
Development of Dl1.72, a Novel Anti-DLL1 Antibody with Anti-Tumor Efficacy against Estrogen Receptor-Positive Breast Cancer

 ,
,  ,
,  , , ,
, , ,  ,
,  ,
,  ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Phage Display Screening, Antibody Generation and Characterization
2.3. Notch Reporter Assay
2.4. Real-Time RT-qPCR
2.5. MCF-7 Cell Growth, Scratch Wound Healing, and Mammosphere Formation Assays
2.6. HUVEC Tube Formation, Cell Growth, and Viability Assays
2.7. Human Breast Tumor Samples
2.8. MCF-7 Breast Cancer Xenograft Model
2.9. Serology
2.10. Histological and Immunohistochemical Analysis
2.11. Analysis of Tumor Cell Apoptosis by TUNEL
2.12. Statistical Analysis
3. Results
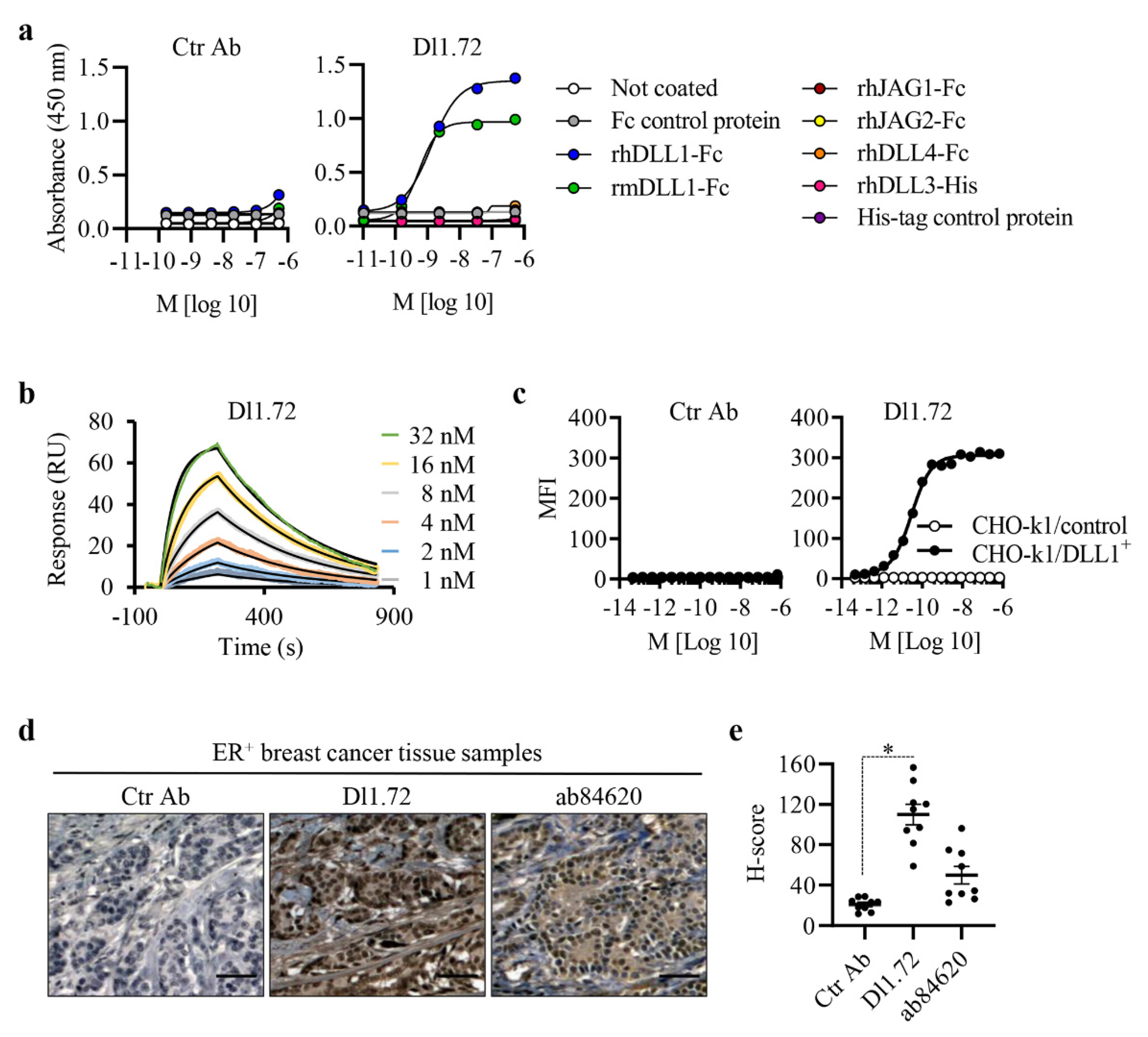
3.1. Characterization of the Generated Anti-DLL1 Dl1.72 Antibody That Specifically Interacts with DLL1
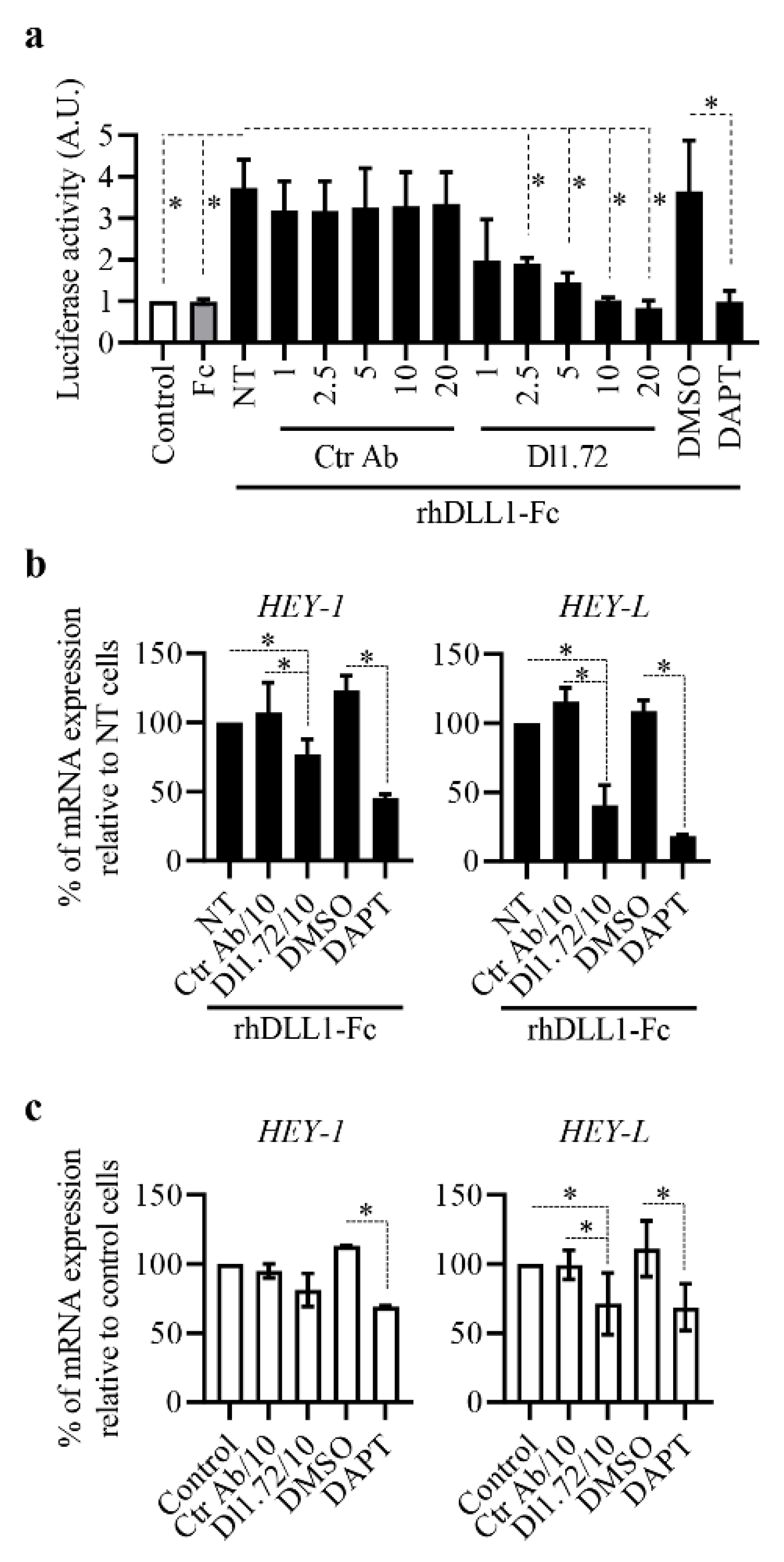
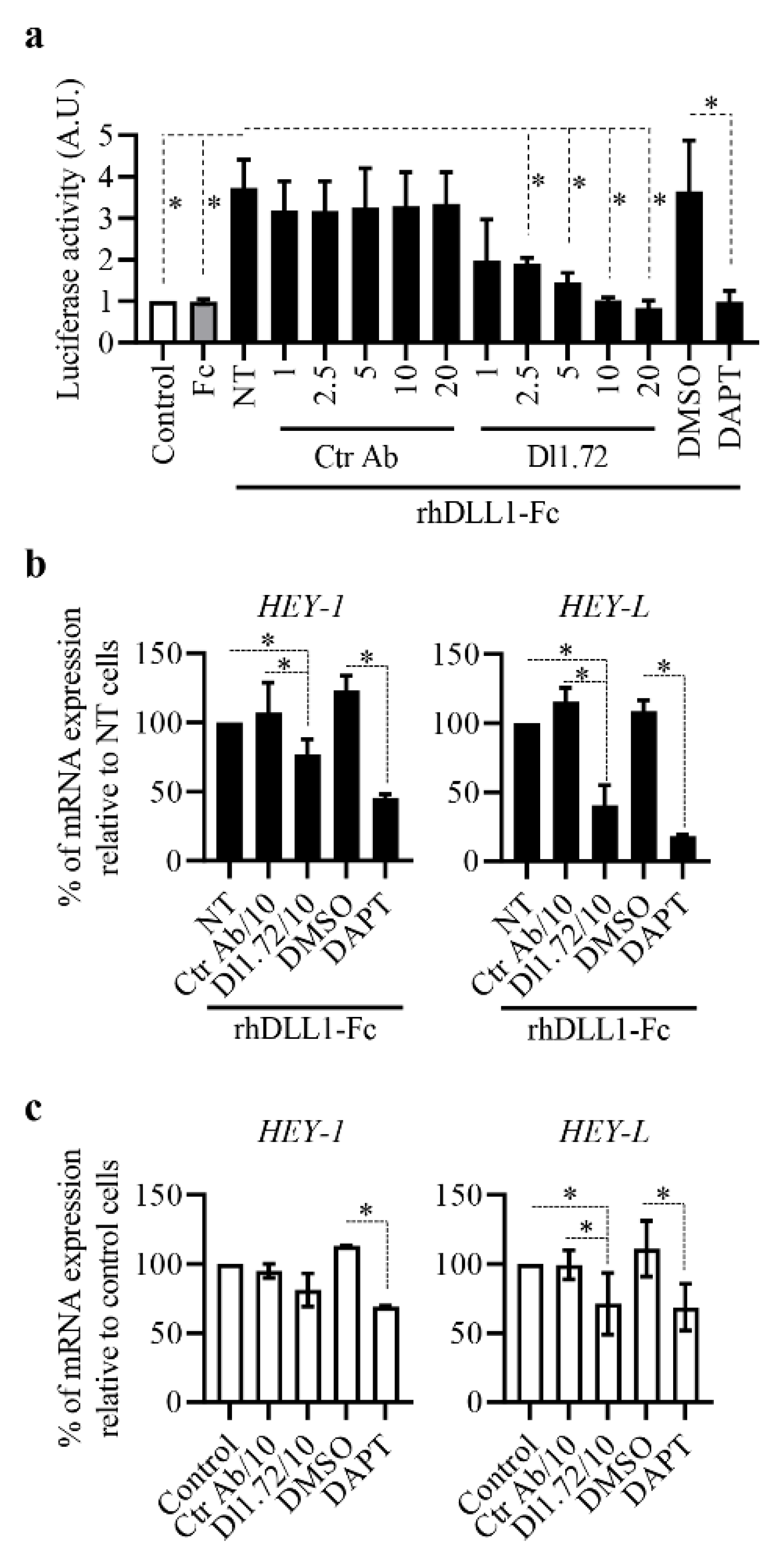
3.2. Anti-DLL1 Dl1.72 Inhibits DLL1-Notch Signaling in ER+ Breast Cancer Cells
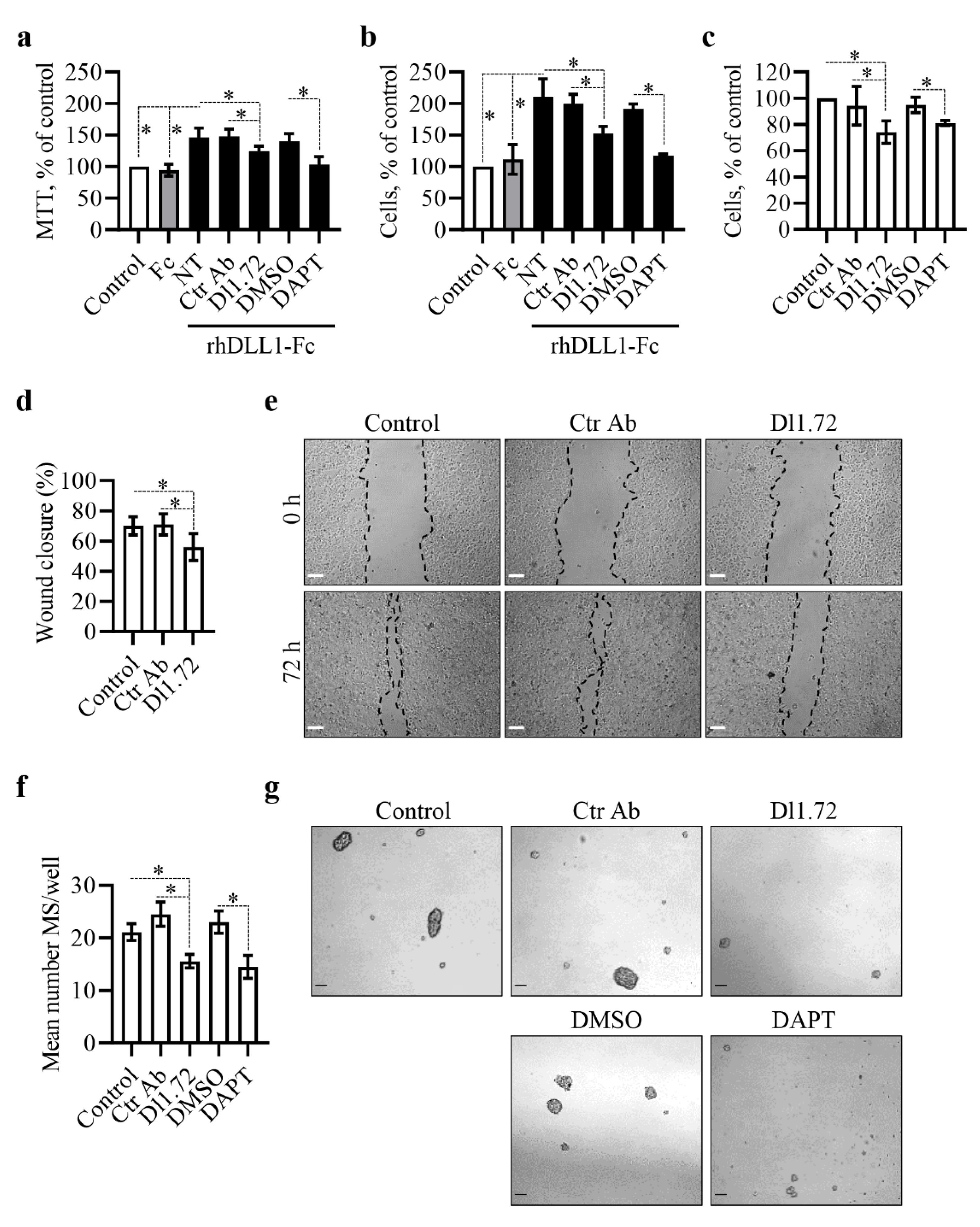
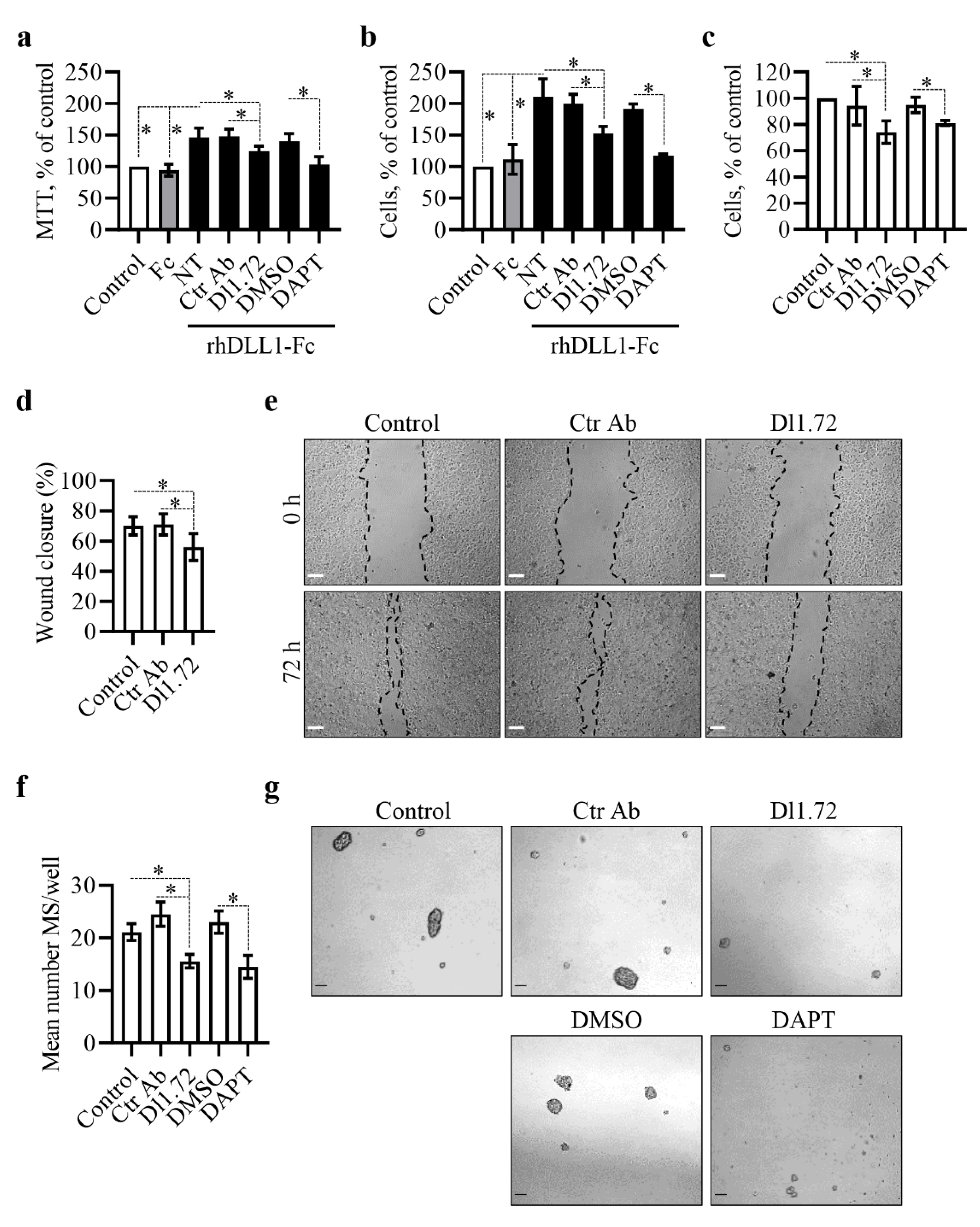
3.3. Anti-DLL1 Dl1.72 Decreases MCF-7 ER+ Breast Cancer Cell Proliferation, Migration and Mammosphere Formation
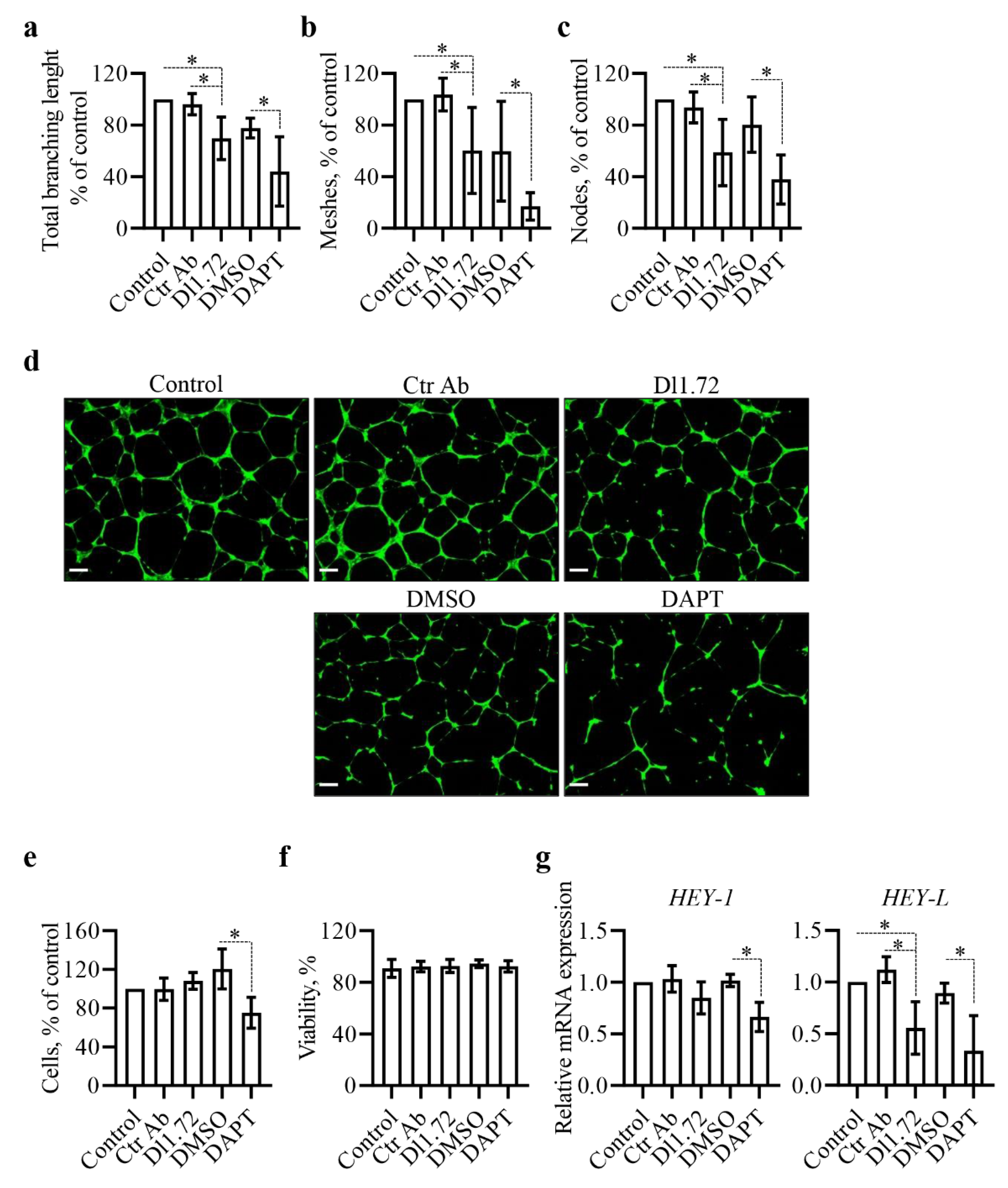
3.4. Anti-DLL1 Dl1.72 Impairs the Angiogenic Potential of Human Vascular Endothelial Cells
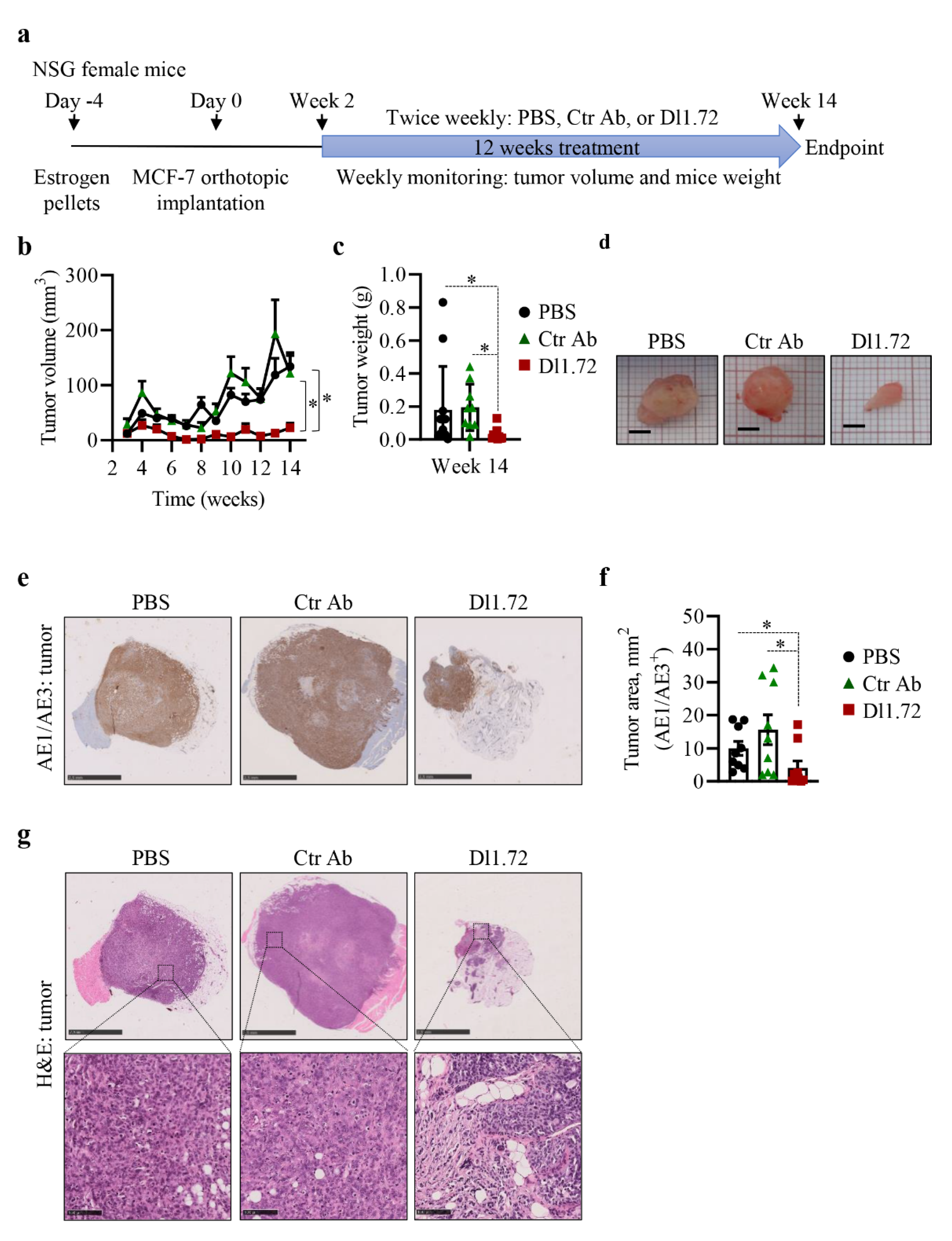
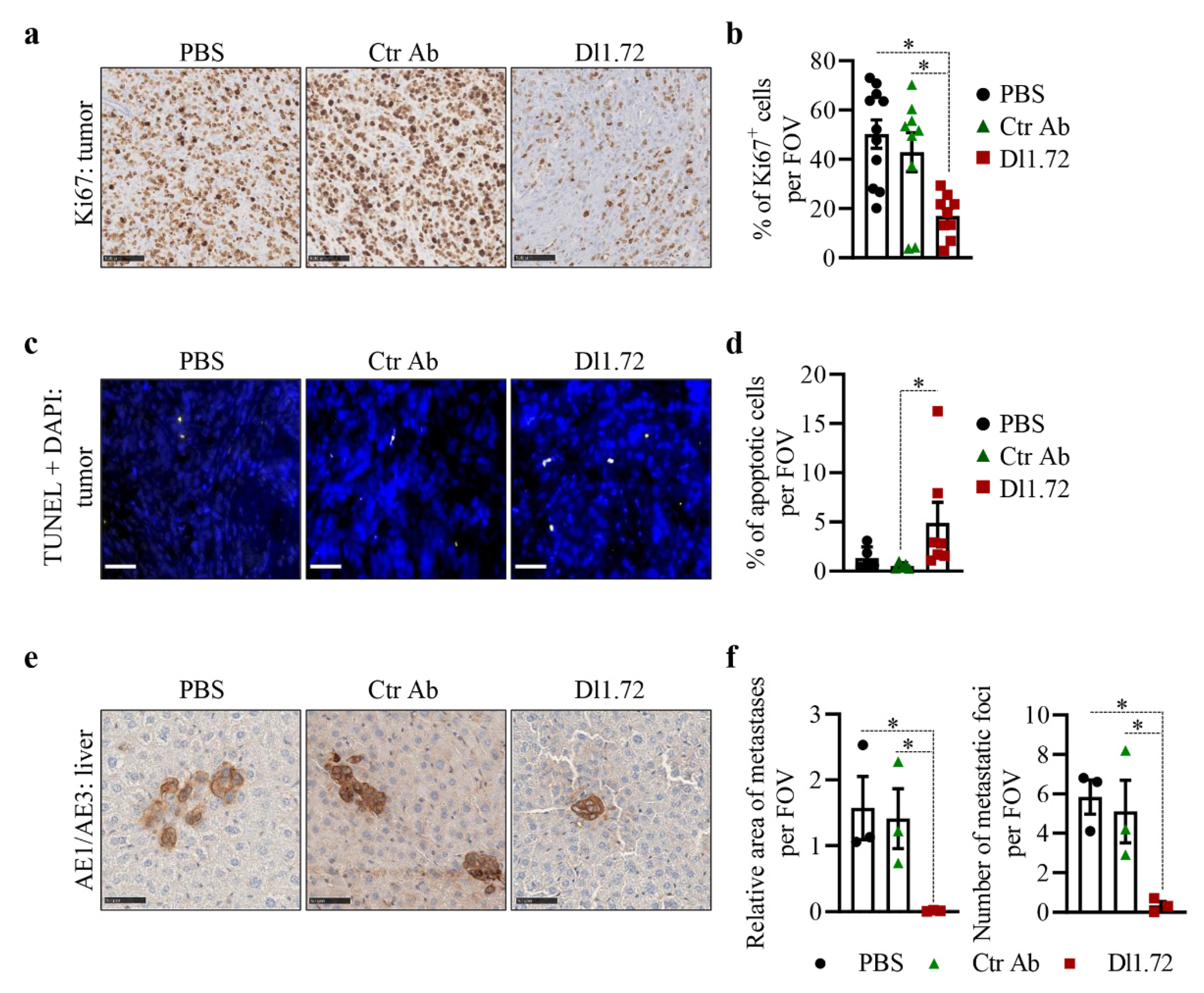
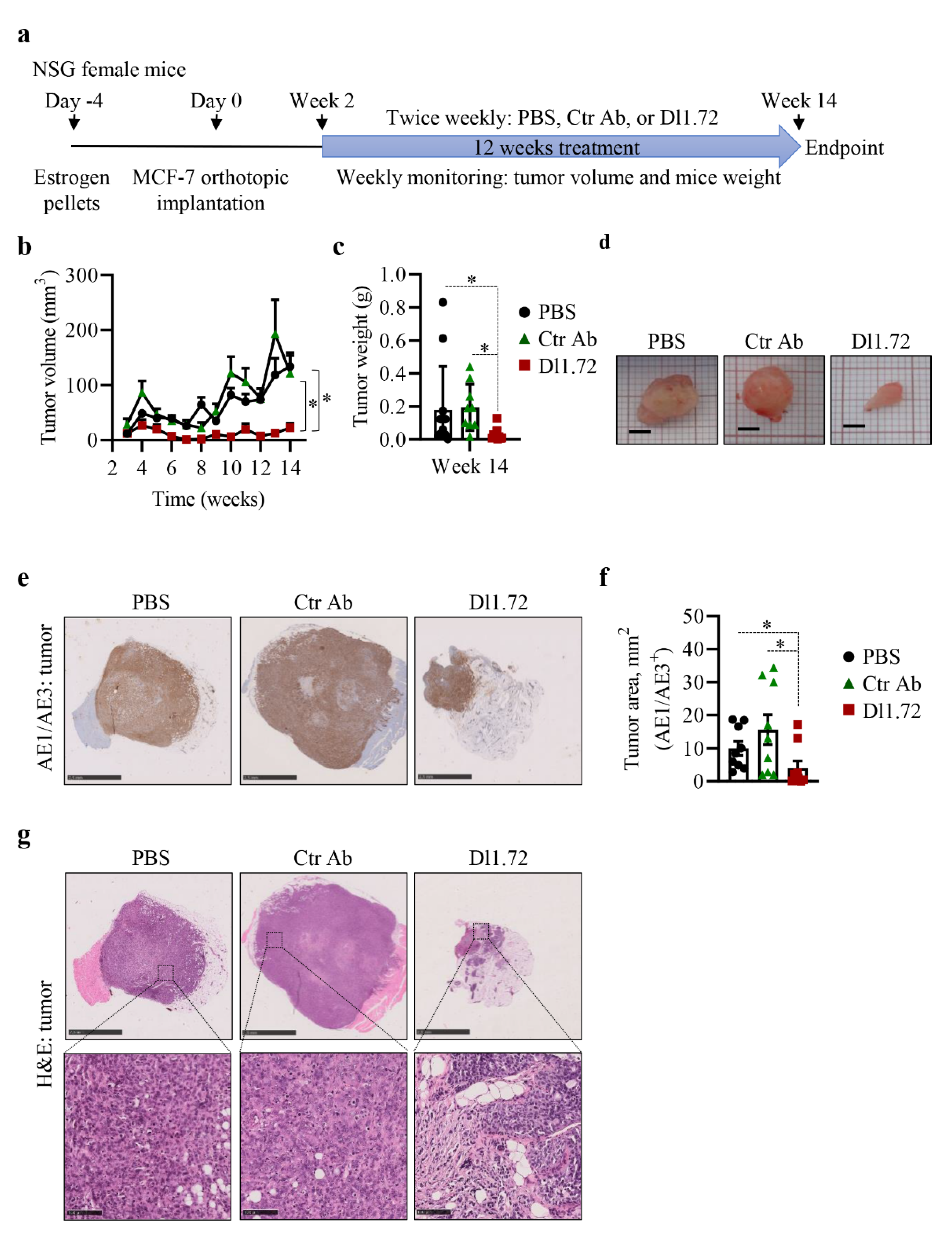
3.5. Anti-DLL1 Dl1.72 Inhibits Tumor Growth in a MCF-7 ER+ Breast Cancer Xenograft Mice Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Mph, K.D.M.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2018, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Bettaieb, A.; Paul, C.; Plenchette, S.; Shan, J.; Chouchane, L.; Ghiringhelli, F. Precision medicine in breast cancer: Reality or utopia? J. Transl. Med. 2017, 15, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Li, J.; Zhu, S.; Wu, J.; Chen, C.; Liu, Q.; Wei, W.; Zhang, Y.; Sun, S. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget 2017, 8, 27990–27996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2017, 186, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Rueda, O.M.; Sammut, S.-J.; Seoane, J.A.; Chin, S.-F.; Caswell-Jin, J.; Callari, M.; Batra, R.; Pereira, B.; Bruna, A.; Ali, H.R.; et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nat. Cell Biol. 2019, 567, 399–404. [Google Scholar] [CrossRef]
- Bai, J.-W.; Wei, M.; Li, J.-W.; Zhang, G.-J. Notch Signaling Pathway and Endocrine Resistance in Breast Cancer. Front. Pharmacol. 2020, 11, 924. [Google Scholar] [CrossRef] [PubMed]
- Bray, S. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Cordle, J.; Johnson, S.; Tay, J.Z.Y.; Roversi, P.; Wilkin, M.B.; De Madrid, B.H.; Shimizu, H.; Jensen, S.; Whiteman, P.; Jin, B.; et al. A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nat. Struct. Mol. Biol. 2008, 15, 849–857. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, N.J.; Church, N.L.; Griffin, M.; Luo, C.S.; Adams, T.; Burgess, A.W. Notch ligand delta-like1: X-ray crystal structure and binding affinity. Biochem. J. 2015, 468, 159–166. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Nandagopal, N.; Santat, L.A.; LeBon, L.; Sprinzak, D.; Bronner, M.E.; Elowitz, M.B. Dynamic Ligand Discrimination in the Notch Signaling Pathway. Cell 2018, 172, 869–880.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pece, S.; Serresi, M.; Santolini, E.; Capra, M.; Hulleman, E.; Galimberti, V.; Zurrida, S.; Maisonneuve, P.; Viale, G.; Di Fiore, P.P. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J. Cell Biol. 2004, 167, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 245–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, B.M.; O’Brien, C.S.; Eyre, R.; Silva, A.; Yu, L.; Sarmiento-Castro, A.; Alférez, D.G.; Spence, K.; Santiago-Gómez, A.; Chemi, F.; et al. Anti-estrogen Resistance in Human Breast Tumors Is Driven by JAG1-NOTCH4-Dependent Cancer Stem Cell Activity. Cell Rep. 2015, 12, 1968–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandi, A.; Chakrabarti, R. The many facets of Notch signaling in breast cancer: Toward overcoming therapeutic resistance. Genes Dev. 2020, 34, 1422–1438. [Google Scholar] [CrossRef]
- Mittal, S.; Subramanyam, D.; Dey, D.; Kumar, R.V.; Rangarajan, A. Cooperation of Notch and Ras/MAPK signaling pathways in human breast carcinogenesis. Mol. Cancer 2009, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Srivastav, R.K.; Wilkes, D.W.; Ross, T.; Kim, S.; Kowalski, J.; Chatla, S.; Zhang, Q.; Nayak, A.; Guha, M.; et al. Estrogen-dependent DLL1-mediated Notch signaling promotes luminal breast cancer. Oncogene 2018, 38, 2092–2107. [Google Scholar] [CrossRef] [Green Version]
- Dias, J.S.; Silva, G.; Lamy, M.; Ferreira, A.; Barbas, A. The Notch ligand DLL1 exerts carcinogenic features in human breast cancer cells. PLoS ONE 2019, 14, e0217002. [Google Scholar] [CrossRef]
- Shui, Y.; Yu, X.; Duan, R.; Bao, Q.; Wu, J.; Yuan, H.; Ma, C. miR-130b-3p inhibits cell invasion and migration by targeting the Notch ligand Delta-like 1 in breast carcinoma. Gene 2017, 609, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nandi, A.; Singh, S.; Regulapati, R.; Li, N.; Tobias, J.W.; Siebel, C.W.; Blanco, M.A.; Klein-Szanto, A.J.; Lengner, C.; et al. Dll1+ quiescent tumor stem cells drive chemoresistance in breast cancer through NF-κB survival pathway. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Durocher, Y.; Perret, S.; Kamen, A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002, 30, E9. [Google Scholar] [CrossRef] [PubMed]
- Sales-Dias, J.; Ferreira, A.; Lamy, M.; Domenici, G.; Monteiro, S.M.; Pires, A.; Lemos, A.R.; Kucheryava, K.; Nobre, L.S.; Sousa, P.M.; et al. Development of antibodies against the notch ligand Delta-Like-1 by phage display with activity against breast cancer cells. New Biotechnol. 2021, 64, 17–26. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, G.; Aboussekhra, A. p16INK4A inhibits the pro-metastatic potentials of osteosarcoma cells through targeting the ERK pathway and TGF-β1. Mol. Carcinog. 2015, 55, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.; Cardoso, B.A.; Belo, H.; Almeida, A.M. Vorinostat induces apoptosis and differentiation in myeloid malignancies: Genetic and molecular mechanisms. PLoS ONE 2013, 8, e53766. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, G. Angiogenesis Analyzer for ImageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2012, 10, 11568. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Kang, Y. Transplantable Mouse Tumor Models of Breast Cancer Metastasis. Methods Mol. Biol. 2015, 1267, 367–380. [Google Scholar] [CrossRef]
- Loureiro, L.R.; Sousa, D.P.; Ferreira, D.; Chai, W.; Lima, L.; Pereira, C.; Lopes, C.B.; Correia, V.G.; Silva, L.M.; Li, C.; et al. Novel monoclonal antibody L2A5 specifically targeting sialyl-Tn and short glycans terminated by alpha-2–6 sialic acids. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Hematoxylin and Eosin Staining of Tissue and Cell Sections. Cold Spring Harb. Protoc. 2008, 2008, pdb-prot4986. [Google Scholar] [CrossRef]
- Casal, D.; Iria, I.; Ramalho, J.S.; Alves, S.; Mota-Silva, E.; Mascarenhas-Lemos, L.; Pontinha, C.; Guadalupe-Cabral, M.; Silva, J.; Ferraz-Oliveira, M.; et al. BD-2 and BD-3 increase skin flap survival in a model of ischemia and Pseudomonas aeruginosa infection. Sci. Rep. 2019, 9, 7854. [Google Scholar] [CrossRef]
- Tuominen, V.J.; Ruotoistenmäki, S.; Viitanen, A.; Jumppanen, M.; Isola, J. ImmunoRatio: A publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR), and Ki-67. Breast Cancer Res. 2010, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Ma, C.; Chen, Z.; Yi, W.; McNutt, M.A.; Wang, Y.; Korteweg, C.; Gu, J. Correlation of Immunoglobulin G Expression and Histological Subtype and Stage in Breast Cancer. PLoS ONE 2013, 8, e58706. [Google Scholar] [CrossRef] [Green Version]
- de Angelis, M.; Francescangeli, F.; Zeuner, A. Breast Cancer Stem Cells as Drivers of Tumor Chemoresistance, Dormancy and Relapse: New Challenges and Therapeutic Opportunities. Cancers 2019, 11, 1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domenici, G.; Aurrekoetxea-Rodríguez, I.; Simões, B.M.; Rábano, M.; Lee, S.Y.; Millán, J.S.; Comaills, V.; Oliemuller, E.; López-Ruiz, J.A.; Zabalza, I.; et al. A Sox2–Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene 2019, 38, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Piva, M.; Domenici, G.; Iriondo, O.; Rábano, M.; Simões, B.M.; Comaills, V.; Barredo, I.; López-Ruiz, J.A.; Zabalza, I.; Kypta, R.; et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 2013, 6, 66–79. [Google Scholar] [CrossRef]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [Green Version]
- Napp, L.C.; Augustynik, M.; Paesler, F.; Krishnasamy, K.; Woiterski, J.; Limbourg, A.; Bauersachs, J.; Drexler, H.; Noble, F.L.; Limbourg, F. Extrinsic Notch Ligand Delta-Like 1 Regulates Tip Cell Selection and Vascular Branching Morphogenesis. Circ. Res. 2012, 110, 530–535. [Google Scholar] [CrossRef] [Green Version]
- DeCicco-Skinner, K.L.; Henry, G.; Cataisson, C.; Tabib, T.; Gwilliam, J.C.; Watson, N.J.; Bullwinkle, E.M.; Falkenburg, L.; O’Neill, R.C.; Morin, A.; et al. Endothelial Cell Tube Formation Assay for the In Vitro Study of Angiogenesis. J. Vis. Exp. 2014, e51312. [Google Scholar] [CrossRef]
- Han, L.; Korangath, P.; Nguyen, N.K.; Diehl, A.; Cho, S.; Teo, W.W.; Cope, L.; Gessler, M.; Romer, L.; Sukumar, S. HEYL Regulates Neoangiogenesis Through Overexpression in Both Breast Tumor Epithelium and Endothelium. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef]
- Mavingire, N.; Campbell, P.; Wooten, J.; Aja, J.; Davis, M.B.; Loaiza-Perez, A.; Brantley, E. Cancer stem cells: Culprits in endocrine resistance and racial disparities in breast cancer outcomes. Cancer Lett. 2020, 500, 64–74. [Google Scholar] [CrossRef]
- Lamy, M.; Ferreira, A.; Dias, J.S.; Braga, S.; Silva, G.; Barbas, A. Notch-out for breast cancer therapies. New Biotechnol. 2017, 39, 215–221. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef]
- Hoey, T.; Yen, W.-C.; Axelrod, F.; Basi, J.; Donigian, L.; Dylla, S.; Fitch-Bruhns, M.; Lazetic, S.; Park, I.-K.; Sato, A.; et al. DLL4 Blockade Inhibits Tumor Growth and Reduces Tumor-Initiating Cell Frequency. Cell Stem Cell 2009, 5, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Bae, Y.; Kasimir-Bauer, S.; Tang, R.; Chen, J.; Ren, G.; Yuan, M.; Esposito, M.; Li, W.; Wei, Y.; et al. Therapeutic Antibody Targeting Tumor- and Osteoblastic Niche-Derived Jagged1 Sensitizes Bone Metastasis to Chemotherapy. Cancer Cell 2017, 32, 731–747.e6. [Google Scholar] [CrossRef] [Green Version]
- Masiero, M.; Li, D.; Whiteman, P.; Bentley, C.; Greig, J.; Hassanali, T.; Watts, S.; Stribbling, S.; Yates, J.; Bealing, E.; et al. Development of Therapeutic Anti-JAGGED1 Antibodies for Cancer Therapy. Mol. Cancer Ther. 2019, 18, 2030–2042. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Paranjape, A.N.; Rangarajan, A.; Dighe, R.R. A Monoclonal Antibody against Human Notch1 Ligand–Binding Domain Depletes Subpopulation of Putative Breast Cancer Stem–like Cells. Mol. Cancer Ther. 2011, 11, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.; Peng, Q.; Jiang, I.; Carroll, C.; Han, G.; Rymer, I.; Lippincott, J.; Zachwieja, J.; Gajiwala, K.; Kraynov, E.; et al. Specific inhibition of Notch1 signaling enhances the antitumor efficacy of chemotherapy in triple negative breast cancer through reduction of cancer stem cells. Cancer Lett. 2013, 328, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Handley, K.F.; Burger, R.; Molin, G.Z.D.; Stagg, R.; Sood, A.K.; Moore, K.N. Demcizumab combined with paclitaxel for platinum-resistant ovarian, primary peritoneal, and fallopian tube cancer: The SIERRA open-label phase Ib trial. Gynecol. Oncol. 2020, 157, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Phannasil, P.; Thuwajit, C.; Warnnissorn, M.; Wallace, J.C.; Macdonald, M.J.; Jitrapakdee, S. Pyruvate Carboxylase Is Up-Regulated in Breast Cancer and Essential to Support Growth and Invasion of MDA-MB-231 Cells. PLoS ONE 2015, 10, e0129848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jana, S.; Krishna, B.M.; Singhal, J.; Horne, D.; Awasthi, S.; Salgia, R.; Singhal, S.S. SOX9: The master regulator of cell fate in breast cancer. Biochem. Pharmacol. 2020, 174, 113789. [Google Scholar] [CrossRef]
- Nakano, Y.; Negishi, N.; Gocho, S.; Mine, T.; Sakurai, Y.; Yazawa, M.; Abe, K.; Yagita, H.; Habu, S.; Kageyama, R.; et al. Disappearance of centroacinar cells in the Notch ligand-deficient pancreas. Genes Cells 2015, 20, 500–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ELISA | Flow Cytometry | SPR | ||
|---|---|---|---|---|
| EC50 (nM) | EC50 (nM) | KD (nM) | ka (s−1) | kd (M−1 s−1) |
| 1.15 ± 0.16 | 2.07 × 10−2 ±1.20 × 10−2 | 4.78 ± 1.05 | 5.44 × 105 ±8.21 × 104 | 2.60 × 10−3 ±2.50 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, G.; Sales-Dias, J.; Casal, D.; Alves, S.; Domenici, G.; Barreto, C.; Matos, C.; Lemos, A.R.; Matias, A.T.; Kucheryava, K.; et al. Development of Dl1.72, a Novel Anti-DLL1 Antibody with Anti-Tumor Efficacy against Estrogen Receptor-Positive Breast Cancer. Cancers 2021, 13, 4074. https://doi.org/10.3390/cancers13164074
Silva G, Sales-Dias J, Casal D, Alves S, Domenici G, Barreto C, Matos C, Lemos AR, Matias AT, Kucheryava K, et al. Development of Dl1.72, a Novel Anti-DLL1 Antibody with Anti-Tumor Efficacy against Estrogen Receptor-Positive Breast Cancer. Cancers. 2021; 13(16):4074. https://doi.org/10.3390/cancers13164074
Chicago/Turabian StyleSilva, Gabriela, Joana Sales-Dias, Diogo Casal, Sara Alves, Giacomo Domenici, Clara Barreto, Carolina Matos, Ana R. Lemos, Ana T. Matias, Khrystyna Kucheryava, and et al. 2021. "Development of Dl1.72, a Novel Anti-DLL1 Antibody with Anti-Tumor Efficacy against Estrogen Receptor-Positive Breast Cancer" Cancers 13, no. 16: 4074. https://doi.org/10.3390/cancers13164074
APA StyleSilva, G., Sales-Dias, J., Casal, D., Alves, S., Domenici, G., Barreto, C., Matos, C., Lemos, A. R., Matias, A. T., Kucheryava, K., Ferreira, A., Moita, M. R., Braga, S., Brito, C., Cabral, M. G., Casalou, C., Barral, D. C., Sousa, P. M. F., Videira, P. A., ... Barbas, A. (2021). Development of Dl1.72, a Novel Anti-DLL1 Antibody with Anti-Tumor Efficacy against Estrogen Receptor-Positive Breast Cancer. Cancers, 13(16), 4074. https://doi.org/10.3390/cancers13164074