
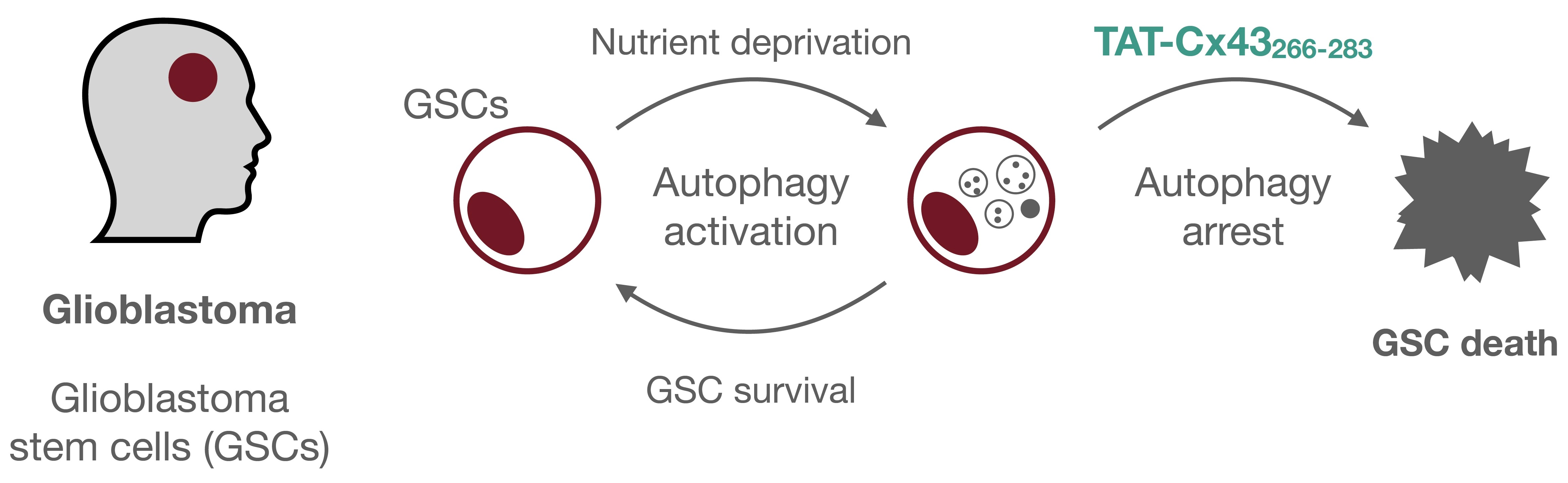
Impairment of Autophagic Flux Participates in the Antitumor Effects of TAT-Cx43266-283 in Glioblastoma Stem Cells

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Glioblastoma Stem Cells
2.2. Treatments
2.3. AlamarBlue Assay
2.4. Human Immunohistochemistry Microarrays
2.5. Immunofluorescence
2.6. MTT Assay
2.7. Patient Survival Analysis
2.8. Time-Lapse Microscopy (Live-Cell)
2.9. Transmission Electron Microscopy
2.10. Western Blotting
2.11. Statistical Analysis
3. Results
3.1. c-Src Activity Is a Relevant Target in Glioblastoma Patients
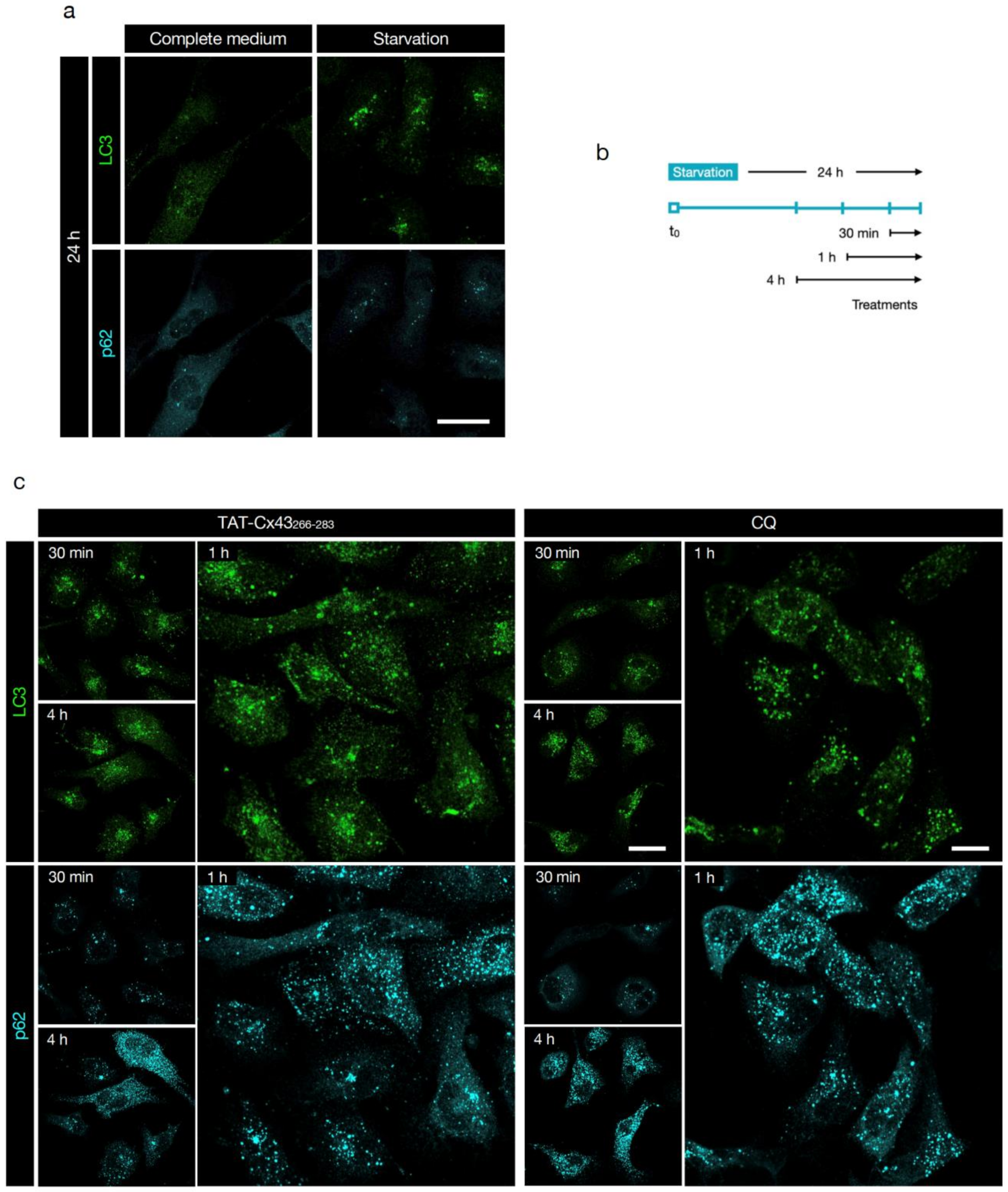
3.2. TAT-Cx43266-283 Blocks Basal Autophagic Flux in GSCs
3.3. TAT-Cx43266-283 Increases Cell Death in Nutrient-Deprived Dormant GSCs
3.4. TAT-Cx43266-283 Blocks Autophagy in Nutrient-Deprived Dormant GSCs
3.5. Autophagy Mediators Are Over-Expressed at the Protein Level in High-Grade Glioma Patients
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; Bent, M.V.D.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabernero, A.; Gangoso, E.; Jaraíz-Rodríguez, M.; Medina, J. The role of connexin43–Src interaction in astrocytomas: A molecular puzzle. Neuroscience 2016, 323, 183–194. [Google Scholar] [CrossRef]
- Frame, M.C. Newest findings on the oldest oncogene; how activated src does it. J. Cell Sci. 2004, 117, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Weissenberger, J.; Steinbach, J.P.; Malin, G.; Spada, S.; Rülicke, T.; Aguzzi, A. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene 1997, 14, 2005–2013. [Google Scholar] [CrossRef] [Green Version]
- González-Sánchez, A.; Jaraíz-Rodríguez, M.; Prieto, M.D.; Herrero-González, S.; Medina, J.M.; Tabernero, A. Connexin43 recruits PTEN and Csk to inhibit c-Src activity in glioma cells and astrocytes. Oncotarget 2016, 7, 49819–49833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangoso, E.; Thirant, C.; Chneiweiss, H.; Medina, J.M.; Tabernero, A. A cell-penetrating peptide based on the interaction between c-Src and connexin43 reverses glioma stem cell phenotype. Cell Death Dis. 2014, 5, e1023. [Google Scholar] [CrossRef] [PubMed]
- Herrero-González, S.; Gangoso, E.; Giaume, C.; Naus, C.C.; Medina, J.M.; Tabernero, A. Connexin43 inhibits the oncogenic activity of c-Src in C6 glioma cells. Oncogene 2010, 29, 5712–5723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soroceanu, L.; Manning, T.J., Jr.; Sontheimer, H. Reduced expression of connexin-43 and functional gap junction coupling in human gliomas. Glia 2001, 33, 107–117. [Google Scholar] [CrossRef]
- Han, X.; Zhang, W.; Yang, X.; Wheeler, C.G.; Langford, C.P.; Wu, L.; Filippova, N.; Friedman, G.; Ding, Q.; Fathallah-Shaykh, H.; et al. The role of Src family kinases in growth and migration of glioma stem cells. Int. J. Oncol. 2014, 45, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaraíz-Rodríguez, M.; Tabernero, M.D.; González-Tablas, M.; Otero, A.; Orfao, A.; Medina, J.M.; Tabernero, A. A Short Region of Connexin43 Reduces Human Glioma Stem Cell Migration, Invasion, and Survival through Src, PTEN, and FAK. Stem Cell Rep. 2017, 9, 451–463. [Google Scholar] [CrossRef]
- Du, J.; Bernasconi, P.; Clauser, K.; Mani, D.R.; Finn, S.; Beroukhim, R.; Burns, M.; Julian, B.; Peng, X.P.; Hieronymus, H.; et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 2008, 27, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Caveney, S.; Kidder, G.M.; Naus, C.C.G. Transfection of C6 glioma-cells with connexin-43 cDNA–analysis of expression, intercellular coupling, and cell-proliferation. Proc. Natl. Acad. Sci. USA 1991, 88, 1883–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.-C.; Xiao, H.-L.; Jiang, X.-F.; Wang, Q.-L.; Li, Y.; Yang, X.-J.; Ping, Y.-F.; Duan, J.J.; Jiang, J.-Y.; Ye, X.-Z.; et al. Connexin 43 Reverses Malignant Phenotypes of Glioma Stem Cells by Modulating E-Cadherin. Stem Cells 2011, 30, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.G.; Elisevich, K.; Zhu, D.G.; Belliveau, D.J.; Delmaestro, R.F. Invivo Growth of C6 Glioma-Cells Transfected with Connexin43 cDNA. Cancer Res. 1992, 52, 4208–4213. [Google Scholar] [PubMed]
- Vivès, E.; Brodin, P.; Lebleu, B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Kanemitsu, M.Y.; Loo, L.W.M.; Simon, S.; Lau, A.F.; Eckhart, W. Tyrosine Phosphorylation of Connexin 43 by v-Src Is Mediated by SH2 and SH3 Domain Interactions. J. Biol. Chem. 1997, 272, 22824–22831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelaz, S.G.; Jaraíz-Rodríguez, M.; Álvarez-Vázquez, A.; Talaverón, R.; García-Vicente, L.; Flores-Hernández, R.; De Cedrón, M.G.; Tabernero, M.; De Molina, A.R.; Lillo, C.; et al. Targeting metabolic plasticity in glioma stem cells in vitro and in vivo through specific inhibition of c-Src by TAT-Cx43266-283. EBioMedicine 2020, 62, 103134. [Google Scholar] [CrossRef] [PubMed]
- Jaraíz-Rodríguez, M.; Talaverón, R.; García-Vicente, L.; Pelaz, S.G.; Domínguez-Prieto, M.; Álvarez-Vázquez, A.; Flores-Hernández, R.; Sin, W.C.; Bechberger, J.; Medina, J.M.; et al. Connexin43 peptide, TAT-Cx43266–283, selectively targets glioma cells, impairs malignant growth, and enhances survival in mouse models in vivo. Neuro-Oncology 2019, 22, 493–504. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Cicchini, M.; Karantza, V.; Xia, B. Molecular Pathways: Autophagy in Cancer—A Matter of Timing and Context. Clin. Cancer Res. 2014, 21, 498–504. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The Role of Autophagy in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2019, 27, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, X.; Wang, N.; Wang, J.; Cao, Y.; Wang, T.; Zhou, X.; Jiao, Y.; Yang, L.; Wang, X.; et al. Autophagy-related gene expression is an independent prognostic indicator of glioma. Oncotarget 2017, 8, 60987–61000. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-W.; Liu, H.-J.; Zhao, Z.; Zhang, Y.; Wang, Z.; Jiang, T.; Bao, Z.-S. Prognostic Correlation of Autophagy-Related Gene Expression-Based Risk Signature in Patients with Glioblastoma. OncoTargets Ther. 2020, 13, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, W.; Xiao, Z.; Guan, G.; Liu, X.; Zhuang, M. A risk signature with four autophagy-related genes for predicting survival of glioblastoma multiforme. J. Cell. Mol. Med. 2020, 24, 3807–3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cj, P.; Hv, E.; Vijayakurup, V.; Menon, G.; Nair, S.; Gopala, S. High LC3/Beclin Expression Correlates with Poor Survival in Glioma: A Definitive Role for Autophagy as Evidenced by In Vitro Autophagic Flux. Pathol. Oncol. Res. 2017, 25, 137–148. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pasqualis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; LaRocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Jiang, T.; Wu, Z. Immunohistochemical assessment of autophagic protein LC3B and p62 levels in glioma patients. Int. J. Clin. Exp. Pathol. 2018, 11, 862–868. [Google Scholar]
- Tamrakar, S.; Yashiro, M.; Kawashima, T.; Uda, T.; Terakawa, Y.; Kuwae, Y.; Ohsawa, M.; Ohata, K. Clinicopathological Significance of Autophagy-related Proteins and its Association With Genetic Alterations in Gliomas. Anticancer Res. 2019, 39, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Akkoc, Y.; Peker, N.; Akcay, A.; Gozuacik, D. Autophagy and Cancer Dormancy. Front. Oncol. 2021, 11, 277. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, L.V.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Rusu, P.; Shao, C.; Neuerburg, A.; Acikgöz, A.A.; Wu, Y.; Zou, P.; Phapale, P.; Shankar, T.S.; Döring, K.; Dettling, S.; et al. GPD1 Specifically Marks Dormant Glioma Stem Cells with a Distinct Metabolic Profile. Cell Stem Cell 2019, 25, 241–257.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shang, Z.; Zhou, Y.; Hu, X.; Chen, Y.; Fan, Y.; Wei, X.; Wu, L.; Liang, Q.; Zhang, J.; et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Briceño, E.; Calderon, A.; Sotelo, J. Institutional experience with chloroquine as an adjuvant to the therapy for glioblastoma multiforme. Surg. Neurol. 2007, 67, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Briceño, E.; Loípez-Gonzaílez, M.A. Adding Chloroquine to Conventional Treatment for Glioblastoma Multiforme. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Rosenfeld, M.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef]
- Compter, I.; Eekers, D.B.P.; Hoeben, A.; Rouschop, K.M.A.; Reymen, B.; Ackermans, L.; Beckervordersantforth, J.; Bauer, N.J.C.; Anten, M.M.; Wesseling, P.; et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: A phase IB trial. Autophagy 2020, 1–9. [Google Scholar] [CrossRef]
- Gump, J.M.; Dowdy, S.F. TAT transduction: The molecular mechanism and therapeutic prospects. Trends Mol. Med. 2007, 13, 443–448. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Blal, H.A.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2017, 27, 233–244. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. Available online: https://www.scirp.org/(S(i43dyn45teexjx455qlt3d2q))/reference/ReferencesPapers.aspx?ReferenceID=1787696 (accessed on 11 May 2021).
- Borcherding, N.A.-O.; Bormann, N.L.; Voigt, A.P.; Zhang, W. TRGAted: A web tool for survival analysis using protein data in the Cancer Genome Atlas. F1000Research 2018, 7, 1235. [Google Scholar] [CrossRef]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.-Y.; Broom, B.M.; Verhaak, R.G.W.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; de Groot, J.; Liu, W.; Gladson, C.L. Targeting SRC in glioblastoma tumors and brain metastases: Rationale and preclinical studies. Cancer Lett. 2010, 298, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Dey, N.; Crosswell, H.E.; De, P.; Parsons, R.; Peng, Q.; Su, J.D.; Durden, D.L. The Protein Phosphatase Activity of PTEN Regulates Src Family Kinases and Controls Glioma Migration. Cancer Res. 2008, 68, 1862–1871. [Google Scholar] [CrossRef] [Green Version]
- Lund, C.V.; Nguyen, M.T.N.; Owens, G.C.; Pakchoian, A.J.; Shaterian, A.; Kruse, C.A.; Eliceiri, B.P. Reduced Glioma Infiltration in Src-deficient Mice. J. Neuro-Oncology 2006, 78, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Barth, S.; Glick, D.; MacLeod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T. How to Interpret LC3 Immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, J.; Abeliovich, H.; Acevedo-Arozena, A.; Adachi, H.; Adams, C.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Araujo, J.; Logothetis, C. Dasatinib: A potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat. Rev. 2010, 36, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianfanelli, V.; De Zio, D.; Di Bartolomeo, S.; Nazio, F.; Strappazzon, F.; Cecconi, F. Ambra1 at a glance. J. Cell Sci. 2015, 128, 2003–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuyuki, S.; Takabayashi, M.; Kawazu, M.; Kudo, K.; Watanabe, A.; Nagata, Y.; Kusama, Y.; Yoshida, K. Detection ofWIPI1mRNA as an indicator of autophagosome formation. Autophagy 2013, 10, 497–513. [Google Scholar] [CrossRef] [Green Version]
- Button, R.W.; Roberts, S.L.; Willis, T.; Hanemann, C.O.; Luo, S. Accumulation of autophagosomes confers cytotoxicity. J. Biol. Chem. 2017, 292, 13599–13614. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.-W.; Cheng, C.; Hackett, C.; Feldman, M.; Houseman, B.T.; Nicolaides, T.; Haas-Kogan, D.; James, C.D.; Oakes, S.A.; Debnath, J.; et al. Akt and Autophagy Cooperate to Promote Survival of Drug-Resistant Glioma. Sci. Signal. 2010, 3, ra81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Honda, T.; Akatsu, A.; Yamaguchi, N.; Yamaguchi, N. The promoting role of lysosome-localized c-Src in autophagosome-lysosome fusion. Cell. Signal. 2020, 75, 109774. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Jorgensen, C.; Pawson, T.; Hedley, D.W. Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. Br. J. Cancer 2008, 99, 1074–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Touny, L.H.; Vieira, A.; Mendoza, A.; Khanna, C.; Hoenerhoff, M.J.; Green, J.E. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J. Clin. Investig. 2013, 124, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.-C.; Aguirre-Ghiso, J.A. ERK1/2 and p38α/β Signaling in Tumor Cell Quiescence: Opportunities to Control Dormant Residual Disease. Clin. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Ghiso, J.A.; Estrada, Y.; Liu, D.; Ossowski, L. ERKMAPK Activity as a Determinant of Tumor Growth and Dormancy; Regulation by p38SAPK. Cancer Res. 2003, 63, 1684. [Google Scholar]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase Receptor and Fibronectin Regulate the ERKMAPK to p38MAPK Activity Ratios That Determine Carcinoma Cell Proliferation or Dormancy In Vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia-Arencibia, M.; Vetrini, F.; Erdin, S.; Huynh, T.; Medina, D.L.; Colella, P.; Sardiello, M.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Colecchia, D.; Strambi, A.; Sanzone, S.; Iavarone, C.; Rossi, M.; Dall’Armi, C.; Piccioni, F.; Di Pianella, A.V.; Chiariello, M. MAPK15/ERK8 stimulates autophagy by interacting with LC3 and GABARAP proteins. Autophagy 2012, 8, 1724–1740. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; Durán, A.; Selloum, M.; Champy, M.-F.; Diez-Guerra, F.J.; Flores, J.M.; Serrano, M.; Auwerx, J.; Diaz-Meco, M.T.; Moscat, J. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006, 3, 211–222. [Google Scholar] [CrossRef]
- Pal, R.; Palmieri, M.; Loehr, J.A.; Li, S.; Abo-Zahrah, R.; Monroe, T.; Thakur, P.B.; Sardiello, M.; Rodney, G.G. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2019, 104, 575–587. [Google Scholar] [CrossRef]
- Button, R.W.; Vincent, J.H.; Strang, C.J.; Luo, S. Dual PI-3 kinase/mTOR inhibition impairs autophagy flux and induces cell death independent of apoptosis and necroptosis. Oncotarget 2016, 7, 5157–5175. [Google Scholar] [CrossRef] [Green Version]
- Pal, R.; Palmieri, M.; Chaudhury, A.; Klisch, T.J.; Di Ronza, A.; Neilson, J.R.; Rodney, G.; Sardiello, M. Src regulates amino acid-mediated mTORC1 activation by disrupting GATOR1-Rag GTPase interaction. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Warn-Cramer, B.J.; Lampe, P.D.; Kurata, W.E.; Kanemitsu, M.Y.; Loo, L.W.; Eckhart, W.; Lau, A.F. Characterization of the Mitogen-activated Protein Kinase Phosphorylation Sites on the Connexin-43 Gap Junction Protein. J. Biol. Chem. 1996, 271, 3779–3786. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiram, G.; Ferber, S.; Ofek, P.; Eldar-Boock, A.; Ben-Shushan, D.; Yeini, E.; Krivitsky, A.; Blatt, R.; Almog, N.; Henkin, J.; et al. Reverting the molecular fingerprint of tumor dormancy as a therapeutic strategy for glioblastoma. FASEB J. 2018, 32, 5835–5850. [Google Scholar] [CrossRef]
- Sánchez-Martín, P.; Komatsu, M. p62/SQSTM1–steering the cell through health and disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Wang, C.; Croce, C.M.; Guan, J.-L. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014, 28, 1204–1216. [Google Scholar] [CrossRef] [Green Version]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The Signaling Adaptor p62 Is an Important NF-κB Mediator in Tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, V.; Hawkins, W.D.; Klionsky, D.J. Watch What You (Self-) Eat: Autophagic Mechanisms that Modulate Metabolism. Cell Metab. 2019, 29, 803–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic Control of Autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [Green Version]
- Lassman, A.B.; Pugh, S.L.; Gilbert, M.R.; Aldape, K.D.; Geinoz, S.; Beumer, J.H.; Christner, S.M.; Komaki, R.; DeAngelis, L.M.; Gaur, R.; et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro-Oncology 2015, 17, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Muir, A.; Heiden, M.G.V. The nutrient environment affects therapy. Science 2018, 360, 962–963. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelaz, S.G.; Ollauri-Ibáñez, C.; Lillo, C.; Tabernero, A. Impairment of Autophagic Flux Participates in the Antitumor Effects of TAT-Cx43266-283 in Glioblastoma Stem Cells. Cancers 2021, 13, 4262. https://doi.org/10.3390/cancers13174262
Pelaz SG, Ollauri-Ibáñez C, Lillo C, Tabernero A. Impairment of Autophagic Flux Participates in the Antitumor Effects of TAT-Cx43266-283 in Glioblastoma Stem Cells. Cancers. 2021; 13(17):4262. https://doi.org/10.3390/cancers13174262
Chicago/Turabian StylePelaz, Sara G., Claudia Ollauri-Ibáñez, Concepción Lillo, and Arantxa Tabernero. 2021. "Impairment of Autophagic Flux Participates in the Antitumor Effects of TAT-Cx43266-283 in Glioblastoma Stem Cells" Cancers 13, no. 17: 4262. https://doi.org/10.3390/cancers13174262
APA StylePelaz, S. G., Ollauri-Ibáñez, C., Lillo, C., & Tabernero, A. (2021). Impairment of Autophagic Flux Participates in the Antitumor Effects of TAT-Cx43266-283 in Glioblastoma Stem Cells. Cancers, 13(17), 4262. https://doi.org/10.3390/cancers13174262