Simple Summary
Cancer of the esophagus is a deadly disease. There are two main subtypes, adenocarcinoma and squamous cell carcinoma, with adenocarcinoma of the esophagus (EAC) being more common in Western countries. Barrett’s esophagus (BE) describes a change in the esophageal surface near the stomach in response to reflux of gastric acid into the esophagus. BE increases the risk of developing EAC, and the incidence of EAC has risen dramatically over recent decades. One likely reason for the poor prognosis of EAC is based on the fact that each tumor has many genes affected by mutations, and most of these genes differ across patients, hampering the efficacy of therapies that target specific cancer driver proteins. In this review, we provide an overview of the gene mutations and gene activity changes in EAC and how these features can be used to divide patients into groups that might have different clinical characteristics.
Abstract
Esophageal adenocarcinoma (EAC) is a deadly disease with limited options for targeted therapy. With the help of next-generation sequencing studies over the last decade, we gained an understanding of the genomic architecture of EAC. The tumor suppressor gene TP53 is mutated in 70 to 80% of tumors followed by genomic alterations in CDKN2A, KRAS, ERBB2, ARID1A, SMAD4 and a long tail of less frequently mutated genes. EAC is characterized by a high burden of point mutations and genomic rearrangements, resulting in amplifications and deletions of genomic regions. The genomic complexity is likely hampering the efficacy of targeted therapies. Barrett’s esophagus (BE), a metaplastic response of the esophagus to gastro-esophageal reflux disease, is the main risk factor for the development of EAC. Almost all EACs are derived from BE. The sequence from BE to EAC provides an opportunity to study the genomic evolution towards EAC. While the overlap of point mutations between BE and EAC within the same patient is, at times, surprisingly low, there is a correlation between the complexity of the genomic copy number profile and the development of EAC. Transcriptomic analyses separated EAC into a basal and a classical subtype, with the basal subtype showing a higher level of resistance to chemotherapy. In this review, we provide an overview of the current knowledge of the genomic and transcriptomic characteristics of EAC and their relevance for the development of the disease and patient care.
1. Introduction
Esophageal cancer is the eighth most common cancer worldwide, and the sixth most common cause of cancer-related death [1]. The most prevalent subtype of esophageal cancer in Western countries is esophageal adenocarcinoma (EAC). EAC has a poor prognosis, with an overall five-year survival rate of 20% and a median overall survival of less than a year due to the lack of targeted therapies and diagnosis usually made at late disease stages [1]. Supporting the importance of disease stage as a prognostic factor, EAC diagnosed at stage I has a five-year survival rate of 80%, decreasing to a dramatic 2% when diagnosed at stage IV, documented recently with a cohort from Northern Ireland [1]. Over the last decade, the incidence of EAC has risen dramatically in Western countries [2], emphasizing the need to improve therapeutic modalities for this tumor type with a poor prognosis. Neoadjuvant radiochemotherapy and perioperative chemotherapy are the standard of care, but the response varies dramatically, ranging from primary resistance to complete response. There are no robust biomarkers that allow predicting which patients will benefit from neoadjuvant therapy. Such biomarkers, however, would be very relevant for the clinic as they would spare non-responders from the toxic treatment. The genomic picture of EAC before and after neoadjuvant chemotherapy has been compared in a small number of studies [3,4,5]. Thus far, no common mechanisms for resistance could be identified.
The genomic architecture of EAC is rather complex, comprising many point mutations and genome structural alterations showing similarities to the chromosome instable group (CIN) of gastric cancer [6]. Overall, there is a need for the identification of patient subgroups based on a combination of molecular and clinical markers that allow a more tailored treatment of EAC patients. In this review, we provide an overview of our current understanding of the molecular characteristics of EAC based on systematic genomic and transcriptomic studies. We connect the molecular findings to clinical relevance and provide a perspective on what to expect in the near future.
2. Genomics of EAC
2.1. Somatic Variations and Copy Number Alterations
Whole genome sequencing (WGS) of a large cohort (n = 129) revealed that the genomic landscape of EAC is driven by large-scale genome alterations. Although point mutations are usually abundant, the recurrence of point mutations in specific genes across patients is low. Instead, Secrier and colleagues reported a dominance of genomic structural variations and copy number alterations [7]. The most frequently altered genes show a higher rate of rearrangements, amplifications or deletions, rather than insertions or deletions of a few base pairs (indels) or nonsynonymous point mutations. These rearranged genes comprise regulators of transcription, signaling and cell communication and include SMYD3 (frequency of 39%), RUNX1 (27%), CTNNA3 (22%), RBFOX1 (21%), CDKN2A and CDKN2B (18%) and CDK14 (16%). Fragile sites such as FHIT (95%) and WWOX (84%) are also recurrently rearranged. Most tumors show evidence for mobile element insertions, with an average of 25 inserts per tumor. Frequently affected genes by mobile element insertions are ERBB4 (5%), CTNNA3 (4%), CTNNA2 (3%), CDH18 (2%) and SOX5 (2%), all of which play important roles in regulating cell signaling, mitosis and adhesion. Amplified genome loci comprise the genes ERBB2, EGFR, RB1, GATA4/6, CCND1 and MDM2. Deletions, which are generally less frequent, include CLDN22 and CDKN2A/2B.
As it has been mentioned, point mutations and indels are abundant but heterogeneous, except for TP53. Nevertheless, seven genes reached significance in the analysis of Secrier and colleagues: TP53 (81%), ARID1A (17%), SMAD4 (16%), CDKN2A (15%), KCNQ3 (12%), CCDC102B (9%) and CYP7B1 (7%). Multiple receptor tyrosine kinases (RTKs) such as ERBB2, EGFR, MET and FGFR are often amplified in EAC tumors. Most samples (91%) have copy number gains in more than one RTK and/or amplifications in genes of the downstream MAPK and PI3K pathways. This may explain the poor results of the several current RTK therapy trials targeting single molecules of these pathways [8,9,10,11,12].
2.1.1. Combining Methods and Expression Levels to Identify Driver Events in EAC
The high heterogeneity of EAC has driven the research community to combine cohorts and data types for systematic analyses with higher sensitivity to find common alterations among patients. Frankell and colleagues analyzed DNA sequencing data from a collection of 551 EACs in total, comprising WGS data of the International Cancer Genome Consortium (ICGC, n = 379) [13], WGS of Nones and colleagues (n = 22) [14] and whole exome sequencing (WES) of Dulak and colleagues (n = 149) [15], together with paired RNA sequencing (RNA-seq) data of 116 patients from the ICGC collection [13]. With a combination of methods detecting recurrent mutations, high-functional impact mutations, mutation clustering and amplification or deletion of genes that are over- or underexpressed, a total of 76 driver genes were suggested. The most frequently altered genes are TP53 (72%), CDKN2A (28%), KRAS (19%), MYC (19%), ERBB2 (18%), GATA4 (15%), SMAD4 (15%), CCND1 (14%), GATA6 (14%), CDK6 (14%), ARID1A (13%) and EGFR (12%) (with GATA4 and GATA6 not meeting the general cancer genome criteria for oncogenes or tumor suppressor genes, respectively (https://cancer.sanger.ac.uk/census, accessed on 2 August 2021). The two driver events GATA4 amplification and SMAD4 mutation are prognostic for poor survival and therefore promising predictors for tailoring clinical care.
Non-coding driver elements discovered by ActiveDriverWGS, which considers functional impact, included regulatory regions of TP53TG1, PTDSS1 and WDR74, which are described to be relevant for other cancer entities [16], and EAC-specific regions of the matrix metalloproteinase MMP24, FTO, MTG2 and histone-encoding HIST1H2BO and HIST1H2AM.
Of 149 amplifications or deletions of genome loci detected by Genomic Identification of Significant Targets in Cancer (GISTIC), only 26% showed relative expression changes in the matched RNA-seq data, though the authors declared the low power to detect small expression differences. Among the altered genes were 17 known oncogenes including ERBB2, KRAS and SMAD4, emphasizing their relevance for EAC.
Ultrahigh copy number amplifications (ploidy-adjusted copy number > 10) are found frequently, likely due to extrachromosomal amplification via double minutes, and have a much stronger effect on expression changes than moderate copy number gains [13,14]. Naturally, but worth mentioning, tumor suppressor genes such as ARID1B are selected for truncating mutations, leading to dysfunctional isoforms, while oncogenes such as ERBB2 are more prone to activating missense mutations. Interestingly, TP53-mutated tumors had more driver genes affected by somatic copy number changes than TP53 wild-type tumors, in agreement with the protein’s prominent role as a key player for DNA integrity.
Underlining the importance of altered TP53 function for EAC, TP53 wild-type tumors are enriched for amplifications of MDM2, which degrades TP53, thus mimicking TP53-mutated tumors [4,13]. Dysfunctional TP53 mutations and MDM2 amplifications rarely appear in the same tumor, compatible with the equivalent functional consequences. Considering the participation of genes in cellular pathways, Frankell and colleagues found a mutation-driven activation of the Wnt pathway (19% of patients) and regulation of the SWI/SNF complex (28%) [13]. By a comparison of different pathways, the authors highlighted co-occurring mutations of TP53 and MYC, GATA6 and SMAD4 and the Wnt and immune pathways, suggesting complementary roles for EAC pathophysiology. On the other hand, mutual exclusivity exists for a) ARID1A and MYC, b) pathways of RTKs and gastrointestinal differentiation and c) the DNA damage repair pathway and SWI/SNF, suggesting that the two members of each pair have similar roles in EAC pathophysiology. Using the Cancer Biomarkers database, the authors found >50% of EAC cases to be potentially sensitive to CDK4/CDK6 inhibitors, based on gene alterations in the related pathways, which could be validated by the first in vitro experiments [13].
2.1.2. Large-Scale Sequencing of EAC Reveals EAC Helper Genes
The high heterogeneity of EAC with low frequencies of specific driver gene alterations makes it challenging to develop therapies. To overcome this problem, a machine learning algorithm called sysSVM has been used to detect recurrent but also rare altered genes contributing to cancer progression for individual patients [17]. Based on their similarity to known cancer genes regarding features including genomic alterations, protein network positions, miRNA associations, gene expression breadth and many more, a list of 952 helper genes has been established that might contribute to the progression of EAC. In accordance with the dominance of SCNAs in EAC, around 80% of the helper genes have copy number gains, accompanied by higher gene expression. Clustering by the proportion of perturbed pathways of the 952 helper genes divides the patient cohort into six subgroups with distinct molecular and clinical features. For example, EAC from subgroup 1 is enriched for the BRCA-related DNA damage response (DDR)-impaired mutational signature S3, and subgroup 4 is prognostic for poor survival. Experimental approaches show that alterations of helper genes increase EAC cell line growth to a similar extent as those of driver genes, underlining the clinical importance of helper genes. Of clinical interest is the fact that in vitro tumor cells develop a dependency on the altered helper genes, making them promising targets to develop patient-specific therapies. Interestingly, damaging alterations are also enriched in helper genes in pre-malignant Barrett’s esophagus (BE) samples and promote cellular proliferation in vitro. Thus, rare helper genes may contribute to the transition of BE to EAC, specifically for individual patients.
2.1.3. Retrospective DNA Sequencing of Short Survivors
Hao and colleagues performed WES with treatment-naïve advanced (metastatic) EAC samples that were stratified into short (n = 20) and long (n = 20) survival [18]. The mutation burden is not different between short and long survivors. The most frequently mutated genes of the entire cohort were reported as TP53 (70%), SMAD4 (22%), LRP1B (20%), FAT4 (18%), ERBB4 (15%), DMD (15%), CACNA1A (12%), SPTA1 (12%), APC (10%) and PIK3CA (10%). Deviating results from previous publications with larger cohorts are likely attributed to the fact that only tumors with the advanced stage 4b were included in this study. Notably, KMT2C alterations were found exclusively in short survivors and were associated with poor prognosis. Of note, short survivors had a higher intratumoral heterogeneity, with more subclones.
In addition to the known copy number losses on chromosomes 4, 9p, 17p and 18 and gains on chromosomes 20, 7p and 8q [6], loss of chromosome 4 was more frequent in the cohort of shorter survivors (95% vs. 40%) and was associated with poor survival in a larger TCGA cohort [18]. Tumors with chromosome 4 loss had a significantly decreased abundance of all major immune cell types, including T cells, suggesting a cold tumor type. Cold tumors are defined as non-immunogenic tumors without or with low infiltration of immune cells, especially T cells, making them invulnerable to immunotherapies.
2.2. Large-Scale Alterations and Genomic Catastrophes
The genomic landscape of EAC is characterized by large-scale alterations also known as genomic catastrophes. Genomic catastrophes can rapidly lead to alterations in many cancer-relevant genes driving cancer development. Chromotripsis is an event described by chromosomes shattered to fragments that are reassembled in a random order and orientation, resulting in a highly rearranged novel chromosome. The frequency of chromotripsis in EAC, at ~32%, is substantially higher compared to most other cancer entities and is accompanied by telomere shortening [7,14,19]. Besides substantial rearrangements of chromosomes, chromotripsis can generate extra-chromosomal double minutes harboring oncogenes. Amplifications in EAC resulting from such double minutes have been reported for oncogenes such as MYC and MDM2 [14]. Chromotripsis in EAC can also lead to the loss of tumor suppressors such as SMAD4 [14]. Interestingly, chromotripsis is associated with TP53 inactivation [14,20]. Thus, the high incidence of chromotripsis in EAC is in accordance with the high recurrence of TP53 mutations or TP53 inhibition by MDM2 amplification. Murugaesu and colleagues found that chromotripsis, such as TP53 mutation, is an early event in EAC [4].
Additional complex genomic events are present in 32% of EACs including kataegis (31%) and focal amplifications (<5 MB: 20%; 5–10 MB: 8%), the breakage-fusion-bridge (BFB) pattern (9%), double minute-like patterns (2%) and subtelomeric BFBs (1%) [7]. BFB results from mitotic failures during anaphase. Sister chromatids with shortened or lost telomeres fuse with one another and are pulled to the opposite poles of the cell by the mitotic spindle, eventually resulting in DNA breakage anywhere between the centromeres. The resulting chromatids again lack telomeres and are prone to repeated BFB cycles in the following cell divisions, which can lead to inverse duplications of genes. In EAC, BFB-driven amplifications were observed for oncogenes such as MDM2, KRAS, RFC3 and VEGFA [14].
Loss of Y Chromosome
Loss of the Y chromosome (LoY) has been observed in various cancer types and even occurs in normal tissue of aging men. However, LoY is particularly frequent in EAC. Fluorescence in situ hybridization analysis of 400 male EACs including lymph node metastases revealed LoY in 52.5% [21]. Intriguingly, LoY was strongly associated with short overall survival, with 19.4 months for LoY and 58.8 months for male EAC patients with the Y chromosome. LoY was an independent prognostic marker but showed a correlation with TP53 mutations, KRAS amplifications, loss of ARID1A and expression of LAG3. It remains unclear whether LoY contributes to the strong sex bias of EAC, with men being seven to nine times more frequently affected by EAC than females.
2.3. Mutational Signatures
In the area of cancer genomics, specific signatures of somatic genome alterations, particularly single-nucleotide variations (SNVs) and indels, have been identified that are described by the type of alteration, e.g., a C-to-T exchange, and the sequence context, i.e., the preceding and following bases [22,23]. Some of these signatures can be explained by specific carcinogens, e.g., tobacco smoke, demonstrating their value for the understanding of tumor development. Six mutational signatures are prominent in EAC tissues: S17A dominated by T > G substitutions in a CTT context known as the hallmark signature for EAC [15,24], a similar signature named S17B with additional T > C substitutions (note historic differences to the latest mutation signature nomenclature at https://cancer.sanger.ac.uk/signatures/, accessed on 2 August 2021), a complex pattern described as being caused by defects in the BRCA1/2 DNA repair pathway (S3), APOBEC-driven hypermutations of C > T in a TCA/TCT context (S2), age-related signature S1 described by C > T mutations in a 5′-CG dinucleotide context (* represents A, C, G, or T) and an S18-like signature with C > A/T substitutions in a GCA/TCT context [7,18]. Interestingly, DNA double-strand breaks described to be associated with mutational signature 3 are late-stage events of the disease [25].
Based on their dominant mutational signature, patients can be clustered into three subgroups called ‘C > A/T dominant’ (driven by S18-like and S1; 31% and 25% in the validation cohort, n = 87), ‘DNA damage repair (DDR)-impaired’ (S3; 15% and 22%) and ‘mutagenic’ (S17A and S17B, 53% and 53%) [7]. The DDR-impaired group has the highest genome instability, while the mutational and neoantigen burden is greatest in the mutagenic subgroup. An increase in defective mutations of genes in the homologous recombination pathway was detected in the DDR-impaired group, whereas the pathway’s key players BRCA1 and BRCA2 themselves are mutated in only 28% of patients within this subgroup. Nevertheless, Nones and colleagues demonstrated that EAC with extreme genomic instability can be driven by somatic BRCA2 mutations accompanied by a prominent BRCA signature [14]. No clinical characteristics including tumor grade, response to therapy and survival are enriched in either of the three subgroups [7]. Instead, these subgroups might be used to stratify patients for specific therapies. Cultured cells with a dominant signature S3 (‘DDR-impaired’) are sensitive to treatment with the PARP inhibitor olaparib, but only when applied together with topoisomerase I inhibitor topotecan. Mitosis checkpoint inhibitors are most effective for cell lines with dominant mutagenic signatures S17A/B. These first results indicate the therapeutic relevance of mutational signatures in EAC, although further studies are needed for validation.
Two APOBEC-driven mutational signatures (S2, S13) are significantly enriched in the longer survival of patients [18]. Further analysis of The Cancer Genome Atlas (TCGA) data showed that higher degrees of the APOBEC signature in patients with elevated APOBEC3B mRNA expression indeed correlate with increased overall survival.
Tumors of patients who received chemotherapy or chemoradiation therapy have similar frequencies of the described mutational signatures compared to treatment-naïve samples. Comparing paired pre- and post-platinum-based chemotherapy tumor samples revealed that the prevalence of mutational signatures largely remains unchanged, except for a decreased proportion of C > T mutations and an increase in C > A mutations in a CpC context [4]. The latter signature is described as a response to platinum treatment, which is in accordance with the patient’s therapy in the described study. Nevertheless, the prominent signatures in EAC generally seem to persist after treatment and are described to be not associated with DNA-damaging chemotherapy or radiation therapy [25].
Instable Microsatellites Are Rare in EAC
Microsatellite instability (MSI) is a mutation signature caused by DNA mismatch repair defects associated with an extremely high load of indels. MSI tumors of a number of cancer entities have a better prognosis which is believed to be due to the larger number of neoantigens allowing the immune system to recognize and eliminate the respective cancer cells. The high level of neoantigens also explains the high response rates to immune checkpoint inhibitor therapies [26,27]. A total of 9 to 21% of gastric cancers and 4% of gastro-esophageal adenocarcinoma are MSI-positive [28,29]. For EAC in general, including more proximal cases (more distant to the gastro-esophageal junction), the frequency of mismatch repair-deficient tumors is only 0.65% [30]. While this is relatively infrequent, it is important to identify this group of patients in the clinic for considering immune checkpoint inhibitor therapy.
2.4. Comparing EAC with Esophageal Squamous Cell Carcinoma and Gastric Cancer
The TCGA research network published a comprehensive analysis of 164 esophageal carcinomas (72 EACs, 90 esophageal squamous cell carcinomas (ESCCs), 2 undifferentiated carcinomas) integrating WES plus shallow WGS, RNA-seq, miRNA sequencing, DNA methylation profiling and proteomics [6]. EAC could be clearly separated from ESCC based on expression profiles alone or together with other omics data layers, emphasizing the importance of considering these cancer types as distinct disease entities driven by cell lineage-specific alterations. Hypermethylation is much more frequent in EAC compared to ESCC. Recurrent mutated genes are rare but distinct for both groups, except for TP53. In addition, somatic copy number alterations are different with EAC-specific recurrent amplifications of VEGFA, ERBB2, GATA6 and CCNE1, and deletions of SMAD4, all of which are absent in ESCC. RTKs and downstream mediators are frequently rearranged in EAC, led by ERBB2 (32%), which is rearranged in only 3% of ESCCs. Dysregulation of the TGF-β pathway, activation of β-catenin and regulation of SWI/SNF complexes is common in EAC, but not in ESCC. Combination of the dataset with data from gastric cancers revealed that EAC is more similar to gastric carcinomas than to ESCC, while ESCC more closely resembles other squamous cell carcinomas [6]. All but one EAC were classified as chromosomal instable (CIN), resembling the CIN subgroup of gastric cancers. Separate analysis of EAC together with CIN gastric cancers showed that hypermethylation compared to normal tissue was strongest in EAC samples and significantly lower in gastric CIN, with decreased methylation towards distal gastric tumors. Hypermethylation in these cancers leads to epigenetic silencing of genes including CDKN2A, MGMT and CHFR. MGMT methylation increases the response to temozolomide treatment [31], and methylation of CHFR sensitizes tumors to dodetaxel and paclitaxel [32], as shown by experimental assays with esophageal cell lines. The authors concluded that EAC and CIN gastric cancers might be considered as a single disease named gastro-esophageal adenocarcinoma.
Next-generation sequencing of 592 target genes in a large cohort of tumors of the upper gastrointestinal tract (1176 EACs, 215 ESCCs, 1951 gastric adenocarcinomas) confirmed that EAC has a more similar molecular profile to gastric adenocarcinoma than to ESCC [33]. Some of the above-described recurrent mutations in EAC were significantly less frequent in ESCC, including APC, ARID1A, CDH1, KRAS, PTEN and SMAD4, while mutations of other genes such as KMT2D, BAP1, CDKN2A and NOTCH1 were found less frequently in EAC. Only few differences were found between EAC and gastric adenocarcinoma, once more supporting the similarity of these adenocarcinomas and demonstrating that EAC and ESCC should be considered as distinct diseases.
2.5. Evolution before and towards Metastasis
As mentioned above, EAC is a highly heterogeneous cancer harboring multiple subclones. Spatial and temporal sampling followed by WES is a powerful tool to determine the genomic evolution of tumors. Somatic SNVs from these analyses are used to generate phylogenetic trees with trunks comprising early events, and branches resembling later stages of tumor progression. In EAC, the vast majority of oncogenic driver mutations appear in the trunks, including TP53 and CDKN2A mutations [4]. Nevertheless, a remarkable amount of putative oncogenic mutations, such as PIK3R1, SMAD4, TLR4 and SLC39A12, appear at branches, are subclonal and thus are later events. It is likely that the recurrence of such mutations is underestimated in reports that are based on the analysis of a single sample per patient. WES of multiple regions within one tumor showed that the majority of non-silent mutations can be detected in only fractions of the regions, with 55% of the mutations being intratumoral heterogeneous [4]. This once again demonstrates the difficulty of molecular diagnostics of EAC based on single small tissue samples, i.e., biopsies [4].
On the other hand, TP53 mutations are clearly early events in EAC. Sequencing samples from different regions of one tumor usually displays clonality of TP53 mutations accompanied by copy neutral loss of heterozygosity (LOH), resulting in the loss of the wild-type allele [4]. TP53 mutations are chronologically followed by increased chromosomal instability (including chromotripsis) and genome doubling, both still early tumor-driving events. Polyploidy is a characteristic feature of EAC cells and recently was discovered to be derived from mitotic slippage due to faulty chromosome attachment to the mitotic spindle [34].
Interestingly, the EAC hallmark mutational signature S17 (T > G in a CTT context) is present at the early stages of tumor progression and decreases during the later stages [4]. S17 arises in cells exposed to gastric acid, and thus it is tempting to speculate that the mutational signature is a consequence of initial gastric reflux, a major risk factor for EAC, but thereafter does not strongly contribute to the progressive intratumoral mutational heterogeneity.
Spatiotemporal sampling of EAC patients with lymph node and distant metastases followed by WGS revealed that multiple subclones of the primary tumor spread to multiple regions in the body [25]. Cells from one subclone can spread to different tissue types, i.e., to the liver and lymph nodes, and one tissue type such as the liver can be infiltrated by cells derived from multiple subclones. Seeding at metastatic sites seems to occur rapidly, demonstrated by relatively few new SNVs with respect to the most recent common ancestor. Most of the recurrent driver SNVs as well as the most frequent CNAs described above appear in the primary tumor at an early time point before spreading [4,25]. Similarly, chromosomal amplifications are early events persisting in the later stages [4]. Nevertheless, after dispersion, genes of the subclones still undergo mutations and rearrangements including driver events such as VEGFA amplification and MAP2K mutation in some patients. Meanwhile, additional genomic alterations in the primary tumor appear, which can be distinct to the events at metastatic sites. The partially disparate genomic composition of metastatic sites and primary tumors emphasizes the problems in finding curative targeted therapies at an advanced stage of the disease. Notably, metastases show a genomic landscape similar to primary EAC with a dominance of copy number variations and large-scale alterations over somatic point mutations. Retrotranspositions of L1 mobile elements are more frequent in metastases, indicating an increased genomic instability. The mode of evolution where the primary tumor progresses to a state comprising multiple different subclones that are able to spread and colonize different or the same metastatic sites seems to be characteristic for EAC as the predominant mechanism and was named ‘clonal diaspora’.
2.6. Genomic Responses to Therapy
Chemotherapies and radiation therapies affect, in particular, highly proliferating cells as these therapies confer stress to the replication process and introduce DNA damage, resulting in increased DNA alterations after erroneous repair. Since EAC has a high rate of relapse after neoadjuvant chemoradiation or perioperative chemotherapy, it is of interest to understand therapy-induced mutations.
WES of 30 paired EACs before and after neoadjuvant chemotherapy with oxaliplatin-5-fluorouracil suggested that the tumors of most good responders pass through a genetic bottleneck resulting in a temporal loss of clonal diversity [3]. Even some poor responders showed evidence for passing through a bottleneck where the tumor regrew before surgery. In 20% of the analyzed tumors, the composition of TP53 mutations changed after treatment, and in some situations, new driver gene mutations post-therapy have been observed.
WGS of ten matched tumors pre- and post-treatment with neoadjuvant chemotherapy combining a platinum-based agent, epirubicin and 5-fluorouracil, as recommended in the United Kingdom at that time, and a second cohort of 62 treatment-naïve and 58 chemotherapy-treated EACs showed no significant differences in SNVs, CNAs, mutational signatures or large-scale genomic alterations after therapy [5]. This is surprising, since these systemic drugs directly affect the DNA integrity and DNA repair mechanisms. The high heterogeneity of EAC with the evolution of distinct subclones within one tumor challenges the comparability of samples before and after treatment. Regions of the genome with LOH before treatment showed clear heterozygosity in samples after treatment, implying that sampling by chance occurred in distinct subclones that might have emerged from a common ancestor clone instead of having evolved from each other.
Similarly, Janjigian and colleagues used a capture-based next-generation sequencing approach to detect somatic mutations, copy number variations and rearrangements of a selection of cancer genes in patients with metastatic stage IV EAC before systemic chemotherapy and correlated the result with response to treatment [35]. Again, samples before and after therapy showed little divergence. No association between defects in homologous recombination-directed repair and survival was observed, and no alterations of known driver genes for homologous recombination repair defects, such as BRCA1/2, correlated with treatment response.
Targeted treatment of HER2 (ERBB2)-positive EAC with trastuzumab in addition to chemotherapy is the standard of care nowadays and results in a prolonged survival of these patients [35]. Overexpression of ERBB2 is routinely discovered by IHC/FISH but can also be robustly detected by next-generation sequencing as it results from amplification of ERBB2 [35]. Alterations of genes involved in the RTK/RAS/PI3K pathway in addition to ERBB2 amplification are predictive for resistance to trastuzumab treatment. This illustrates the opportunity in the genomic characterization of patients to stratify them for specific treatment regimens. Interestingly, sequencing of paired tumors pre- and post-trastuzumab treatment revealed that 16% of patients lose the ERBB2 amplification during disease progression upon treatment, illustrating a selection for an ERBB2-independent subclone as a resistance mechanism of trastuzumab-treated EAC. Furthermore, post-trastuzumab tumors had a deletion of ERBB2 exon 16, resulting in a hyperphosphorylated ERBB2 isoform that is described to be resistant against ERBB2-targeted therapies in cancer models [35,36].
2.7. Genomic Evolution from BE to EAC
In the majority of cases, the development of EAC is triggered by gastro-esophageal reflux disease (GERD), which leads to a replacement of the squamous epithelium by a columnar epithelium [37]. The resulting still benign metaplasia termed Barrett’s esophagus (BE) increases the risk for EAC by 10- to 50-fold [38,39]. However, only 3.5% of individuals progress from BE to EAC during their lifetime [40]. It is therefore desirable to identify biomarkers for the progression of BE to EAC. An accepted model for the development of EAC is the following sequence of events: BE > low-grade dysplasia > high-grade dysplasia > EAC. Despite a benign histology, BE already often contains mutations that have been reported in cancer, e.g., alterations of APC, CDKN2A (p16) and TP53 [41,42]. To understand the determinants of this sequence, several studies have investigated the genomic evolution of BE to EAC.
WGS of BE/EAC pairs of 23 patients displayed surprisingly little overlap between somatic SNVs identified in BE and paired EAC tissue of the same patient, with less than 20% overlap in 57% of the cases [42]. A higher overlap was observed between dysplastic BE and EAC. The study revealed an increase in somatic SNVs and copy number alterations in EAC compared to BE. This suggests that the progressive genomic instability that defines the clonal relatedness between BE and derived EAC is lower compared to the relatedness between dysplasia and EAC. In a similar study, analysis of WES of 25 BE/EAC pairs led to a model of two genomic trajectories of how BE transforms [43]: in trajectory 1, a gradual accumulation of alterations frequently affecting TP53, CDKN2A and SMAD4 leads to dysplasia and genomic instability, where, finally, oncogene amplification results in EAC. In trajectory 2, early TP53 loss is followed by genome doubling which, in turn, leads to genomic instability, oncogene amplification and EAC. Whole genome doubling in cancer is a mechanism that protects cancer cells from the deleterious effects of somatic alterations and allows for extensive LOH. These studies highlight the progressive genomic instability as a key mechanism for the development of EAC.
Martinez and colleagues investigated the genomic copy number profile of individual crypts and entire biopsies of four BE patients who progressed to EAC and of four patients who did not progress [44]. The study indicated that most BE segments (the entire part of the esophagus that is metaplastic) are clonal, with similar numbers and inferred rates of alterations for individual crypts and entire biopsies. Genome doubling and high levels of somatic CNAs were detectable in most individuals who later developed EAC four years before progression, whereas somatic CNA levels remained low in most non-progressors. Multi-region analysis suggested that BE forms from the clonal expansion of a single founder, rather than from polyclonal (trans-) differentiation of multiple lineages. This raises hope that the genomic analysis of biopsies can provide a representative genomic picture of BE, a prerequisite to stratifying patients into high- and low-risk groups, once robust genomic biomarkers can be established.
In an attempt to find such biomarkers, shallow WGS of 777 biopsies sampled from 88 patients in BE surveillance over a period of up to 15 years showed that genomic signals can distinguish progressive from stable disease even 10 years before histopathological transformation [45]. The selection of copy number profiles as a relatively abstract biomarker for progression to EAC reflects the difficulties to establish alterations of single genes for risk stratification. At the same time, genome-wide biomarkers provide an opportunity that should be considered in the clinical context.
The Oesophageal Cancer Clinical and Molecular Stratification Consortium integrated several ‘omics’ layers to define differences and similarities between BE and EAC. The consortium performed DNA methylation analyses of 150 BE and 285 EAC tissues and combined these data with transcriptome and genomic data, resulting in four molecular subtypes [46]. Subtypes 1 and 4 consisted almost exclusively of EAC, subtype 2 of BE and subtype 3 of both groups but enriched for EAC. Subtype 1 was characterized by DNA methylation, a high mutation burden and mutations in the cell cycle and RTK signaling pathway genes. Subtype 2 showed gene expression patterns associated with metabolic processes. Subtype 3 displayed no methylation changes compared to normal tissue but also immune cell infiltration, and subtype 4 was characterized by DNA hypomethylation associated with genome rearrangements and amplification of CCNE1. EAC cases of subtype 2 and subtype 3 had the highest and lowest survival probabilities, respectively, indicating that the molecular signature is of prognostic value.
The largest next-generation sequencing-based genome studies are listed in Table 1, and the main molecular characteristics of EAC are summarized in Figure 1.

Table 1.
Main genome next-generation sequencing studies of EAC.
Figure 1.
Schematic representation of characteristic molecular features of EAC. Top, sequence of histological states towards EAC. Bottom, list of molecular categories and key genes that are typically altered. GERD, gastro-esophageal reflux disease; ITH, intratumoral heterogeneity; RTK, receptor tyrosine kinase; SCNA, somatic genome copy number alteration; SNV: single-nucleotide variant; WGD, whole genome doubling.
3. Transcriptomics of EAC
The stratification of EACs into molecular subtypes by gene expression profiling methods provides new opportunities for understanding the molecular characteristics of EACs and developing possible therapeutic strategies. By analyzing gene expression profiling data of three independent EAC cohorts, two expression patterns could be defined [47]. Genes that were overexpressed in subtype 1 (basal subtype) were enriched in biological processes including epithelial cell differentiation, keratinocyte differentiation and the KEGG pathway basal cell carcinoma. Subtype 2 (classical subtype) showed a more similar expression pattern to BE. Correlating the subtypes with therapy response suggested subtype 1 to be more chemotherapy-resistant.
Integrating genomic and transcriptomic data of advanced EAC for risk stratification in the clinical context of 20 short vs. 20 long survivors, Hao and colleagues discovered novel molecular features for prognosticating overall survival [18]. The genomic analysis revealed alterations of the epigenetic modifier KMT2C exclusively in the short survivors together with a higher level of intratumor heterogeneity, whereas the APOBEC mutation signature was enriched in longer survivors. By clustering RNA sequencing data of 33 specimens of these patients, the authors identified three clusters, with cluster 1 mainly composed of tumors from long survivors and cluster 3 with tumors from short survivors. Tumors of cluster 1 showed a significantly increased expression of multiple immune-related markers such as MPO, FCN1, CD200 and LEF1. Cluster 3 showed high expression of tumor promoters MAP3K13, MECOM and JAK2, predicting worse survival. MAP3K13 upregulation has been reported to correlate with a poor outcome in tumor progression [48,49].
Highly significant expression changes in 17 known cancer genes such as ERBB2, KRAS and SMAD4 were observed by analyzing the RNA sequencing data of 116 EACs, showing a correlation with a high degree of chromosomal instability [13]. The genomic landscape of driver events comprises mutations and CNAs in oncogenes and tumor suppressor genes. Copy number loss was not necessarily associated with a reduced expression of the tumor suppressor genes ARID1A and CDH11 but instead was associated with loss of heterozygosity. The expression levels of CDKN2A compared to normal tissue suggest that CDKN2A is generally activated in EAC and returns to normal levels when deleted. Some genes showed overexpression or downregulation without genomic aberrations, for example, overexpression of MYC. GATA4, GATA6 and MUC6, being involved in the differentiated phenotype of gastrointestinal tissue, were downregulated and may be lost during dedifferentiation observed in cancer [13].
3.1. RNA Sequencing of the Tumor Microenvironment in EAC
Li and colleagues focused, in their study, on characterizing the stroma microenvironment in a mixed cohort of EAC and ESCC, as an activated stroma and the extracellular matrix play a significant role in tumor initiation, progression and metastasis [50]. In their study based on previously published genomic and transcriptomic data with a training (n = 182) and a validation cohort (n = 227), the authors identified genes that were correlated with stromal elements. Based on their estimation of stromal activation, the authors could divide their cohorts into two subgroups, with subgroup 2 consisting of patients with high stromal activity, associated with a high tumor stage and increased stromal cell infiltration. Subgroup 2 showed worse survival. The identification of the stromal marker genes MMP11, COL6A2, COL1A2, CTHRC1, FAP and LUM that are involved in cancer-associated fibroblast (CAF) function highlighted the crucial role of CAFs in the tumor stroma of subgroup 2.
Changes in the immune profile during BE-to-EAC progression have been identified by RNA-sequencing of 65 BE/dysplasia/EAC samples [51]. A subset of chemokines and cytokines, in particular, IL6 and CXCL8, increased during BE progression to EAC. While high-grade dysplasia showed increased immune cell populations, EAC was immune-poor, with a rise in the immune checkpoint protein PD-L1 and a loss of CD8+ T cells providing a supporting rationale for recent efforts to investigate immune checkpoint inhibitor therapies for EAC.
3.2. Single-Cell RNA Sequencing of EAC
Single-cell transcriptomics is a major advancement in the field of RNA sequencing technologies. Conventional approaches such as bulk RNA sequencing lack the resolution to deconvolute the contributions of subpopulations that may be identified as cell types or states [52,53,54]. Using this technology, studying the tumor microenvironment (TME) on a single cell scale has been a focus of current research for many cancer entities, such as melanoma [55], breast cancer [56], lung cancer [57] and colorectal carcinoma [58]. The TME has been implied to play various roles in cancer progression, such as promoting metastasis, facilitating immune evasion, fostering angiogenesis and conveying therapeutic resistance [59]. By characterization of the different cell types and states within the TME, it is hoped to find new therapeutic approaches to halt cancer progression [59]. Several components of the TME of EAC have been found to correlate with patient outcomes. For example, CAFs have been reported to promote invasion by secreting periostin that activates the PI3K/Akt pathway by interaction with tumor cell integrins [60]. On the other hand, the infiltration of lymphocytes was found to be associated with improved cancer-specific survival as seen in other solid cancers [61].
Thus far, single-cell sequencing studies of the TME of EAC are pending. However, Owen and colleagues could demonstrate the accessibility of esophageal tissue to single-cell RNA sequencing [62]. It was possible to discriminate cell type differences in BE compared to normal tissue samples of the esophagus, stomach and duodenum, providing evidence of the similarity of BE and the esophageal submucosal glands. Furthermore, differentiated expression profiles of different cell types abundant in BE could be identified. This is of particular interest because of the association of BE with the genesis of EAC, as discussed above [62].
There is still a way to go in using single-cell RNA sequencing in patient diagnostics, since costs remain high and interpretation of results is time-consuming. A way to efficiently enhance the resolution of bulk RNA sequencing is ‘FACS-seq’. As we demonstrated previously, using a fluorescence-activated cell sorting (FACS) step before performing bulk RNA sequencing, it is possible to single out fibroblasts, leukocytes and epithelial or tumor cells. Thus, features of EAC and its tumor microenvironment become accessible to analysis in patient diagnostics, eventually extending patient stratification for personalized therapy options [63]. Major transcriptomic findings are summarized in Table 2.
Table 2.
Major transcriptomic findings in next-generation sequencing studies of EAC.
4. Clinical Relevance of Molecular Characteristics
As illustrated above, EAC is a genetically complex disease with a high mutational burden and extensive chromosomal instability [13,17,64]. Remarkably, in EAC, not only alterations of classical driver genes such as TP53, CDKN2A, SMARCA4, ARID1A, SMAD4, ERBB2 or PIK3CA but also those of helper genes such as ABI2 and NCOR2, which are mutated only at a low frequency in single patients, contribute to tumor development [17,65,66].
In the context of such a heterogeneous disease, the successful transfer of genetic characteristics into clinical care is difficult and the prognosis of EAC remains devastating, resulting in an overall 5-year survival of about 20% [1,67]. Despite the development of multimodal treatment regimens based on a combination of either neoadjuvant chemoradiation [68] or perioperative chemotherapy [69] plus radical surgical resection, about one third of all patients show only little therapeutic response, with the majority of tumor cells still viable after neoadjuvant treatment [70]. Biomarkers are needed to identify patients at the time of diagnosis who are unlikely to benefit from therapy in order to spare them side effects. Interestingly, genome-wide sequencing approaches could demonstrate that almost half of the patients with EAC harbor somatic mutations in frequently alternated cancer pathways that are biomarkers or putative targets for therapy [15]. Nevertheless, only a few personalized therapeutic options based on specific molecular characteristics have made their way into the clinical routine.
Nowadays, patients with ERBB2-positive metastasized gastric or esophagogastric adenocarcinoma qualify for a combination of chemotherapy and the monoclonal cytotoxic ERBB2 antibody trastuzumab, improving the patients’ survival compared to chemotherapeutic treatment alone [71]. Recent studies could demonstrate an even better outcome in such cohorts by adding immunotherapeutic treatment via PD-1 inhibition (monoclonal antibody: pembrolizumab) to this regime of ERBB2 blockade and chemotherapy [72,73]. However, most data were derived from mixed cohorts of both entities, adenocarcinoma of a gastric or an esophagogastric junction origin. Of note, in large EAC-restricted cohorts, ERBB2 positivity was associated with a survival advantage [74,75].
Other targeted therapies are still the subject of studies with ambivalent results. As 30–60% of metastasized patients with esophageal cancer have overexpression of VEGFA [76], the monoclonal antibody bevacizumab was utilized in combination with chemotherapy for first-line therapy in patients with adenocarcinoma of the esophagogastric junction (an entity overlapping with EAC) within the international AVAGAST study. Although the survival benefit was not significant, it demonstrated different ethnic therapy responses. While Asian patients did not respond at all, Caucasian populations showed marginally improved survival under treatment [10]. The combined VEGF2/ERBB2 inhibitor ramucirumab has now been approved for second-line treatment of EAC, either as monotherapy or combined with chemotherapy such as Abraxane based on studies such as REGARD, RAINBOW or RAINFALL [77,78,79]. Inhibition of EGFR via monoclonal antibodies such as cetuximab or panitumumab in patients with esophageal cancer did not lead to improved survival in different analyses [9,11,12]. This approach has therefore not been considered as effective EAC therapy thus far. Similarly, MET inhibition via inhibition of its downstream target HGF with the antibody rilotumumab has not yet been successful in clinical studies [8]. Table 3 summarizes the current targeted approaches for EAC within the clinical routine.
Table 3.
Current approaches for targeted therapy in EAC (and other adenocarcinomas of the upper gastrointestinal tract) based on molecular characteristics.
Besides these current clinical limitations, knowledge of molecular characteristics provides an opportunity for future personalized treatments. Different mutational signatures of the tumors might result in deviating therapeutic regimens as Secrier and colleagues proposed [7]. Patients with dominant C > A/T mutational patterns associated with aging processes might benefit from conventional chemotherapy (in combination with ERBB2 inhibition) since the tumor’s genome is more stable. Patients with prevalent defects within those genes responsible for homologous recombination-mediated DNA repair might receive radiation combined with PARP inhibition as these tumors are vulnerable to DNA-damaging treatments and cannot repair the therapy-induced DNA damage sufficiently. Furthermore, the authors proposed another subgroup of patients with a dominant T > G mutational signature resulting in a high mutational burden and the presence of a high load of neoantigens. In these cases, immunotherapies such as checkpoint inhibition targeting CTLA4 and PD-1\PD-L1 might be promising [7].
Another central aspect of clinical interest is the estimation of putative tumor growth/shrinking under therapy. Besides classical histopathological features, e.g., lymphatic metastasis, locally advanced growth or tumor cell death under neoadjuvant therapy, molecular specifications detected in endoscopic biopsies and surgical specimens might help to improve risk stratification. Recent studies of combined strategies including targeted sequencing, screening for promoter methylation and WES in EAC patients revealed different response profiles associated with deviating histopathological features [4,80]. Increased copy number variations, mutations and amplifications of CSMD1 or ETV4 as well as CpG island promoter methylation correlated with a favorable histopathological response [81], while alterations in SMARC4 or SMURF1 were associated with a worse tumor response [81]. Interestingly, early during the evolution of EAC, genomic instability occurs and is relatively stable and preserved throughout the course under therapeutic pressure such as chemotherapy. Therefore, the resulting gene amplification may represent suitable candidates for targeted treatments [4]. However, such analyses and classifications have not made their way into the daily routine.
The upcoming clinical relevance of molecular tumor characteristics and usage of innovative genomic technologies is not only limited to therapeutic decisions and evaluation of treatment response but might also play an important role in early detection as well as surveillance of EAC and dysplastic BE as precursor lesions. Thus far, diagnostics rely on clinical parameters with esophagogastroduodenoscopy as the gold standard for detection since no other markers are available. Utilizing novel sequencing techniques provides an opportunity for primary diagnosis as well as surveillance during follow-up. The cytosponge technique, also known as ‘pill-on-a-string’, is a non-invasive method for the detection of abnormal epithelial cells in the esophagus. In 2014, Weaver and colleagues demonstrated that WGS and amplicon-based resequencing of 112 EACs, as well as 109 samples of patients with either non-dysplastic BE or high-grade dysplasia of the esophageal mucosa derived from cytosponge sampling, were able to identify significant transition points in the sequential development of the malignancy [24]. Somatic mutations within TP53 only occurred in dysplastic BE and EAC, while SMAD4 mutations indicated the transformation from dysplastic mucosa to early EAC. Therefore, this non-endoscopic approach might offer novel screening options for patients with dysplastic alterations at risk of early tumorigenesis. Recent analyses using the cytosponge technique strengthen these data, emphasizing the fact that the sampling bias of conventional endoscopic biopsies due to intratumoral heterogeneity and different tumor clones becomes obsolete [42,82]. Nevertheless, targeted sequencing of samples taken during routine endoscopic biopsy surveillance in BE patients was also able to identify progression to EAC based on the presence of TP53 mutations. Progressors from dysplastic BE to EAC showed increased TP53 mutations compared to ‘stable’ BE patients [83].
Finally, the transfer of knowledge gained from molecular characteristics as well as genomic profiling of EAC into clinical care is still unsatisfactory. However, more and more molecular data have become available, bearing a high potential for improved patient stratification. Therefore, the clinical importance of molecular characteristics in EAC will certainly continue to grow in the near future.
5. Conclusions
EAC is still a cancer with a poor outcome due to the very limited options for targeted therapies inhibiting RTKs and biomarker-based rationales for treatment concepts such as inhibitors of immune checkpoints, PARP and vascularization. It is likely that the lack of inhibitors of oncogenic drivers other than ERBB2 is due to the complex genomic architecture of EAC, with multiple putative driver alterations per tumor. The problem is illustrated by a model for smoking-related lung adenocarcinoma, where the smoking-induced high mutation load results in a high number of usually weak driver mutations in contrast to non-smokers with a low mutation load, who only develop lung cancer in the unlikely situation when one of the few mutations hits a strong driver such as EGFR [84]. It is plausible that a tumor that is driven by one strong driver against which a targeted drug is available responds better compared to a tumor that is driven by many (weak) drivers where inhibition of one of them only marginally reduces the overall pro-proliferative signaling. Therefore, a focus of current translational research on EAC may be on the identification of biomarkers that help to predict treatment response to therapies other than inhibitors of RTKs and related pro-proliferative signaling. Profiles such as the degree of genome instability, mutation load, stroma features and the immune repertoire, but also the germline genetic risk profile [85], should be systematically assessed and correlated with histopathological and clinical characteristics, particularly treatment response, to stratify EAC patients for more tailored therapies. Single-cell sequencing approaches are promising concepts to understand EAC biology and to identify cell type-specific biomarkers. Integrating the various data layers using machine learning provides an opportunity to define clinically relevant patient groups that cannot be easily described by one biomarker. Overall, we have gained an understanding of the EAC genomics over recent years and require integrative molecular concepts to translate molecular and clinical information into patient benefits.
Author Contributions
S.H., C.J., M.C.W., O.V.C., C.A., P.S.P. and A.M.H. wrote the original draft manuscript and prepared the figures; Y.Z., R.B. and A.Q. reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the German Research Foundation ‘Esophageal adenocarcinoma: understanding the molecular basis of differential treatment response’ (418074181) and ‘Predictability in evolution-Predicting patterns of adaptation to radio-chemotherapy in cancer’ (CRC 1310, SP8), the Wilhelm Sander Foundation ‘Räumlich-transkriptomische und funktionelle Analyse der Interaktion von Tumorzellen und cancer associated fibroblasts (CAFs) bei Adenokarzinomen des Ösophagus’ (2020.119.1), the Federal Ministry of Education and Research (BMBF) ‘Deep Insight-Integrating germline and somatic genetic profiles through machine learning to understand esophageal cancer etiology’ (031L0267B) and the German Cancer Aid ‘National Network Genomic Medicine Lung Cancer’ (70114428).
Acknowledgments
Figure 1 and graphical abstract were created using Inkscape 1.1 software (Inkscape Project, 2021; Inkscape, available at: https://inkscape.org, accessed on 25 August 2021). The authors acknowledge the free provision of two icons by Servier Medical Art which were used for the graphical abstract [86].
Conflicts of Interest
A.M.H. receives research funding from Dracen Pharmaceuticals Inc. Other authors declare no conflict of interest.
References
- Coleman, H.G.; Xie, S.H.; Lagergren, J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.J.; Li, X.; Galipeau, P.C.; Vaughan, T.L. Barrett’s Oesophagus and Oesophageal Adenocarcinoma: Time for a new synthesis. Nat. Rev. Cancer 2010, 10, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Findlay, J.M.; Castro-Giner, F.; Makino, S.; Rayner, E.; Kartsonaki, C.; Cross, W.; Kovac, M.; Ulahannan, D.; Palles, C.; Gillies, R.S.; et al. Differential Clonal Evolution in Oesophageal Cancers in Response to Neo-adjuvant Chemotherapy. Nat. Commun. 2016, 7, 11111. [Google Scholar] [CrossRef] [PubMed]
- Murugaesu, N.; Wilson, G.A.; Birkbak, N.J.; Watkins, T.; McGranahan, N.; Kumar, S.; Abbassi-Ghadi, N.; Salm, M.; Mitter, R.; Horswell, S.; et al. Tracking the Genomic Evolution of Esophageal Adenocarcinoma through Neoadjuvant Chemotherapy. Cancer Discov. 2015, 5, 821–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noorani, A.; Bornschein, J.; Lynch, A.G.; Secrier, M.; Achilleos, A.; Eldridge, M.; Bower, L.; Weaver, J.M.J.; Crawte, J.; Ong, C.A.; et al. A Comparative Analysis of Whole Genome Sequencing of Esophageal Adenocarcinoma Pre- and Post-chemotherapy. Genome Res 2017, 27, 902–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Oesophageal Carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational Signatures in Esophageal Adenocarcinoma Define Etiologically Distinct Subgroups with Therapeutic Relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef] [Green Version]
- Catenacci, D.V.T.; Tebbutt, N.C.; Davidenko, I.; Murad, A.M.; Al-Batran, S.E.; Ilson, D.H.; Tjulandin, S.; Gotovkin, E.; Karaszewska, B.; Bondarenko, I.; et al. Rilotumumab Plus Epirubicin, Cisplatin, and Capecitabine as First-line Therapy in Advanced MET-positive Gastric or Gastro-oesophageal junction cancer (RILOMET-1): A Randomised, Double-blind, Placebo-controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1467–1482. [Google Scholar] [CrossRef]
- Huang, Z.H.; Ma, X.W.; Zhang, J.; Li, X.; Lai, N.L.; Zhang, S.X. Cetuximab for Esophageal Cancer: An Updated Meta-analysis of Randomized Controlled Trials. BMC. Cancer 2018, 18, 1170. [Google Scholar] [CrossRef]
- Ohtsu, A.; Shah, M.A.; Van Cutsem, E.; Rha, S.Y.; Sawaki, A.; Park, S.R.; Lim, H.Y.; Yamada, Y.; Wu, J.; Langer, B.; et al. Bevacizumab in Combination with Chemotherapy as First-line Therapy in Advanced Gastric Cancer: A randomized, double-blind, Placebo-controlled Phase III Study. J. Clin. Oncol. 2011, 29, 3968–3976. [Google Scholar] [CrossRef]
- Tew, W.P.; Kelsen, D.P.; Ilson, D.H. Targeted Therapies for Esophageal Cancer. Oncologist 2005, 10, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Waddell, T.; Chau, I.; Cunningham, D.; Gonzalez, D.; Okines, A.F.; Okines, C.; Wotherspoon, A.; Saffery, C.; Middleton, G.; Wadsley, J.; et al. Epirubicin, Oxaliplatin, and Capecitabine with or without panitumumab for Patients with Previously Untreated Advanced Oesophagogastric Cancer (REAL3): A Randomised, Open-label Phase 3 Trial. Lancet Oncol. 2013, 14, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Frankell, A.M.; Jammula, S.; Li, X.; Contino, G.; Killcoyne, S.; Abbas, S.; Perner, J.; Bower, L.; Devonshire, G.; Ococks, E.; et al. The landscape of Selection in 551 Esophageal Adenocarcinomas Defines Genomic Biomarkers for the Clinic. Nat. Genet. 2019, 51, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Nones, K.; Waddell, N.; Wayte, N.; Patch, A.M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic Catastrophes Frequently Arise in Esophageal Adenocarcinoma and Drive Tumorigenesis. Nat. Commun. 2014, 5, 5224. [Google Scholar] [CrossRef] [Green Version]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and Whole-genome sequencing of Esophageal Adenocarcinoma Identifies Recurrent Driver Events and Mutational Complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef]
- Rheinbay, E.; Nielsen, M.M.; Abascal, F.; Wala, J.A.; Shapira, O.; Tiao, G.; Hornshoj, H.; Hess, J.M.; Juul, R.I.; Lin, Z.; et al. Analyses of Non-coding Somatic Drivers in 2658 Cancer Whole Genomes. Nature 2020, 578, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Mourikis, T.P.; Benedetti, L.; Foxall, E.; Temelkovski, D.; Nulsen, J.; Perner, J.; Cereda, M.; Lagergren, J.; Howell, M.; Yau, C.; et al. Patient-specific Cancer Genes Contribute to Recurrently Perturbed Pathways and Establish Therapeutic Vulnerabilities in Esophageal Adenocarcinoma. Nat. Commun. 2019, 10, 3101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, D.; He, S.; Harada, K.; Pizzi, M.P.; Lu, Y.; Guan, P.; Chen, L.; Wang, R.; Zhang, S.; Sewastjanow-Silva, M.; et al. Integrated Genomic Profiling and Modelling for Risk Stratification in Patients with Advanced Oesophagogastric Adenocarcinoma. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer Patterns of Somatic Copy Number Alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stutz, A.M.; Zichner, T.; Weischenfeldt, J.; Jager, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome Sequencing of Pediatric Medulloblastoma Links Catastrophic DNA Rearrangements with TP53 Mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeser, H.; Wolwer, C.B.; Alakus, H.; Chon, S.H.; Zander, T.; Buettner, R.; Hillmer, A.M.; Bruns, C.J.; Schroeder, W.; Gebauer, F.; et al. Y Chromosome Loss is a Frequent Event in Barrett’s Adenocarcinoma and Associated with Poor Outcome. Cancers 2020, 12, 1743. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, J.M.J.; Ross-Innes, C.S.; Shannon, N.; Lynch, A.G.; Forshew, T.; Barbera, M.; Murtaza, M.; Ong, C.J.; Lao-Sirieix, P.; Dunning, M.J.; et al. Ordering of Mutations in Preinvasive Disease Stages of Esophageal Carcinogenesis. Nat. Genet. 2014, 46, 837–843. [Google Scholar] [CrossRef]
- Noorani, A.; Li, X.; Goddard, M.; Crawte, J.; Alexandrov, L.B.; Secrier, M.; Eldridge, M.D.; Bower, L.; Weaver, J.; Lao-Sirieix, P.; et al. Genomic Evidence Supports a Clonal Diaspora Model for Metastases of Esophageal Adenocarcinoma. Nat. Genet. 2020, 52, 74–83. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive Molecular Characterization of Clinical Responses to PD-1 Inhibition in Metastatic Gastric Cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- Polom, K.; Marano, L.; Marrelli, D.; De Luca, R.; Roviello, G.; Savelli, V.; Tan, P.; Roviello, F. Meta-Analysis of Microsatellite Instability in Relation to Clinicopathological Characteristics and Overall Survival in Gastric Cancer. Br. J. Surg. 2018, 105, 159–167. [Google Scholar] [CrossRef]
- Krzykala, B.; Turut, P.; Darrason, G.; Madelain, J.; Dumont, P. Epidemiology of Choroidal Folds. Bull. Soc. Ophtalmol. Fr. 1989, 89, 295–296, 299–300. [Google Scholar] [PubMed]
- Hewitt, L.C.; Inam, I.Z.; Saito, Y.; Yoshikawa, T.; Quaas, A.; Hoelscher, A.; Bollschweiler, E.; Fazzi, G.E.; Melotte, V.; Langley, R.E.; et al. Epstein-Barr virus and Mismatch Repair Deficiency Status Differ between Oesophageal and Gastric Cancer: A Large Multi-centre Study. Eur. J. Cancer 2018, 94, 104–114. [Google Scholar] [CrossRef]
- Hasina, R.; Surati, M.; Kawada, I.; Arif, Q.; Carey, G.B.; Kanteti, R.; Husain, A.N.; Ferguson, M.K.; Vokes, E.E.; Villaflor, V.M.; et al. O-6-methylguanine-deoxyribonucleic Acid Methyltransferase Methylation Enhances Response to Temozolomide Treatment in Esophageal Cancer. J. Carcinog. 2013, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.; Liu, Y.; Gao, D.; Linghu, E.; Brock, M.V.; Yin, D.; Zhan, Q.; Herman, J.G.; Guo, M. Methylation of CHFR Sensitizes Esophageal Squamous Cell Cancer to Docetaxel and Paclitaxel. Genes Cancer 2015, 6, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; Puccini, A.; Xiu, J.; Raghavan, D.; Lenz, H.J.; Korn, W.M.; Shields, A.F.; Philip, P.A.; Marshall, J.L.; Goldberg, R.M. Comparative Molecular Analyses of Esophageal Squamous Cell Carcinoma, Esophageal Adenocarcinoma, and Gastric Adenocarcinoma. Oncologist 2018, 23, 1319–1327. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.J.; Li, X.; Jammula, S.; Devonshire, G.; Lindon, C.; Fitzgerald, R.C.; D’Avino, P.P. Evidence that Polyploidy in Esophageal Adenocarcinoma Originates from Mitotic Slippage Caused by Defective Chromosome Attachments. Cell Death Differ. 2021, 28, 2179–2193. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Sanchez-Vega, F.; Jonsson, P.; Chatila, W.K.; Hechtman, J.F.; Ku, G.Y.; Riches, J.C.; Tuvy, Y.; Kundra, R.; Bouvier, N.; et al. Genetic Predictors of Response to Systemic Therapy in Esophagogastric Cancer. Cancer Discov. 2018, 8, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Castiglioni, F.; Tagliabue, E.; Campiglio, M.; Pupa, S.M.; Balsari, A.; Menard, S. Role of Exon-16-Deleted HER2 in Breast Carcinomas. Endocr. Relat. Cancer 2006, 13, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, Y.; Al-Kaabi, A.; Shaheen, N.J.; Chak, A.; Blum, A.; Souza, R.F.; Di Pietro, M.; Iyer, P.G.; Pech, O.; Fitzgerald, R.C.; et al. Barrett Oesophagus. Nat. Rev. Dis. Primers. 2019, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Zeki, S.; Fitzgerald, R.C. The Use of Molecular Markers in Predicting Dysplasia and Guiding Treatment. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, N.; Ransohoff, D.F. Gastroesophageal reflux, barrett esophagus, and esophageal cancer: Scientific review. JAMA 2002, 287, 1972–1981. [Google Scholar] [CrossRef]
- Jankowski, J.; Barr, H.; Wang, K.; Delaney, B. Diagnosis and Management of Barrett’s Oesophagus. BMJ 2010, 341, c4551. [Google Scholar] [CrossRef] [Green Version]
- Leedham, S.J.; Preston, S.L.; McDonald, S.A.; Elia, G.; Bhandari, P.; Poller, D.; Harrison, R.; Novelli, M.R.; Jankowski, J.A.; Wright, N.A. Individual Crypt Genetic Heterogeneity and the Origin of Metaplastic Glandular Epithelium in Human Barrett’s Oesophagus. Gut 2008, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross-Innes, C.S.; Becq, J.; Warren, A.; Cheetham, R.K.; Northen, H.; O’Donovan, M.; Malhotra, S.; di Pietro, M.; Ivakhno, S.; He, M.; et al. Whole-genome Sequencing Provides New Insights into the Clonal Architecture of Barrett’s Esophagus and Esophageal Adenocarcinoma. Nat. Genet. 2015, 47, 1038–1046. [Google Scholar] [CrossRef]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired Exome Analysis of Barrett’s Esophagus and Adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef] [Green Version]
- Martinez, P.; Mallo, D.; Paulson, T.G.; Li, X.; Sanchez, C.A.; Reid, B.J.; Graham, T.A.; Kuhner, M.K.; Maley, C.C. Evolution of Barrett’s Esophagus through Space and Time at Single-crypt and Whole-biopsy Levels. Nat. Commun. 2018, 9, 794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killcoyne, S.; Gregson, E.; Wedge, D.C.; Woodcock, D.J.; Eldridge, M.D.; de la Rue, R.; Miremadi, A.; Abbas, S.; Blasko, A.; Kosmidou, C.; et al. Genomic Copy Number Predicts Esophageal Cancer Years before Transformation. Nat. Med. 2020, 26, 1726–1732. [Google Scholar] [CrossRef]
- Jammula, S.; Katz-Summercorn, A.C.; Li, X.; Linossi, C.; Smyth, E.; Killcoyne, S.; Biasci, D.; Subash, V.V.; Abbas, S.; Blasko, A.; et al. Identification of Subtypes of Barrett’s Esophagus and Esophageal Adenocarcinoma Based on DNA Methylation Profiles and Integration of Transcriptome and Genome Data. Gastroenterology 2020, 158, 1682–1697. [Google Scholar] [CrossRef]
- Guo, X.; Tang, Y.; Zhu, W. Distinct Esophageal Adenocarcinoma Molecular Subtype has Subtype-Specific Gene Expression and Mutation Patterns. BMC. Genom. 2018, 19, 769. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Li, X.; Cui, K.; Liu, C.; Wu, M.; Prochownik, E.V.; Li, Y. The MAP3K13-TRIM25-FBXW7alpha Axis Affects c-Myc Protein Stability and Tumor Development. Cell Death Differ. 2020, 27, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Edwards, Z.C.; Trotter, E.W.; Torres-Ayuso, P.; Chapman, P.; Wood, H.M.; Nyswaner, K.; Brognard, J. Survival of Head and Neck Cancer Cells Relies upon LZK Kinase-Mediated Stabilization of Mutant p53. Cancer Res. 2017, 77, 4961–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zeng, Z.; Jiang, X.; Zhang, N.; Gao, Y.; Luo, Y.; Sun, W.; Li, S.; Ren, J.; Gong, Y.; et al. Stromal Microenvironment Promoted Infiltration in Esophageal Adenocarcinoma and Squamous Cell Carcinoma: A multi-cohort gene-based analysis. Sci. Rep. 2020, 10, 18589. [Google Scholar] [CrossRef] [PubMed]
- Lagisetty, K.H.; McEwen, D.P.; Nancarrow, D.J.; Schiebel, J.G.; Ferrer-Torres, D.; Ray, D.; Frankel, T.L.; Lin, J.; Chang, A.C.; Kresty, L.A.; et al. Immune Determinants of Barrett’s Progression to Esophageal Adenocarcinoma. JCI. Insight 2021, 6, e143888. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The Technology and Biology of Single-cell RNA Sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amezquita, R.A.; Lun, A.T.L.; Becht, E.; Carey, V.J.; Carpp, L.N.; Geistlinger, L.; Marini, F.; Rue-Albrecht, K.; Risso, D.; Soneson, C.; et al. Orchestrating Single-cell Analysis with Bioconductor. Nat. Methods 2020, 17, 137–145. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Z.; Roden, D.L.; Wang, C.; Holliday, H.; Harvey, K.; Cazet, A.S.; Murphy, K.J.; Pereira, B.; Al-Eryani, G.; Bartonicek, N.; et al. Stromal Cell Diversity Associated with Immune Evasion in Human Triple-negative Breast Cancer. EMBO J. 2020, 39, e104063. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.I.; Suh, Y.L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA Sequencing Demonstrates the Molecular and Cellular Reprogramming of Metastatic Lung Adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference Component Analysis of Single-cell Transcriptomes Elucidates Cellular Heterogeneity in Human Colorectal Tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Hanahan, D. and Coussens, L.M.; Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.J.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated Fibroblasts Predict Poor Outcome and Promote Periostin-dependent Invasion in Oesophageal Adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef]
- Noble, F.; Mellows, T.; McCormick Matthews, L.H.; Bateman, A.C.; Harris, S.; Underwood, T.J.; Byrne, J.P.; Bailey, I.S.; Sharland, D.M.; Kelly, J.J.; et al. Tumour Infiltrating Lymphocytes Correlate with Improved Survival in Patients with Oesophageal Adenocarcinoma. Cancer Immunol. Immunother. 2016, 65, 651–662. [Google Scholar] [CrossRef]
- Owen, R.P.; White, M.J.; Severson, D.T.; Braden, B.; Bailey, A.; Goldin, R.; Wang, L.M.; Ruiz-Puig, C.; Maynard, N.D.; Green, A.; et al. Single Cell RNA-seq Reveals Profound Transcriptional Similarity between Barrett’s Oesophagus and Oesophageal Submucosal Glands. Nat. Commun. 2018, 9, 4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, M.; Plum, P.S.; Velazquez Camacho, O.; Folz-Donahue, K.; Thelen, M.; Garcia-Marquez, I.; Wolwer, C.; Busker, S.; Wittig, J.; Franitza, M.; et al. Cell Type-specific Transcriptomics of Esophageal Adenocarcinoma as a Scalable Alternative for Single Cell Transcriptomics. Mol. Oncol. 2020, 14, 1170–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Loga, K.; Woolston, A.; Punta, M.; Barber, L.J.; Griffiths, B.; Semiannikova, M.; Spain, G.; Challoner, B.; Fenwick, K.; Simon, R.; et al. Extreme Intratumour Heterogeneity and Driver Evolution in Mismatch Repair Deficient Gastro-oesophageal Cancer. Nat. Commun. 2020, 11, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repana, D.; Nulsen, J.; Dressler, L.; Bortolomeazzi, M.; Venkata, S.K.; Tourna, A.; Yakovleva, A.; Palmieri, T.; Ciccarelli, F.D. The Network of Cancer Genes (NCG): A Comprehensive Catalogue of Known and Candidate Cancer Genes from Cancer Sequencing Screens. Genome Biol. 2019, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Contino, G.; Vaughan, T.L.; Whiteman, D.; Fitzgerald, R.C. The Evolving Genomic Landscape of Barrett’s Esophagus and Esophageal Adenocarcinoma. Gastroenterology 2017, 153, 657–673 e1. [Google Scholar] [CrossRef] [PubMed]
- Rustgi, A.K.; El-Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef]
- van Hagen, P.; Hulshof, M.C.; van Lanschot, J.J.; Steyerberg, E.W.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.; Richel, D.J.; Nieuwenhuijzen, G.A.; Hospers, G.A.; Bonenkamp, J.J.; et al. Preoperative Chemoradiotherapy for Esophageal or Junctional Cancer. N. Engl. J. Med. 2012, 366, 2074–2084. [Google Scholar] [CrossRef] [Green Version]
- Al-Batran, S.E.; Hartmann, J.T.; Hofheinz, R.; Homann, N.; Rethwisch, V.; Probst, S.; Stoehlmacher, J.; Clemens, M.R.; Mahlberg, R.; Fritz, M.; et al. Biweekly Fluorouracil, Leucovorin, Oxaliplatin, and Docetaxel (FLOT) for Patients with Metastatic Adenocarcinoma of the Stomach or Esophagogastric Junction: A phase II trial of the Arbeitsgemeinschaft Internistische Onkologie. Ann. Oncol. 2008, 19, 1882–1887. [Google Scholar] [CrossRef]
- den Bakker, C.M.; Smit, J.K.; Bruynzeel, A.M.E.; van Grieken, N.C.T.; Daams, F.; Derks, S.; Cuesta, M.A.; Plukker, J.T.M.; van der Peet, D.L. Non Responders to Neoadjuvant Chemoradiation for Esophageal Cancer: Why Better Prediction is Necessary. J. Thorac. Dis. 2017, 9, S843–S850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in Combination with Chemotherapy Versus Chemotherapy Alone for Treatment of HER2-positive Advanced Gastric or Gastro-oesophageal Junction Cancer (ToGA): A Phase 3, Open-label, Randomised Controlled Trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Shah, M.A.; Kojima, T.; Hochhauser, D.; Enzinger, P.; Raimbourg, J.; Hollebecque, A.; Lordick, F.; Kim, S.B.; Tajika, M.; Kim, H.T.; et al. Efficacy and Safety of Pembrolizumab for Heavily Pretreated Patients With Advanced, Metastatic Adenocarcinoma or Squamous Cell Carcinoma of the Esophagus: The Phase 2 KEYNOTE-180 Study. JAMA. Oncol. 2019, 5, 546–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janjigian, Y.Y.; Maron, S.B.; Chatila, W.K.; Millang, B.; Chavan, S.S.; Alterman, C.; Chou, J.F.; Segal, M.F.; Simmons, M.Z.; Momtaz, P.; et al. First-line Pembrolizumab and Trastuzumab in HER2-positive Oesophageal, Gastric, or Gastro-oesophageal Junction Cancer: An Open-label, Single-arm, Phase 2 Trial. Lancet Oncol. 2020, 21, 821–831. [Google Scholar] [CrossRef]
- Yoon, H.H.; Shi, Q.; Sukov, W.R.; Wiktor, A.E.; Khan, M.; Sattler, C.A.; Grothey, A.; Wu, T.T.; Diasio, R.B.; Jenkins, R.B.; et al. Association of HER2/ErbB2 Expression and Gene Amplification with Pathologic Features and Prognosis in Esophageal Adenocarcinomas. Clin. Cancer Res. 2012, 18, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Plum, P.S.; Gebauer, F.; Kramer, M.; Alakus, H.; Berlth, F.; Chon, S.H.; Schiffmann, L.; Zander, T.; Buttner, R.; Holscher, A.H.; et al. HER2/neu (ERBB2) expression and Gene Amplification Correlates with Better Survival in Esophageal Adenocarcinoma. BMC. Cancer 2019, 19, 38. [Google Scholar] [CrossRef] [Green Version]
- Kleespies, A.; Guba, M.; Jauch, K.W.; Bruns, C.J. Vascular Endothelial Growth Factor in Esophageal Cancer. J. Surg. Oncol. 2004, 87, 95–104. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab Monotherapy for Previously Treated Advanced Gastric or gastro-Oesophageal Junction Adenocarcinoma (REGARD): An international, Randomised, Multicentre, Placebo-Controlled, Phase 3 Trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab Plus Paclitaxel Versus Placebo Plus Paclitaxel in Patients with Previously Treated Advanced Gastric or Gastro-oesophageal Junction Adenocarcinoma (RAINBOW): A Double-blind, Randomised Phase 3 Trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Shitara, K.; Di Bartolomeo, M.; Lonardi, S.; Al-Batran, S.E.; Van Cutsem, E.; Ilson, D.H.; Alsina, M.; Chau, I.; Lacy, J.; et al. Ramucirumab with Cisplatin and Fluoropyrimidine as First-line Therapy in Patients with Metastatic Gastric or Junctional Adenocarcinoma (RAINFALL): A Double-blind, Randomised, Placebo-controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 420–435. [Google Scholar] [CrossRef]
- de Klerk, L.K.; Goedegebuure, R.S.A.; van Grieken, N.C.T.; van Sandick, J.W.; Cats, A.; Stiekema, J.; van der Kaaij, R.T.; Farina Sarasqueta, A.; van Engeland, M.; Jacobs, M.; et al. Molecular Profiles of Response to Neoadjuvant Chemoradiotherapy in Oesophageal Cancers to Develop Personalized Treatment Strategies. Mol. Oncol. 2021, 15, 901–914. [Google Scholar] [CrossRef]
- Sutherland, S. Burned adolescents’ descriptions of their coping strategies. Heart Lung 1988, 17, 150–157. [Google Scholar] [PubMed]
- Gregson, E.M.; Bornschein, J.; Fitzgerald, R.C. Genetic Progression of Barrett’s Oesophagus to Oesophageal Adenocarcinoma. Br. J. Cancer 2016, 115, 403–410. [Google Scholar] [CrossRef]
- Stachler, M.D.; Camarda, N.D.; Deitrick, C.; Kim, A.; Agoston, A.T.; Odze, R.D.; Hornick, J.L.; Nag, A.; Thorner, A.R.; Ducar, M.; et al. Detection of Mutations in Barrett’s Esophagus Before Progression to High-Grade Dysplasia or Adenocarcinoma. Gastroenterology 2018, 155, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Nahar, R.; Zhai, W.; Zhang, T.; Takano, A.; Khng, A.J.; Lee, Y.Y.; Liu, X.; Lim, C.H.; Koh, T.P.T.; Aung, Z.W.; et al. Elucidating the Genomic Architecture of Asian EGFR-Mutant Lung Adenocarcinoma through Multi-region Exome Sequencing. Nat. Commun. 2018, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Gharahkhani, P.; Fitzgerald, R.C.; Vaughan, T.L.; Palles, C.; Gockel, I.; Tomlinson, I.; Buas, M.F.; May, A.; Gerges, C.; Anders, M.; et al. Genome-wide Association Studies in Oesophageal Adenocarcinoma and Barrett’s Oesophagus: A Large-scale Meta-analysis. Lancet Oncol. 2016, 17, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- SERVIER, L.L. Servier Medical Art. Available online: https://smart.servier.com (accessed on 29 June 2021).
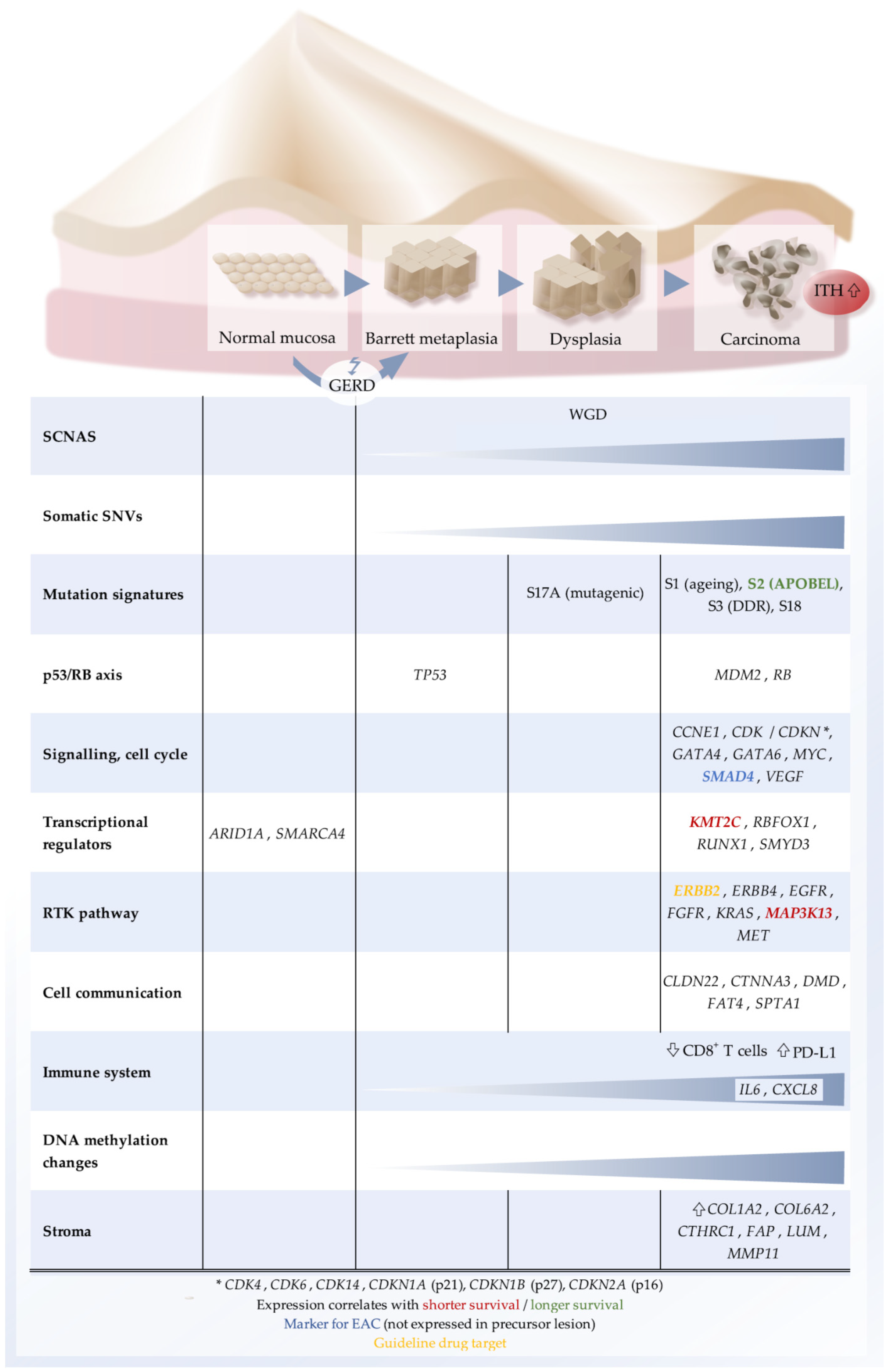
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).