131.e4. [Google Scholar] [CrossRef]
- Sever, R.; Glass, C.K. Signaling by nuclear receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a016709. [Google Scholar] [CrossRef] [Green Version]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Culig, Z.; Santer, F.R. Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev. 2014, 33, 413–427. [Google Scholar] [CrossRef]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen Signaling in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a030452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2019, 22, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heemers, H.V. Targeting Androgen Receptor Action for Prostate Cancer Treatment: Does the Post-Receptor Level Provide Novel Opportunities? Int. J. Biol. Sci. 2014, 10, 576–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saranyutanon, S.; Srivastava, S.K.; Pai, S.; Singh, S.; Singh, A.P. Therapies Targeted to Androgen Receptor Signaling Axis in Prostate Cancer: Progress, Challenges, and Hope. Cancers 2019, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Ku, S.Y.; Gleave, M.E.; Beltran, H. Towards precision oncology in advanced prostate cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of Advanced Prostate Cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef]
- Mansinho, A.; Macedo, D.; Fernandes, I.; Costa, L. Castration-Resistant Prostate Cancer: Mechanisms, Targets and Treatment. Adv. Exp. Med. Biol. 2018, 1096, 117–133. [Google Scholar] [CrossRef]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The Androgen Receptor Induces a Distinct Transcriptional Program in Castration-Resistant Prostate Cancer in Man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.R.; Azeem, W.; Hellem, M.R.; Marvyin, K.; Hua, Y.; Qu, Y.; Li, L.; Lin, B.; Ke, X.I.S.; Øyan, A.M.; et al. Context dependent regulatory patterns of the androgen receptor and androgen receptor target genes. BMC Cancer 2016, 16, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.J.; Kim, J.; Yu, J. Androgen receptor genomic regulation. Transl. Androl. Urol. 2013, 2, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Gritsina, G.; Gao, W.Q.; Yu, J. Transcriptional repression by androgen receptor: Roles in castration-resistant prostate cancer. Asian J. Androl. 2019, 21, 215–223. [Google Scholar] [CrossRef]
- Nassar, Z.D.; Mah, C.Y.; Dehairs, J.; Burvenich, I.J.; Irani, S.; Centenera, M.M.; Helm, M.; Shrestha, R.K.; Moldovan, M.; Don, A.S.; et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. Elife 2020, 9, e54166. [Google Scholar] [CrossRef]
- Clocchiatti, A.; Ghosh, S.; Procopio, M.G.; Mazzeo, L.; Bordignon, P.; Ostano, P.; Goruppi, S.; Bottoni, G.; Katarkar, A.; Levesque, M.; et al. Androgen receptor functions as transcriptional repressor of cancer-associated fibroblast activation. J. Clin. Investig. 2018, 128, 5531–5548. [Google Scholar] [CrossRef] [Green Version]
- Boulkroun, S.; Le Moellic, C.; Blot-Chabaud, M.; Farman, N.; Courtois-Coutry, N. Expression of androgen receptor and androgen regulation of NDRG2 in the rat renal collecting duct. Pflügers Arch. 2005, 451, 388–394. [Google Scholar] [CrossRef]
- Mah, C.Y.; Nassar, Z.D.; Swinnen, J.V.; Butler, L.M. Lipogenic effects of androgen signaling in normal and malignant prostate. Asian J. Urol. 2020, 7, 258–270. [Google Scholar] [CrossRef]
- Putluri, N.; Shojaie, A.; Vasu, V.T.; Nalluri, S.; Vareed, S.K.; Putluri, V.; Vivekanandan-Giri, A.; Byun, J.; Pennathur, S.; Sana, T.R.; et al. Metabolomic profiling reveals a role for androgen in activating amino acid metabolism and methylation in prostate cancer cells. PLoS ONE 2011, 6, e21417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, Z.; Hämäläinen, J.; Loikkanen, I.; Moilanen, A.M.; Hirvikoski, P.; Väisänen, T.; Paavonen, T.K.; Vaarala, M.H. Sorbitol dehydrogenase expression is regulated by androgens in the human prostate. Oncol. Rep. 2010, 23, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Centenera, M.M.; Swinnen, J.V. Androgen control of lipid metabolism in prostate cancer: Novel insights and future applications. Endocr. Relat. Cancer 2016, 23, R219–R227. [Google Scholar] [CrossRef]
- Dang, Q.; Chen, Y.A.; Hsieh, J.T. The dysfunctional lipids in prostate cancer. Am. J. Clin. Exp. Urol. 2019, 7, 273–280. [Google Scholar]
- Gangoiti, P.; Camacho, L.; Arana, L.; Ouro, A.; Granado, M.H.; Brizuela, L.; Casas, J.; Fabrias, G.; Abad, J.L.; Delgado, A.; et al. Control of metabolism and signaling of simple bioactive sphingolipids: Implications in disease. Prog. Lipid. Res. 2010, 49, 316–334. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortazar, A.R.; Torrano, V.; Martin-Martin, N.; Caro-Maldonado, A.; Camacho, L.; Hermanova, I.; Guruceaga, E.; Lorenzo-Martin, L.F.; Caloto, R.; Gomis, R.R.; et al. CANCERTOOL: A Visualization and Representation Interface to Exploit Cancer Datasets. Cancer Res. 2018, 78, 6320–6328. [Google Scholar] [CrossRef] [Green Version]
- Thorek, D.L.; Evans, M.J.; Carlsson, S.V.; Ulmert, D.; Lilja, H. Prostate-specific kallikrein-related peptidases and their relation to prostate cancer biology and detection. Established relevance and emerging roles. Thromb. Haemost. 2013, 110, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrano, V.; Valcarcel-Jimenez, L.; Cortazar, A.R.; Liu, X.; Urosevic, J.; Castillo-Martin, M.; Fernandez-Ruiz, S.; Morciano, G.; Caro-Maldonado, A.; Guiu, M.; et al. The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nat. Cell Biol. 2016, 18, 645–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchiroud, L.; Eichner, L.J.; Shaw, R.J.; Auwerx, J. Transcriptional coregulators: Fine-tuning metabolism. Cell Metab. 2014, 20, 26–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinsky, G.V.; Glinskii, A.B.; Stephenson, A.J.; Hoffman, R.M.; Gerald, W.L. Gene expression profiling predicts clinical outcome of prostate cancer. J. Clin. Investig. 2004, 113, 913–923. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapointe, J.; Li, C.; Higgins, J.P.; van de Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Dhanasekaran, S.M.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007, 39, 41–51. [Google Scholar] [CrossRef]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [CrossRef] [PubMed] [Green Version]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.-J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Coss, C.C.; Bauler, M.; Narayanan, R.; Miller, D.D.; Dalton, J.T. Alanine aminotransferase regulation by androgens in non-hepatic tissues. Pharm. Res. 2012, 29, 1046–1056. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merscher, S.; Fornoni, A. Podocyte pathology and nephropathy–sphingolipids in glomerular diseases. Front. Endocrinol. 2014, 5, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Runkle, C.; Jin, H.J.; Yu, J.; Li, J.; Yang, X.; Kuzel, T.; Lee, C.; Yu, J. CCN3/NOV gene expression in human prostate cancer is directly suppressed by the androgen receptor. Oncogene 2014, 33, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosse, A.; Bartsch, S.; Baniahmad, A. Androgen receptor-mediated gene repression. Mol. Cell Endocrinol. 2012, 352, 46–56. [Google Scholar] [CrossRef]
- Wilson, S.; Qi, J.; Filipp, F.V. Refinement of the androgen response element based on ChIP-Seq in androgen-insensitive and androgen-responsive prostate cancer cell lines. Sci. Rep. 2016, 6, 32611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, N.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun. 2018, 9, 4080. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Gomez-Munoz, A. The Role of Ceramide 1-Phosphate in Tumor Cell Survival and Dissemination. Adv. Cancer Res. 2018, 140, 217–234. [Google Scholar] [CrossRef]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Tousignant, K.D.; Rockstroh, A.; Taherian Fard, A.; Lehman, M.L.; Wang, C.; McPherson, S.J.; Philp, L.K.; Bartonicek, N.; Dinger, M.E.; Nelson, C.C.; et al. Lipid Uptake Is an Androgen-Enhanced Lipid Supply Pathway Associated with Prostate Cancer Disease Progression and Bone Metastasis. Mol. Cancer Res. 2019, 17, 1166–1179. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Gao, S.; Barrett, D.; Ahmed, M.; Han, D.; Macoska, J.A.; He, H.H.; Cai, C. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene 2018, 37, 710–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centenera, M.M.; Scott, J.S.; Machiels, J.; Nassar, Z.D.; Miller, D.C.; Zininos, I.; Dehairs, J.; Burvenich, I.J.G.; Zadra, G.; Chetta, P.; et al. ELOVL5 is a critical and targetable fatty acid elongase in prostate cancer. Cancer Res. 2021, 81, 1704–1718. [Google Scholar] [CrossRef]
- Abudurexiti, M.; Zhu, W.; Wang, Y.; Wang, J.; Xu, W.; Huang, Y.; Zhu, Y.; Shi, G.; Zhang, H.; Zhu, Y.; et al. Targeting CPT1B as a potential therapeutic strategy in castration-resistant and enzalutamide-resistant prostate cancer. Prostate 2020, 80, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Audet-Walsh, É.; Vernier, M.; Yee, T.; Laflamme, C.; Li, S.; Chen, Y.; Giguère, V. SREBF1 Activity Is Regulated by an AR/mTOR Nuclear Axis in Prostate Cancer. Mol. Cancer Res. 2018, 16, 1396–1405. [Google Scholar] [CrossRef] [Green Version]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Gangoiti, P.; Granado, M.H.; Alonso, A.; Goñi, F.M.; Gómez-Muñoz, A. Implication of ceramide, ceramide 1-phosphate and sphingosine 1-phosphate in tumorigenesis. Transl. Oncogenomics 2008, 3, 81–98. [Google Scholar] [PubMed]
- Rivera, I.-G.; Ordoñez, M.; Presa, N.; Gangoiti, P.; Gomez-Larrauri, A.; Trueba, M.; Fox, T.; Kester, M.; Gomez-Muñoz, A. Ceramide 1-phosphate regulates cell migration and invasion of human pancreatic cancer cells. Biochem. Pharmacol. 2016, 102, 107–119. [Google Scholar] [CrossRef]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.C.; Yu, J.; Runkle, C.; Wu, L.; Hu, M.; Wu, D.; Liu, J.S.; Wang, Q.; Qin, Z.S.; Yu, J. Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012, 22, 322–331. [Google Scholar] [CrossRef] [Green Version]
- Fong, K.W.; Zhao, J.C.; Kim, J.; Li, S.; Yang, Y.A.; Song, B.; Rittie, L.; Hu, M.; Yang, X.; Perbal, B.; et al. Polycomb-Mediated Disruption of an Androgen Receptor Feedback Loop Drives Castration-Resistant Prostate Cancer. Cancer Res. 2017, 77, 412–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, K.R.; Chang, C.W.; Tan, S.K.; Yang, C.; Hong, S.Z.; Sng, N.Y.; Cheung, E. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. Embo. J. 2012, 31, 2810–2823. [Google Scholar] [CrossRef] [PubMed]
- Wiederschain, D.; Susan, W.; Chen, L.; Loo, A.; Yang, G.; Huang, A.; Chen, Y.; Caponigro, G.; Yao, Y.M.; Lengauer, C.; et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 2009, 8, 498–504. [Google Scholar] [CrossRef]
- Ugalde-Olano, A.; Egia, A.; Fernandez-Ruiz, S.; Loizaga-Iriarte, A.; Zuniga-Garcia, P.; Garcia, S.; Royo, F.; Lacasa-Viscasillas, I.; Castro, E.; Cortazar, A.R.; et al. Methodological aspects of the molecular and histological study of prostate cancer: Focus on PTEN. Methods 2015, 77, 25–30. [Google Scholar] [CrossRef]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol. Cell 2005, 19, 631–642. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camacho, L.; Zabala-Letona, A.; Cortazar, A.R.; Astobiza, I.; Dominguez-Herrera, A.; Ercilla, A.; Crespo, J.; Viera, C.; Fernández-Ruiz, S.; Martinez-Gonzalez, A.; et al. Identification of Androgen Receptor Metabolic Correlome Reveals the Repression of Ceramide Kinase by Androgens. Cancers 2021, 13, 4307. https://doi.org/10.3390/cancers13174307
Camacho L, Zabala-Letona A, Cortazar AR, Astobiza I, Dominguez-Herrera A, Ercilla A, Crespo J, Viera C, Fernández-Ruiz S, Martinez-Gonzalez A, et al. Identification of Androgen Receptor Metabolic Correlome Reveals the Repression of Ceramide Kinase by Androgens. Cancers. 2021; 13(17):4307. https://doi.org/10.3390/cancers13174307
Chicago/Turabian StyleCamacho, Laura, Amaia Zabala-Letona, Ana R. Cortazar, Ianire Astobiza, Asier Dominguez-Herrera, Amaia Ercilla, Jana Crespo, Cristina Viera, Sonia Fernández-Ruiz, Ainara Martinez-Gonzalez, and et al. 2021. "Identification of Androgen Receptor Metabolic Correlome Reveals the Repression of Ceramide Kinase by Androgens" Cancers 13, no. 17: 4307. https://doi.org/10.3390/cancers13174307
APA StyleCamacho, L., Zabala-Letona, A., Cortazar, A. R., Astobiza, I., Dominguez-Herrera, A., Ercilla, A., Crespo, J., Viera, C., Fernández-Ruiz, S., Martinez-Gonzalez, A., Torrano, V., Martín-Martín, N., Gomez-Muñoz, A., & Carracedo, A. (2021). Identification of Androgen Receptor Metabolic Correlome Reveals the Repression of Ceramide Kinase by Androgens. Cancers, 13(17), 4307. https://doi.org/10.3390/cancers13174307