
Epigenetic Alterations Upstream and Downstream of p53 Signaling in Colorectal Carcinoma



Abstract
Simple Summary
Abstract
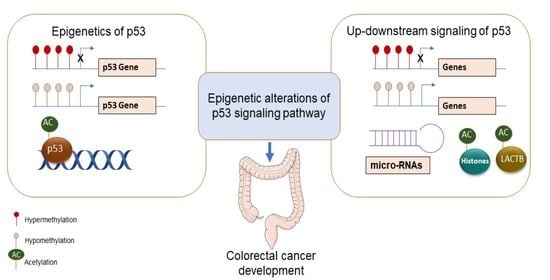
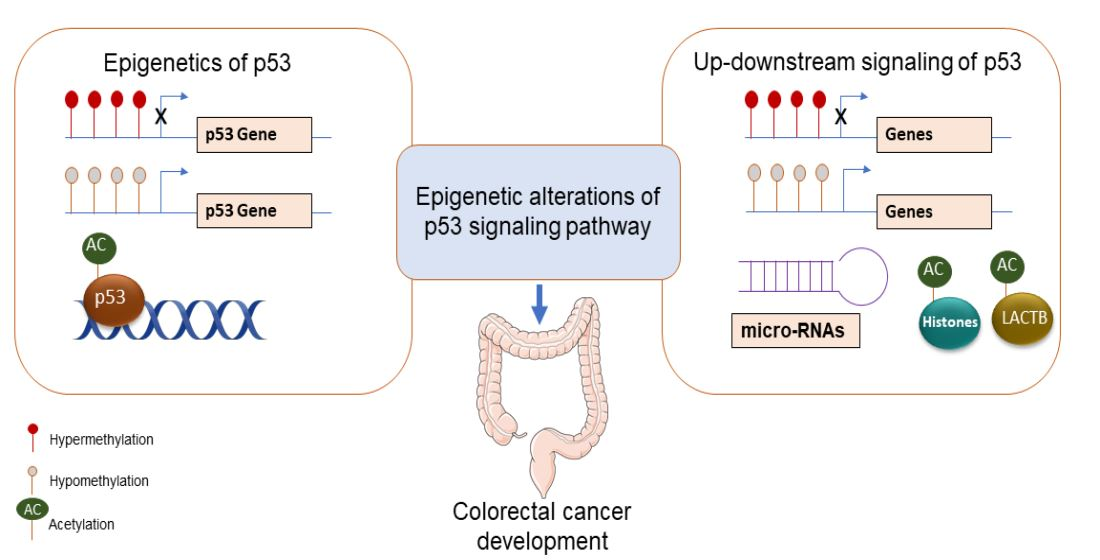
1. Introduction
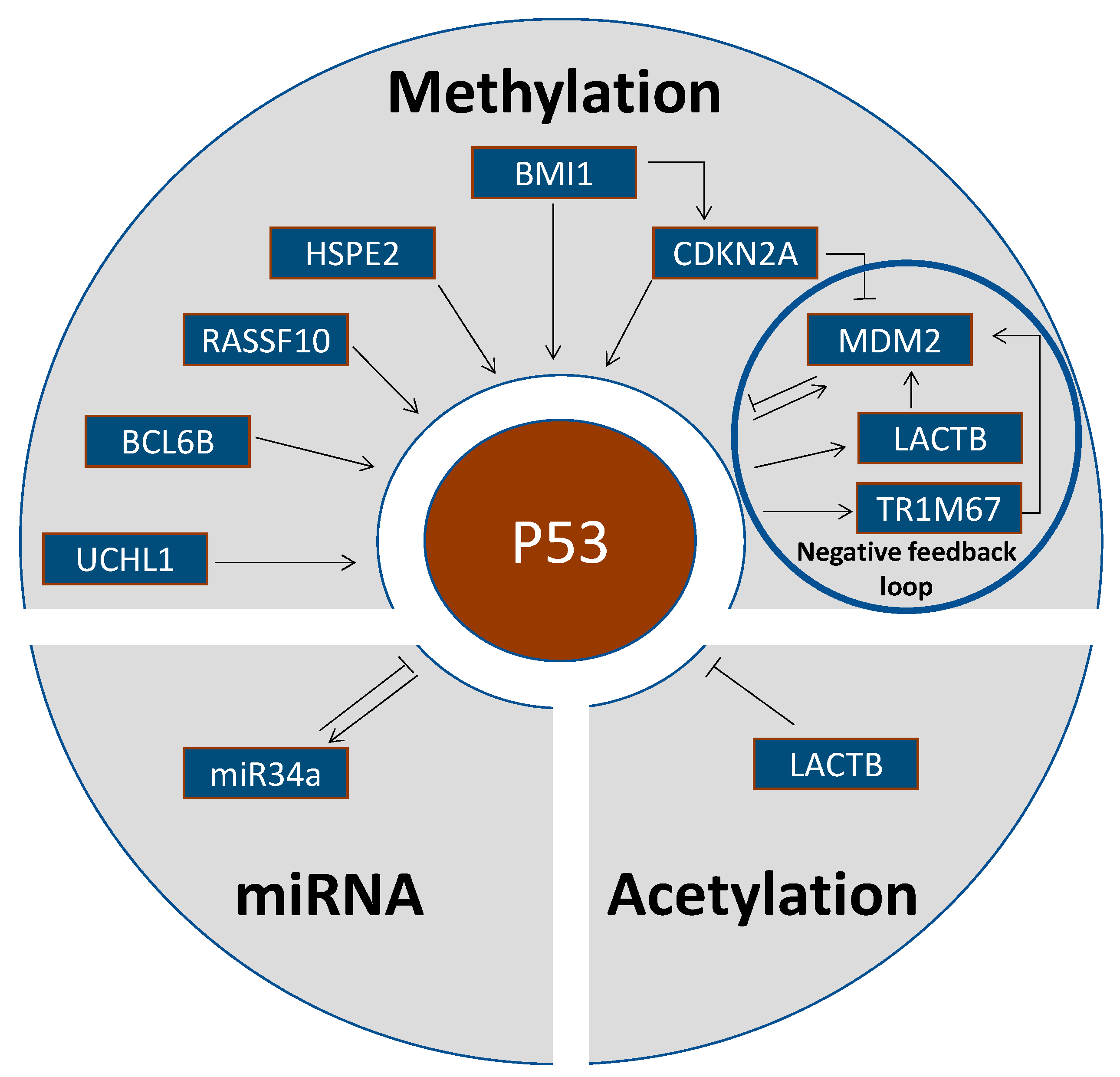
2. Epigenetic Alterations Upstream of p53
2.1. Methylation Status of p53-Activating Genes
2.2. Methylation Status of p53-Inhibiting Genes
2.3. Methylation Status of Other Genes
2.4. Acetylation Status of p53-Inhibiting Genes
2.5. P53 Regulation by Micro-RNAs

{kind=link}
{kind=link}
{kind=link}
| Gene Name | Epigenetic Event | Cellular Function | Reference |
|---|---|---|---|
| Lactamase β (LACTB) | LACTB promoter methylation and histone deacetylation | Activation of CRC cell proliferation, migration, and invasion in HCT116 and HCT8 cells. Inhibited CRC xenograft growth and metastasis in vivo | Zeng et al., 2018 [50] |
| Deregulation of P53 | Abnormalities of miRNA expression | p53 effects (e.g., cell cycle arrest and apoptosis) were phenocopied in HCT-116 cells | Nugent et al., 2011 [76] |
| O6-methylguanine DNA methyltransferase | Aberrant methylation of APC, MGMT, hMLH1, P16, N33 | High levels of microsatellite instability in 208 CRC patients | Suehiro et al., 2008 [43] |
| Inhibitor of growth 2 (ING2) | ING2 is a cofactor of p300 for p53 acetylation | Positive regulation of p53-mediated replicative senescence in young fibroblasts | Pedeux, et al., 2020 [77] |
| NAD+-dependent protein deacetylase SIRT1 | Deacetylation of p53 | Decrease of cell proliferation and induction of apoptosis in vivo and in CRC patients | Stünkel et al., 2007 [78] |
3. Epigenetic Alterations at p53 as Target Site
3.1. Methylation of the TP53 Gene
3.2. Acetylation of p53
| Gene | Gene Name | Gene Function | Epigenetic Event | Reference |
|---|---|---|---|---|
| TP53 | Tumor suppressor 53 | Tumor suppressor | Methylation inside and outside of exonic CG sequences was correlated with point mutations. Cigarette smoking increased the occurrence of methylation-associated mutations. | Kouidou et al., 2006 [79] |
| TP53 | DNA methylation, histone modifications, chromatin remodeling, and non-coding RNAs were significantly associated with colitis-related carcinogenesis and tumor progression. | Saraggi et al., 2017 [80] | ||
| POLE | Polymerase ε | Involved in DNA repair and chromosomal DNA replication. Methylated cytosines (5mCs) are frequently mutated. POLE exonuclease domain mutations increase 5mC mutagenesis and a mutator phenotype. | Highly methylated CpGs in TP53 contain mutation hotspots in POLE-mutated CRC | Poulos et al., 2017 [82] |
| PHF2 | PHD finger protein 2 | Tumor suppressor. Histone demethylase. PHF2 demethylates the repressive H3K9-Me2 mark in chromatin to induce p53 transcription. | PHF2 was downregulated in CRC and PHF2 correlated with p21 in cancers expressing functional p53. | Lee et al., 2015 [83] |
| LSD1 | LSD1demethylated K370me2 (dimethylated Lys370) of p53, which cannot bind to DNA and is inactivated. LSD1 inhibits p53-mediated apoptosis. | Huang et al., 2007 [84] | ||
| LSD1 | Histone lysine-specific demethylase 1 | Lysine-specific demethylases that removes histone methylations catalyzed by histone methyltransferases. | LSD1-knockout in HCT116 cells did not increase H3K4me2 (dimethyl-H3K4) or change stability or function of p53. | Jin et al., 2013 [85] |
| LSD2/KDM1B | Lysine-specific histone demethylase 1B | Oncogene. LSD2 directly binds to TP53 and represses p53 expression via H3K4me2 demethylation at the TP53 promoter. | LSD2 bound and inhibited p53 activity in CRC. | Cai et al., 2020 [86] |
| NRDC | Nardilysin, N-arginine dibasic convertase | NRDC is a dimethyl-H3K4 binding protein. | Nuclear NRDC bound to histone deacetylase 1 (HDAC1) and inhibited HDAC1 recruitment to the TP53 promoter and p53 acetylation. | Li et al., 2012; Kanda et al., 2018 [87,88] |
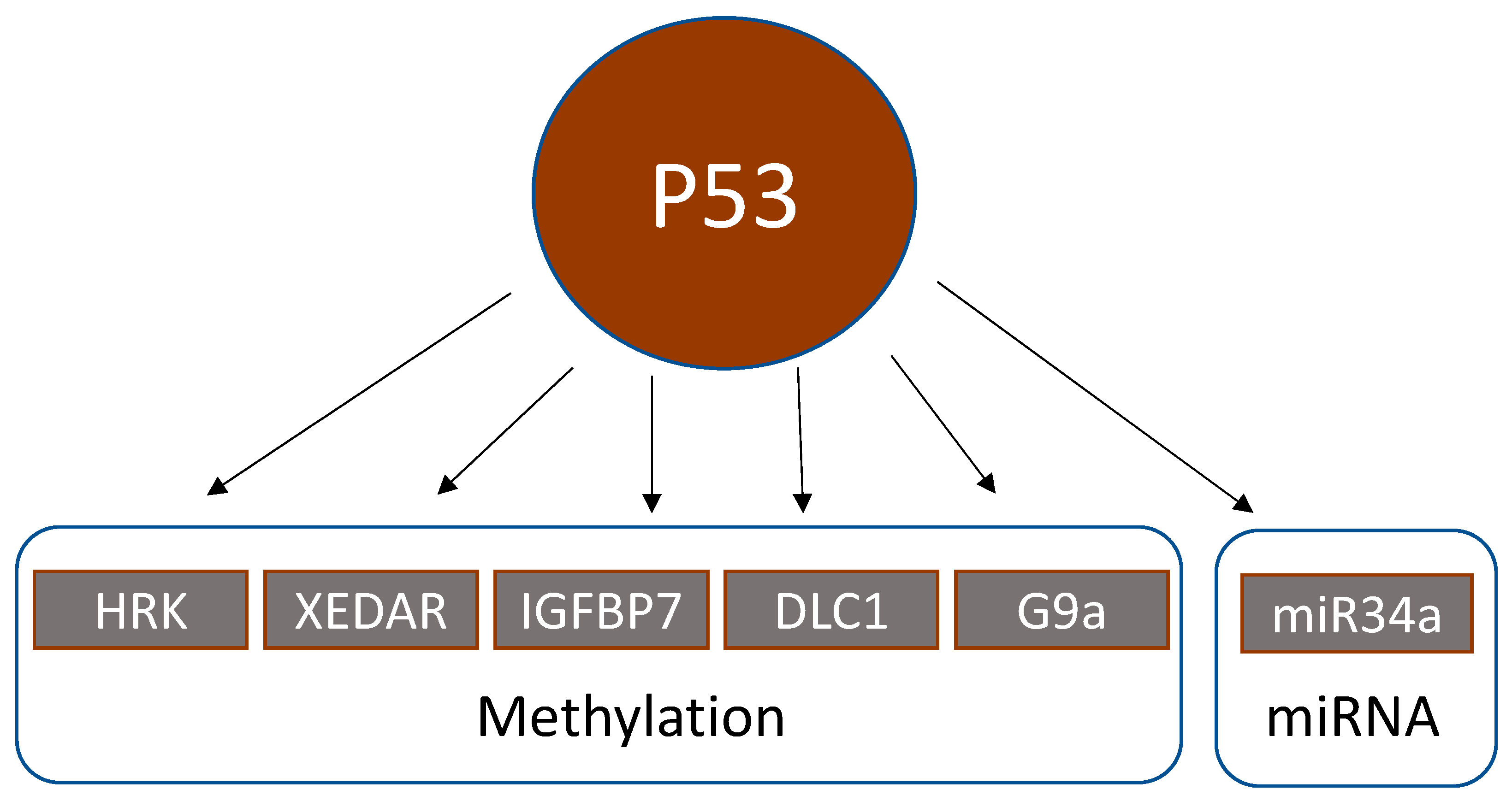
4. Epigenetic Alterations Downstream of p53
4.1. Methylation Status of p53 Downstream Genes
4.2. Regulation of p53 by Acetylation
4.3. Regulation of Micro-RNAs by p53
| Gene | Gene Name | Gene Function | Epigenetic Event | Reference |
|---|---|---|---|---|
| HRK | BCL-2-interacting protein | Proapoptotic p53 target protein, whose expression is reversible by methylation inhibitors (5-aza-deoxycytidine) and further enhanced by histone deacetylase inhibitors (trichostatin A, depsipeptide) | HRK promoter methylation significantly correlated with wildtype p53 in 58 CRC. | Obata et al., 2003 [89] |
| XEDAR | X-linked ectodermal dysplasia receptor | XEDAR is a member of the tumor necrosis factor receptor family. P53 upregulates XEDAR expression through two p53-binding sites within intron 1 of the XEDAR gene. Inactivation of XEDAR results in enhanced cell adhesion and spreading, and resistance to p53-induced apoptosis. | XEDAR downregulation by hypermethylation or TP53 mutations in CRC cell lines and biopsies | Tanikawa et al., 2009 [90] |
| IGFBP7 | Insulin-like growth factor-binding protein 7 | P53 induces expression of IGFBP7 upon binding to a p53 response element in intron 1 of IGFBP7. | IGFBP7 methylation was significantly higher in 83 CRC biopsies than in normal colonic tissue. 5-Aza-2′-deoxycytidine restored p53-induced IGFBP7 expression. IGFBP7 methylation significantly correlated with wildtype p53. | Suzuki et al., 2010 [91] |
| DLC1 | Deleted in liver cancer 1 | Tumor suppressor. The functional DLC1-i4 promoter is activated by wild-type p53. | DLC1 isoform 4 (DLC1-i4) was methylated in 2/4 CRC cell lines (=50%). Demethylation with 5-aza-2′-deoxycytidine restored DLC-i4 expression. | Low et al., 2011 [92] |
| G9a/KMT1C/EHMT2 | Lysine methyltransferase 1C, euchromatic histone methyltransferase 2 | Histone methyltransferase that catalyzes mono- and dimethylation of histone H3K9 | G9a dimethylated p53 at lysine 373. This transactivated PLK1 expression and increased proliferation | Zhang et al., 2018 [93] |
| miR-34b/c | Micro-RNA 34b/c | p53 activates the transcription of miR-34 family members. The miR-34b/c CpG island is a bidirectional promoter, regulating the expression of both miR-34b/c and B-cell translocation gene 4 (BTG4). Methylation of this promoter silenced BTG4 transcription. | miR-34b/c CpG island methylation in 100/111 (=90%) CRC | Toyota et al., 2008; Navarro and Lieberman, 2015 [98,99] |
5. Epigenetic Alterations and Clinical Outcome upon Standard Chemotherapy of CRC
6. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Gayon, J. From Mendel to epigenetics: History of genetics. C. R. Biol. 2016, 339, 225–230. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G.P. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Edwards, J.R.; Yarychkivska, O.; Boulard, M.; Bestor, T.H. DNA methylation and DNA methyltransferases. Epigenet. Chromatin 2017, 10, 23. [Google Scholar] [CrossRef]
- Fatemi, M.; Pao, M.M.; Jeong, S.; Gal-Yam, E.N.; Egger, G.; Weisenberger, D.J.; Jones, P.A. Footprinting of mammalian promoters: Use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005, 33, e176. [Google Scholar] [CrossRef]
- Chatterjee, R.; Vinson, C. CpG methylation recruits sequence specific transcription factors essential for tissue specific gene expression. BBA-Gene Regul. Mech. 2012, 1819, 763–770. [Google Scholar] [CrossRef]
- Muller, F.; Tora, L. Chromatin and DNA sequences in defining promoters for transcription initiation. BBA-Gene Regul. Mech. 2014, 1839, 118–128. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.N.; Ma, X.L.; Mo, F.; Yang, S.Y.; Han, J.H.; Wei, X.W. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. CSH Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA Translation and Stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef]
- Kanwal, R.; Gupta, K.; Gupta, S. Cancer Epigenetics: An Introduction. Methods Mol. Biol. 2015, 1238, 3–25. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2019, 11, 267–276. [Google Scholar] [CrossRef]
- Raskov, H.; Soby, J.H.; Troelsen, J.; Bojesen, R.D.; Gogenur, I. Driver Gene Mutations and Epigenetics in Colorectal Cancer. Ann. Surg. 2020, 271, 75–85. [Google Scholar] [CrossRef] [PubMed]
- De’ Angelis, G.L.; Bottarelli, L.; Azzoni, C.; De’ Angelis, N.; Leandro, G.; Di Mario, F.; Gaiani, F.; Negri, F. Microsatellite instability in colorectal cancer. Acta Bio-Med. Atenei Parm. 2018, 89, 97–101. [Google Scholar] [CrossRef]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef]
- Toyota, M.; Suzuki, H. Epigenetic drivers of genetic alterations. Adv. Genet. 2010, 70, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef]
- Tessitore, A.; Cicciarelli, G.; Del Vecchio, F.; Gaggiano, A.; Verzella, D.; Fischietti, M.; Vecchiotti, D.; Capece, D.; Zazzeroni, F.; Alesse, E. MicroRNAs in the DNA Damage/Repair Network and Cancer. Int. J. Genom. 2014, 2014, 820248. [Google Scholar] [CrossRef]
- Wang, Y.P.; Lei, Q.Y. Metabolic recoding of epigenetics in cancer. Cancer Commun. 2018, 38, 25. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.J.; Min, B.H.; Ryu, K.J.; Kim, K.M.; Chang, D.K.; Kim, J.J.; Rhee, J.C.; Kim, Y.H. The role of the CpG island methylator phenotype on survival outcome in colon cancer. Gut Liver 2015, 9, 202–207. [Google Scholar] [CrossRef][Green Version]
- Advani, S.M.; Advani, P.; DeSantis, S.M.; Brown, D.; VonVille, H.M.; Lam, M.; Loree, J.M.; Mehrvarz Sarshekeh, A.; Bressler, J.; Lopez, D.S.; et al. Clinical, Pathological, and Molecular Characteristics of CpG Island Methylator Phenotype in Colorectal Cancer: A Systematic Review and Meta-analysis. Transl. Oncol. 2018, 11, 1188–1201. [Google Scholar] [CrossRef]
- Cha, Y.; Kim, S.Y.; Yeo, H.Y.; Baek, J.Y.; Choi, M.K.; Jung, K.H.; Dong, S.M.; Chang, H.J. Association of CHFR Promoter Methylation with Treatment Outcomes of Irinotecan-Based Chemotherapy in Metastatic Colorectal Cancer. Neoplasia 2019, 21, 146–155. [Google Scholar] [CrossRef]
- Yagi, K.; Akagi, K.; Hayashi, H.; Nagae, G.; Tsuji, S.; Isagawa, T.; Midorikawa, Y.; Nishimura, Y.; Sakamoto, H.; Seto, Y.; et al. Three DNA methylation epigenotypes in human colorectal cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 21–33. [Google Scholar] [CrossRef]
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007, 50, 113–130. [Google Scholar] [CrossRef]
- Yu, J.; Tao, Q.; Cheung, K.F.; Jin, H.; Poon, F.F.; Wang, X.; Li, H.Y.; Cheng, Y.Y.; Rocken, C.; Ebert, M.P.A.; et al. Epigenetic identification of ubiquitin carboxyl-terminal hydrolase L1 as a functional tumor suppressor and biomarker for hepatocellular carcinoma and other digestive tumors. Hepatology 2008, 48, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Cao, B.P.; Zhang, M.Y.; Linghu, E.Q.; Zhan, Q.M.; Brock, M.V.; Herman, J.G.; Mao, G.P.; Guo, M.Z. Epigenetic silencing BCL6B induced colorectal cancer proliferation and metastasis by inhibiting P53 signaling. Am. J. Cancer Res. 2015, 5, 651–662. [Google Scholar] [PubMed]
- Jin, Y.; Cao, B.; Zhang, M.; Zhan, Q.; Herman, J.G.; Yu, M.; Guo, M. RASSF10 suppresses hepatocellular carcinoma growth by activating P53 signaling and methylation of RASSF10 is a docetaxel resistant marker. Genes Cancer 2015, 6, 231–240. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, C.X.; Shi, C.; Zhang, J.Y.; Qian, T.; Wang, Z.; Ma, R.; Wu, J.Z.; Jiang, F.; Feng, J.F. Hypermethylation of heparanase 2 promotes colorectal cancer proliferation and is associated with poor prognosis. J. Transl. Med. 2021, 19, 98. [Google Scholar] [CrossRef] [PubMed]
- Maynard, M.A.; Ferretti, R.; Hilgendorf, K.I.; Perret, C.; Whyte, P.; Lees, J.A. Bmi1 is required for tumorigenesis in a mouse model of intestinal cancer. Oncogene 2014, 33, 3742–3747. [Google Scholar] [CrossRef]
- Esteller, M.; Tortola, S.; Toyota, M.; Capella, G.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 2000, 60, 129–133. [Google Scholar]
- Esteller, M.; Cordon-Cardo, C.; Corn, P.G.; Meltzer, S.J.; Pohar, K.S.; Watkins, D.N.; Capella, G.; Peinado, M.A.; Matias-Guiu, X.; Prat, J.; et al. p14(ARF) silencing by promoter hypermethylation mediates abnormal intracellular localization of MDM2. Cancer Res. 2001, 61, 2816–2821. [Google Scholar] [PubMed]
- Nyiraneza, C.; Sempoux, C.; Detry, R.; Kartheuser, A.; Dahan, K. Hypermethylation of the 5′ CpG island of the p14ARF flanking exon 1beta in human colorectal cancer displaying a restricted pattern of p53 overexpression concomitant with increased MDM2 expression. Clin. Epigenet. 2012, 4, 9. [Google Scholar] [CrossRef]
- Suehiro, Y.; Wong, C.W.; Chirieac, L.R.; Kondo, Y.; Shen, L.; Webb, C.R.; Chan, Y.W.; Chan, A.S.; Chan, T.L.; Wu, T.T.; et al. Epigenetic-genetic interactions in the APC/WNT, RAS/RAF, and P53 pathways in colorectal carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 2560–2569. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stamos, J.L.; Weis, W.I. The beta-Catenin Destruction Complex. CSH Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Esteller, M.; Risques, R.A.; Toyota, M.; Capella, G.; Moreno, V.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Promoter hypermethylation of the DNA repair gene O-6-methylguanine-DNA methyltransferase is associated with the presence of G:C to A: T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res. 2001, 61, 4689–4692. [Google Scholar] [PubMed]
- Deng, G.; Kakar, S.; Tanaka, H.; Matsuzaki, K.; Miura, S.; Sleisenger, M.H.; Kim, Y.S. Proximal and distal colorectal cancers show distinct gene-specific methylation profiles and clinical and molecular characteristics. Eur. J. Cancer 2008, 44, 1290–1301. [Google Scholar] [CrossRef]
- Alonso, S.; Dai, Y.C.; Yamashita, K.; Horiuchi, S.; Dai, T.; Matsunaga, A.; Sanchez-Munoz, R.; Bilbao-Sieyro, C.; Daz-Chico, J.C.; Chernov, A.V.; et al. Methylation of MGMT and ADAMTS14 in normal colon mucosa: Biomarkers of a field defect for cancerization preferentially targeting elder African-Americans. Oncotarget 2015, 6, 3420–3431. [Google Scholar] [CrossRef] [PubMed]
- Bonavita, E.; Gentile, S.; Rubino, M.; Maina, V.; Papait, R.; Kunderfranco, P.; Greco, C.; Feruglio, F.; Molgora, M.; Laface, I.; et al. PTX3 Is an Extrinsic Oncosuppressor Regulating Complement-Dependent Inflammation in Cancer. Cell 2015, 160, 700–714. [Google Scholar] [CrossRef]
- Liu, T.; Wang, X.; Hu, W.Q.; Fang, Z.; Jin, Y.; Fang, X.D.; Miao, Q.R. Epigenetically Down-Regulated Acetyltransferase PCAF Increases the Resistance of Colorectal Cancer to 5-Fluorouracil. Neoplasia 2019, 21, 557–570. [Google Scholar] [CrossRef]
- Zeng, K.X.; Chen, X.X.; Hu, X.X.; Liu, X.X.; Xu, T.; Sun, H.L.; Pan, Y.Q.; He, B.S.; Wang, S.K. LACTB, a novel epigenetic silenced tumor suppressor, inhibits colorectal cancer progression by attenuating MDM2-mediated p53 ubiquitination and degradation. Oncogene 2018, 37, 5534–5551. [Google Scholar] [CrossRef]
- Wang, S.Y.; Zhang, Y.Q.; Huang, J.Z.; Wong, C.C.; Zhai, J.N.; Li, C.G.; Wei, G.F.; Zhao, L.Y.; Wang, G.P.; Wei, H.; et al. TRIM67 Activates p53 to Suppress Colorectal Cancer Initiation and Progression. Cancer Res. 2019, 79, 4086–4098. [Google Scholar] [CrossRef]
- Toyota, M.; Shen, L.; Ohe-Toyota, M.; Hamilton, S.R.; Sinicrope, F.A.; Issa, J.P.J. Aberrant methylation of the Cyclooxygenase 2 CpG island in colorectal tumors. Cancer Res. 2000, 60, 4044–4048. [Google Scholar]
- Toyota, M.; Ohe-Toyota, M.; Ahuja, N.; Issa, J.P.J. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc. Natl. Acad. Sci. USA 2000, 97, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Konishi, K.; Issa, J.P.J. Targeting aberrant chromatin structure in colorectal carcinomas. Cancer J. 2007, 13, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Kaller, M.; Liffers, S.T.; Oeljeklaus, S.; Kuhlmann, K.; Roh, S.; Hoffmann, R.; Warscheid, B.; Hermeking, H. Genome-wide Characterization of miR-34a Induced Changes in Protein and mRNA Expression by a Combined Pulsed SILAC and Microarray Analysis. Mol. Cell. Proteom. 2011, 10, M111.010462. [Google Scholar] [CrossRef]
- Vogt, M.; Munding, J.; Gruner, M.; Liffers, S.T.; Verdoodt, B.; Hauk, J.; Steinstraesser, L.; Tannapfel, A.; Hermeking, H. Frequent concomitant inactivation of miR-34a and miR-34b/c by CpG methylation in colorectal, pancreatic, mammary, ovarian, urothelial, and renal cell carcinomas and soft tissue sarcomas. Virchows Arch. 2011, 458, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Zhao, Y.; Feng, Z. MicroRNA Control of p53. J. Cell. Biochem. 2017, 118, 7–14. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Wang, X.; Wu, R.; Lin, M.; Laddha, S.V.; Yang, Q.; Chan, C.S.; Feng, Z. MicroRNA-339-5p inhibits colorectal tumorigenesis through regulation of the MDM2/p53 signaling. Oncotarget 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Noorolyai, S.; Baghbani, E.; Maleki, L.A.; Kojabad, A.B.; Shanehbandi, D.; Shahgoli, V.K.; Mokhtarzadeh, A.; Baradaran, B. Restoration of miR-193a-5p and miR-146 a-5p Expression Induces G1 Arrest in Colorectal Cancer through Targeting of MDM2/p53. Adv. Pharm. Bull. 2020, 10, 130–134. [Google Scholar] [CrossRef]
- Chen, X.X.; Zeng, K.X.; Xu, M.; Liu, X.X.; Hu, X.X.; Xu, T.; He, B.S.; Pan, Y.Q.; Sun, H.L.; Wang, S.K. P53-induced miR-1249 inhibits tumor growth, metastasis, and angiogenesis by targeting VEGFA and HMGA2. Cell Death Dis. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.J.; Zhao, J.H.; Wu, C.W.; Zhang, L.J.; Liu, X.D.; Kang, W.; Leung, W.W.; Zhang, N.; Chan, F.K.L.; Sung, J.J.Y.; et al. Tumor Suppressor Functions of miR-133a in Colorectal Cancer. Mol. Cancer Res. 2013, 11, 1051–1060. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Kaller, M.; Hermeking, H. Repression of c-Kit by p53 is mediated by miR-34 and is associated with reduced chemoresistance, migration and stemness. Oncotarget 2013, 4, 1399–1415. [Google Scholar] [CrossRef]
- Shi, X.L.; Kaller, M.; Rokavec, M.; Kirchner, T.; Horst, D.; Hermeking, H. Characterization of a p53/miR-34a/CSF1R/STAT3 Feedback Loop in Colorectal Cancer. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 391–418. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Jackstadt, R.; Siemens, H.; Li, H.H.; Kirchner, T.; Hermeking, H. p53-induced miR-15a/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2014, 74, 532–542. [Google Scholar] [CrossRef]
- Ma, Q.Y.; Wang, X.Y.; Li, Z.; Li, B.S.; Ma, F.L.; Peng, L.; Zhang, Y.; Xu, A.G.; Jiang, B. microRNA-16 represses colorectal cancer cell growth in vitro by regulating the p53/survivin signaling pathway. Oncol. Rep. 2013, 29, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Dashzeveg, N.; Yoshida, K. Tumor suppressor p53 induces miR-1915 processing to inhibit Bcl-2 in the apoptotic response to DNA damage. FEBS J. 2014, 281, 2937–2944. [Google Scholar] [CrossRef]
- Wang, G.; Cao, X.; Lai, S.; Luo, X.; Feng, Y.; Wu, J.; Ning, Q.; Xia, X.; Wang, J.; Gong, J.; et al. Altered p53 regulation of miR-148b and p55PIK contributes to tumor progression in colorectal cancer. Oncogene 2015, 34, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, A.; Valvo, C.; Fabrizi, E.; di Martino, S.; Biffoni, M.; Runci, D.; Forte, S.; De Maria, R.; Ricci-Vitiani, L. Analysis of the combined action of miR-143 and miR-145 on oncogenic pathways in colorectal cancer cells reveals a coordinate program of gene repression. Oncogene 2013, 32, 4806–4813. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Mullany, L.E.; Wolff, R.K.; Sakoda, L.C.; Samowitz, W.S.; Herrick, J.S. The p53-signaling pathway and colorectal cancer: Interactions between downstream p53 target genes and miRNAs. Genomics 2019, 111, 762–771. [Google Scholar] [CrossRef]
- Laudato, S.; Patil, N.; Abba, M.L.; Leupold, J.H.; Benner, A.; Gaiser, T.; Marx, A.; Allgayer, H. P53-induced miR-30e-5p inhibits colorectal cancer invasion and metastasis by targeting ITGA6 and ITGB1. Int. J. Cancer 2017, 141, 1879–1890. [Google Scholar] [CrossRef]
- Zhang, P.L.; Zuo, Z.G.; Wu, A.H.; Shang, W.J.; Bi, R.C.; Jin, Q.K.; Wu, J.B.; Jiang, L. miR-600 inhibits cell proliferation, migration and invasion by targeting p53 in mutant p53-expressing human colorectal cancer cell lines. Oncol. Lett. 2017, 13, 1789–1796. [Google Scholar] [CrossRef]
- Kwak, B.; Kim, D.U.; Kim, T.O.; Kim, H.S.; Kim, S.W. MicroRNA-552 links Wnt signaling to p53 tumor suppressor in colorectal cancer. Int. J. Oncol. 2018, 53, 1800–1808. [Google Scholar] [CrossRef]
- Maqbool, R.; Lone, S.N.; Ul Hussain, M. Post-transcriptional regulation of the tumor suppressor p53 by a novel miR-27a, with implications during hypoxia and tumorigenesis. Biochem. J. 2016, 473, 3597–3610. [Google Scholar] [CrossRef]
- Wang, L.; Yu, P.W. miR-300 promotes proliferation and EMT-mediated colorectal cancer migration and invasion by targeting p53. Oncol. Rep. 2016, 36, 3225–3232. [Google Scholar] [CrossRef]
- Liu, F.; Wang, X.D. miR-150-5p represses TP53 tumor suppressor gene to promote proliferation of colon adenocarcinoma. Sci. Rep. 2019, 9, 6740. [Google Scholar] [CrossRef]
- Nugent, M.; Miller, N.; Kerin, M.J. MicroRNAs in colorectal cancer: Function, dysregulation and potential as novel biomarkers. Ejso 2011, 37, 649–654. [Google Scholar] [CrossRef]
- Pedeux, R.; Sengupta, S.; Shen, J.C.; Demidov, O.N.; Saito, S.; Onogi, H.; Kumamoto, K.; Wincovitch, S.; Garfield, S.H.; McMenamin, M.; et al. ING2 regulates the onset of replicative senescence by induction of p300-dependent p53 acetylation. Mol. Cell. Biol. 2005, 25, 6639–6648. [Google Scholar] [CrossRef] [PubMed]
- Stunkel, W.; Peh, B.K.; Tan, Y.C.; Nayagam, V.M.; Wang, X.; Salto-Tellez, M.; Ni, B.; Entzeroth, M.; Wood, J. Function of the SIRT1 protein deacetylase in cancer. Biotechnol. J. 2007, 2, 1360–1368. [Google Scholar] [CrossRef]
- Kouidou, S.; Malousi, A.; Maglaveras, N. Methylation and repeats in silent and nonsense mutations of p53. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 2006, 599, 167–177. [Google Scholar] [CrossRef]
- Saraggi, D.; Fassan, M.; Mescoli, C.; Scarpa, M.; Valeri, N.; Michielan, A.; D’Inca, R.; Rugge, M. The molecular landscape of colitis-associated carcinogenesis. Dig. Liver Dis. 2017, 49, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wei, L.; Devbhandari, S.; Zhang, T.; Xiang, J.; Remus, D.; Zhao, X. DNA polymerase epsilon relies on a unique domain for efficient replisome assembly and strand synthesis. Nat. Commun. 2020, 11, 2437. [Google Scholar] [CrossRef]
- Poulos, R.C.; Olivier, J.; Wong, J.W.H. The interaction between cytosine methylation and processes of DNA replication and repair shape the mutational landscape of cancer genomes. Nucleic Acids Res. 2017, 45, 7786–7795. [Google Scholar] [CrossRef]
- Lee, K.H.; Park, J.W.; Sung, H.S.; Choi, Y.J.; Kim, W.H.; Lee, H.S.; Chung, H.J.; Shin, H.W.; Cho, C.H.; Kim, T.Y.; et al. PHF2 histone demethylase acts as a tumor suppressor in association with p53 in cancer. Oncogene 2015, 34, 2897–2909. [Google Scholar] [CrossRef]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.H.; Hanigan, C.L.; Wu, Y.; Wang, W.; Park, B.H.; Wosterf, P.M.; Casero, R.A. Loss of LSD1 (lysine-specific demethylase 1) suppresses growth and alters gene expression of human colon cancer cells in a p53- and DNMT1 (DNA methyltransferase 1)-independent manner. Biochem. J. 2013, 449, 459–468. [Google Scholar] [CrossRef]
- Cai, S.X.; Wang, J.S.; Zeng, W.; Cheng, X.F.; Liu, L.H.; Li, W.H. Lysine-specific histone demethylase 1B (LSD2/KDM1B) represses p53 expression to promote proliferation and inhibit apoptosis in colorectal cancer through LSD2-mediated H3K4me2 demethylation. Aging 2020, 12, 14990–15001. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chu, M.Y.; Wang, S.S.; Chan, D.; Qi, S.K.; Wu, M.; Zhou, Z.L.; Li, J.W.; Nishi, E.; Qin, J.; et al. Identification and Characterization of Nardilysin as a Novel Dimethyl H3K4-binding Protein Involved in Transcriptional Regulation. J. Biol. Chem. 2012, 287, 10089–10098. [Google Scholar] [CrossRef] [PubMed]
- Kanda, K.; Sakamoto, J.; Matsumoto, Y.; Ikuta, K.; Goto, N.; Morita, Y.; Ohno, M.; Nishi, K.; Eto, K.; Kimura, Y.; et al. Nardilysin controls intestinal tumorigenesis through HDAC1/p53-dependent transcriptional regulation. JCI Insight 2018, 3, e91316. [Google Scholar] [CrossRef]
- Obata, T.; Toyota, M.; Satoh, A.; Sasaki, Y.; Ogi, K.; Akino, K.; Suzuki, H.; Murai, M.; Kikuchi, T.; Mita, H.; et al. Identification of HRK as a target of epigenetic inactivation in colorectal and gastric cancer. Clin. Cancer Res. 2003, 9, 6410–6418. [Google Scholar]
- Tanikawa, C.; Furukawa, Y.; Yoshida, N.; Arakawa, H.; Nakamura, Y.; Matsuda, K. XEDAR as a putative colorectal tumor suppressor that mediates p53-regulated anoikis pathway. Oncogene 2009, 28, 3081–3092. [Google Scholar] [CrossRef]
- Suzuki, H.; Igarashi, S.; Nojima, M.; Maruyama, R.; Yamamoto, E.; Kai, M.; Akashi, H.; Watanabe, Y.; Yamamoto, H.; Sasaki, Y.; et al. IGFBP7 is a p53-responsive gene specifically silenced in colorectal cancer with CpG island methylator phenotype. Carcinogenesis 2010, 31, 342–349. [Google Scholar] [CrossRef]
- Low, J.S.W.; Tao, Q.; Ng, K.M.; Goh, H.K.; Shu, X.S.; Woo, W.L.; Ambinder, R.F.; Srivastava, G.; Shamay, M.; Chan, A.T.C.; et al. A novel isoform of the 8p22 tumor suppressor gene DLC1 suppresses tumor growth and is frequently silenced in multiple common tumors. Oncogene 2011, 30, 1923–1935. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.F.; Shen, Y.Y.; He, P.X.; Ding, J.; Chen, Y. G9a stimulates CRC growth by inducing p53 Lys373 dimethylation-dependent activation of Plk1. Theranostics 2018, 8, 2884–2895. [Google Scholar] [CrossRef]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.; Lieberman, J. miR-34 and p53: New Insights into a Complex Functional Relationship. PLoS ONE 2015, 10, e0132767. [Google Scholar] [CrossRef]
- Toyota, M.; Suzuki, H.; Sasaki, Y.; Maruyama, R.; Imai, K.; Shinomura, Y.; Tokino, T. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 2008, 68, 4123–4132. [Google Scholar] [CrossRef]
- Puccini, A.; Berger, M.D.; Naseem, M.; Tokunaga, R.; Battaglin, F.; Cao, S.; Hanna, D.L.; McSkane, M.; Soni, S.; Zhang, W.; et al. Colorectal cancer: Epigenetic alterations and their clinical implications. BBA-Rev. Cancer 2017, 1868, 439–448. [Google Scholar] [CrossRef]
- Grady, W.M.; Yu, M.; Markowitz, S.D. Epigenetic Alterations in the Gastrointestinal Tract: Current and Emerging Use for Biomarkers of Cancer. Gastroenterology 2021, 160, 690–709. [Google Scholar] [CrossRef]
- Barchitta, M.; Maugeri, A.; Li Destri, G.; Basile, G.; Agodi, A. Epigenetic Biomarkers in Colorectal Cancer Patients Receiving Adjuvant or Neoadjuvant Therapy: A Systematic Review of Epidemiological Studies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef]
- Lam, K.; Pan, K.; Linnekamp, J.F.; Medema, J.P.; Kandimalla, R. DNA methylation based biomarkers in colorectal cancer: A systematic review. Biochim. Biophys. Acta 2016, 1866, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Okugawa, Y.; Grady, W.M.; Goel, A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology 2015, 149, 1204–1225.e12. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Casanova, A.; Costa-Fraga, N.; Bao-Caamano, A.; Lopez-Lopez, R.; Muinelo-Romay, L.; Diaz-Lagares, A. Epigenetic Landscape of Liquid Biopsy in Colorectal Cancer. Front. Cell Dev. Biol. 2021, 9, 622459. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.A.; Wu, C.; Yu, M.; Gourgioti, G.; Wirtz, R.; Raptou, G.; Gkakou, C.; Kotoula, V.; Pentheroudakis, G.; Papaxoinis, G.; et al. Evaluation of CpG Island Methylator Phenotype as a Biomarker in Colorectal Cancer Treated with Adjuvant Oxaliplatin. Clin. Colorectal Cancer 2016, 15, 164–169. [Google Scholar] [CrossRef][Green Version]
- Gallois, C.; Taieb, J.; Le Corre, D.; Le Malicot, K.; Tabernero, J.; Mulot, C.; Seitz, J.F.; Aparicio, T.; Folprecht, G.; Lepage, C.; et al. Prognostic Value of Methylator Phenotype in Stage III Colon Cancer Treated with Oxaliplatin-Based Adjuvant Chemotherapy. Clin. Cancer Res. 2018, 24, 4745–4753. [Google Scholar] [CrossRef]
- Cha, Y.; Kim, K.J.; Han, S.W.; Rhee, Y.Y.; Bae, J.M.; Wen, X.; Cho, N.Y.; Lee, D.W.; Lee, K.H.; Kim, T.Y.; et al. Adverse prognostic impact of the CpG island methylator phenotype in metastatic colorectal cancer. Br. J. Cancer 2016, 115, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Shiovitz, S.; Bertagnolli, M.M.; Renfro, L.A.; Nam, E.; Foster, N.R.; Dzieciatkowski, S.; Luo, Y.X.; Lao, V.V.; Monnat, R.J.; Emond, M.J.; et al. CpG Island Methylator Phenotype Is Associated with Response to Adjuvant Irinotecan-Based Therapy for Stage III Colon Cancer. Gastroenterology 2014, 147, 637–645. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, K.H.; Shin, S.J.; Lee, K.Y.; Kim, T.I.; Kim, N.K.; Rha, S.Y.; Roh, J.K.; Ahn, J.B. p16 Hypermethylation and KRAS Mutation Are Independent Predictors of Cetuximab Plus FOLFIRI Chemotherapy in Patients with Metastatic Colorectal Cancer. Cancer Res. Treat. 2016, 48, 208–215. [Google Scholar] [CrossRef]
- Zhang, X.F.; Shimodaira, H.; Soeda, H.; Komine, K.; Takahashi, H.; Ouchi, K.; Inoue, M.; Takahashi, M.; Takahashi, S.; Ishioka, C. CpG island methylator phenotype is associated with the efficacy of sequential oxaliplatin- and irinotecan-based chemotherapy and EGFR-related gene mutation in Japanese patients with metastatic colorectal cancer. Int. J. Clin. Oncol. 2016, 21, 1091–1101. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Wang, D.; Feng, M.; Wu, X. Epigenetically regulated gene expression profiles reveal four molecular subtypes with prognostic and therapeutic implications in colorectal cancer. Brief. Bioinform. 2021, 22, bbaa309. [Google Scholar] [CrossRef] [PubMed]
- Condelli, V.; Calice, G.; Cassano, A.; Basso, M.; Rodriquenz, M.G.; Zupa, A.; Maddalena, F.; Crispo, F.; Pietrafesa, M.; Aieta, M.; et al. Novel Epigenetic Eight-Gene Signature Predictive of Poor Prognosis and MSI-Like Phenotype in Human Metastatic Colorectal Carcinomas. Cancers 2021, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed]


| Gene | Gene Name | Gene Function | Epigenetic Event | Reference |
|---|---|---|---|---|
| UCHL1 | ubiquitin carboxyl-terminal hydrolase L1 | Tumor suppressor. Carboxyl-terminal ubiquitin hydrolase regulating cellular ubiquitin levels | UCHL1 methylation in 22/31 CRC (=71%). UCHL1 binds to and stabilizes p53 by the ubiquitination pathway, UCHL1 demethylation caused growth inhibition, G2/M arrest and induction of apoptosis. | Yu et al., 2008 [35] |
| BCL6B | B-Cell CLL/Lymphoma 6 Member B | Tumor suppressor. BCL6B activates p53 signaling and induces apoptosis. | BCL6B was methylated in 81/102 CRC (=79%). | Hu et al., 2015 [36] |
| RASSF10 | Ras-association domain family member 10 | RASSF10 activates p53 signaling and sensitizes to docetaxel. RASSF10 demethylation induced apoptosis and inhibited proliferation. | RASSF10 was methylated in 54/89 CRC (=61%) and was positively associated with tumor stage and metastasis. | Jin et al., 2015 [37] |
| HPSE2 | Heparanase 2 | Tumor suppressor. HPSE2 regulates p53 signaling and G1 cell cycle arrest. | HPSE2 hypermethylation correlated with shorter survival times of CRC patients. | Zhang et al., 2021 [38] |
| BMI1 | B Lymphoma Mo-MLV insertion region 1 homologue | Proto-oncogene. BMI1 is an epigenetic repressor by chromatin remodeling. p14ARF is silenced by BMI1. | p14ARF and wild-type p53 were upregulated in BMI1-mutant CRC. p14ARF is required for the induction of p53 and apoptosis. | Maynard et al., 2014 [39] |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A | CDKN2A/p14ARF antagonizes MDM2-dependent p53 degradation. | p14ARF hypermethylation was increased in tumors with wildtype compared to mutated p53 without statistical significance. | Esteller et al., 2000 [40] |
| p14ARF promoter demethylation was associated with nuclear MDM2 expression (active state), p14ARF promoter hypermethylation with cytosolic MDM2 expression (inactive state). | Nuclear MDM2 expression was associated with p14ARF promoter demethylation in 33 CRC. | Esteller et al., 2001b [41] | ||
| Tumor suppressor. Regulates p53 protein stability by interaction with MDM2. p14ARF promoter hypermethylation abrogates wild-type p53 activity. | CDKN2A promoter hypermethylation was significantly correlated with restricted p53 overexpression and MDM2 overexpression. Epigenetic silencing of CDKN2A is a deregulating mechanism of the p53-MDM2-p14ARF pathway in CRC exhibiting restricted p53 overexpression. | Nyiraneza et al., 2012 [42] | ||
| APC | Adenomatous Polyposis Coli Protein | APC inhibits β-catenin, and β-catenin overexpression increases the TP53 mutation rate. | APC hypermethylation was associated with p53 mutation in 208 CRC. | Suehiro et al., 2008; Stamos and Weis, 2013 [43,44] |
| MGMT | O6-methylguanine-DNA methyltransferase | MGMT downregulation by promoter hypermethylation predisposes to p53 mutations. MGMT hypermethylation is associated with G > A mutations in TP53. | MGMT promoter hypermethylation was significantly correlated to G:C > A:T transition p53 mutations in 314 CRC. | Esteller et al., 2001a [45] |
| MGMT methylation was associated with G > A mutations in the TP53 gene. | Deng et al., 2008 [46] | |||
| No association between MGMT methylation and TP53 mutations in 261 CRC biopsies from Afro-American patients | Alonso et al., 2015 [47] | |||
| PTX3 | Pentraxin 3 | Tumor suppressor. PTX3 deficiency increases susceptibility to carcinogenesis by increasing DNA damage, p53 mutations, and inactivation of p53 downstream signaling (Mdm2, Bax, and Cdkn1a /p21). | Increasing PTX3 promoter methylation from normal colon epithelium to adenomas and CRC. | Bonavita et al., 2015 [48] |
| PCAF | P300/CBP-associated factor | Trimethylated histone H3K27 binding in the PCAF promoter attenuated its transcription. | Decreased PCAF impairs the acetylation of p53 and attenuates the p53-dependent transcription of p21, which results in the increased cyclin D1 expression and Retinobla-stoma 1 phosphorylation as well as increased resistance to 5-fluorouracil. | Liu et al., 2019 [49] |
| LACTB | Lactamase β | Tumor suppressor. LACTB binds to the p53 C terminus to inhibit p53 degradation by MDM2. | LACTB was significantly downregulated in CRC due to promoter methylation and histone deacetylation. | Zeng et al., 2018 [50] |
| TRIM67 | Tripartite motif containing 67 | Tumor suppressor. Upon stress, p53 binds to TRIM67 promoter and upregulates TRIM67 expression in a TRIM67/p53 self-amplifying loop. TRIMP67 interacts with the p53 C-terminus to inhibit p53 degradation by MDM2. | 108/138 CRC (=79%) downregulated TRIMP67 expression due to promoter methylation. Demethylation by treatment with 5-aza-2′-deoxycytidine restored TRIM67 expression in CRC cells. | Wang et al., 2019 [51] |
| COX2 | Cyclooxygenase 2 | Role in CRC progression. Converts arachidonic acid to prostaglandins | Methylation in 12/93 (=13%) CRC and 7/50 (=14%) colorectal ademonas. COX2 methylation was inversely related to p53 mutations. Functional relationship unclear | Toyota et al., 2000a [52] |
| CIMP | CpG island methylator phenotype | High levels of DNA methylation may predispose to carcinogenesis. | TP53 mutations in 10/41 (=24%) CIMP-positive CRC compared to 30/46 (=60%) CIMP-negative cases. Functional relationship unclear | Toyota et al., 2000b [53] |
| CIMP | CpG island methylator phenotype | CIMP correlated with wildtype p53. Functional relationship unclear | Konishi et al., 2007 [54] | |
| miR-34a | Micro-RNA 34a | miR-34a acts as translational repressor. | miR-34a activates p53 by inhibiting its acetylation by MTA2 and HDAC1. | Kaller et al., 2011 [55] |
| miR-34a methylation correlates with wild-type p53 in CRC and other tumor types. | Vogt et al., 2011 [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomicic, M.T.; Dawood, M.; Efferth, T. Epigenetic Alterations Upstream and Downstream of p53 Signaling in Colorectal Carcinoma. Cancers 2021, 13, 4072. https://doi.org/10.3390/cancers13164072
Tomicic MT, Dawood M, Efferth T. Epigenetic Alterations Upstream and Downstream of p53 Signaling in Colorectal Carcinoma. Cancers. 2021; 13(16):4072. https://doi.org/10.3390/cancers13164072
Chicago/Turabian StyleTomicic, Maja T., Mona Dawood, and Thomas Efferth. 2021. "Epigenetic Alterations Upstream and Downstream of p53 Signaling in Colorectal Carcinoma" Cancers 13, no. 16: 4072. https://doi.org/10.3390/cancers13164072
APA StyleTomicic, M. T., Dawood, M., & Efferth, T. (2021). Epigenetic Alterations Upstream and Downstream of p53 Signaling in Colorectal Carcinoma. Cancers, 13(16), 4072. https://doi.org/10.3390/cancers13164072