
Therapeutic Strategies Targeting Tumor Suppressor Genes in Pancreatic Cancer

 ,
,  ,
, 
Abstract
:Simple Summary
Abstract

1. Introduction
2. BRCA1/2 Tumor Suppressor Gene—PARP Inhibitors
2.1. Clinical Trials of Inhibitors of PDAC
2.2. Other PARP Inhibitors for the Treatment of Pancreatic Cancer
2.2.1. Veliparib
2.2.2. Talazoparib (MDV3800 or BMN 673)
2.2.3. Rucaparib (NCT02042378)
3. TP53 Tumor Suppressor Gene
3.1. Gain-of-Function Mutant p53 (mutp53)
3.1.1. Hotspots of mutp53
3.1.2. Restoring wtp53 Function
CP-31398
STIMA-1
PRIMA-1 and APR-246
p53R3
PK083 and PK7088
RITA
Chetomin
Phenethyl Isothiocyanate
3.1.3. Zinc-Based Therapy
NSC319726/ZMC1
COTI-2
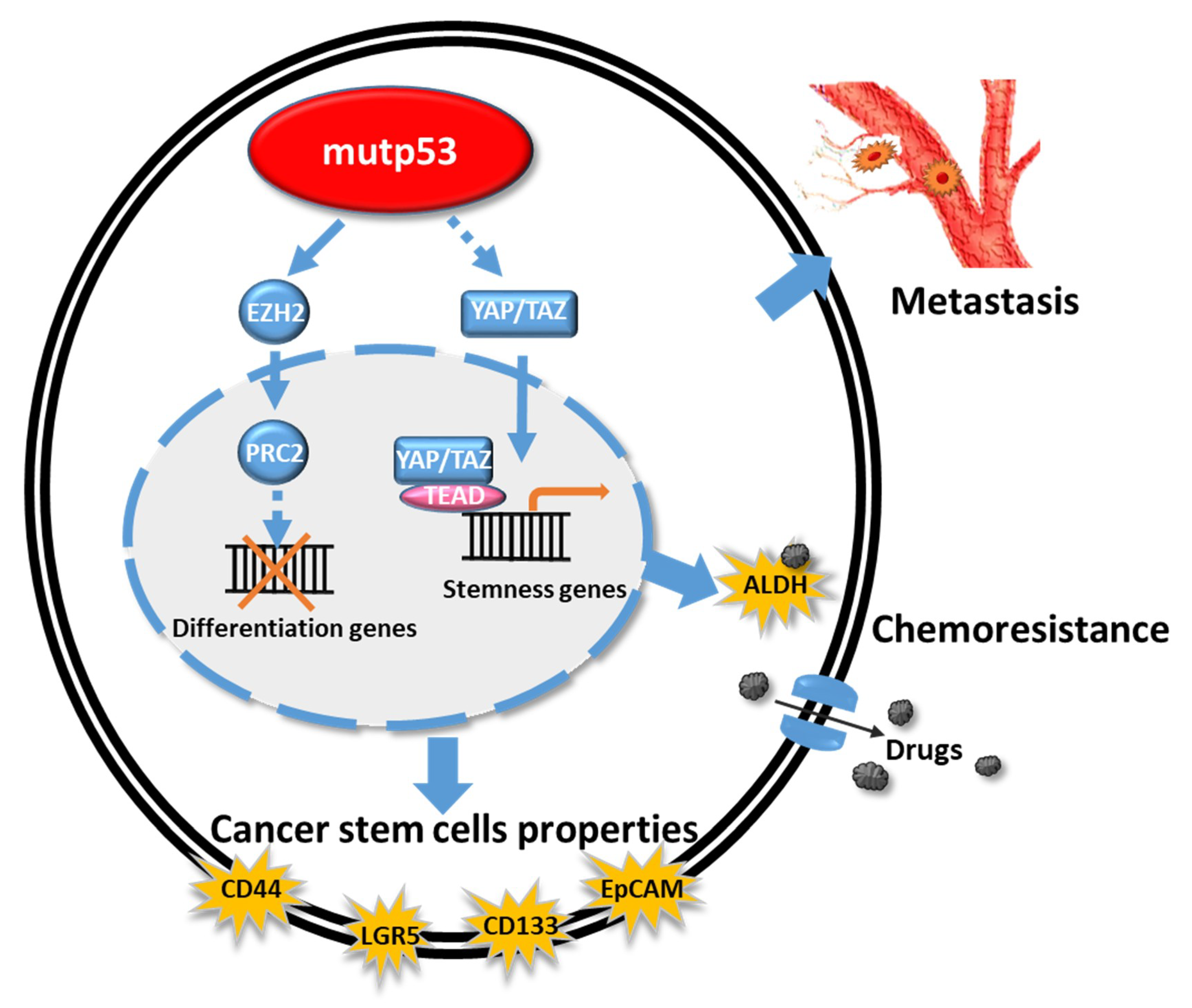
4. GOF mutTP53 Proteins and Cancer Stem Cell Phenotypes
5. Targeting Mutant p53 Protein Stability
5.1. Geldanamycin
5.2. Ganetespib
5.3. Alvespimycin Plus SAHA
6. Synthetic Lethality of p53 Loss
7. Immunoregulation of the Microenvironment of PDAC
7.1. PD-1/PD-L1 in PDAC
7.2. Mutant p53 GOF Mechanisms via the Shedding of the Tumor-Promoting Secretome (Including Exosomes)
7.3. Neoepitopes from mutp53 Proteins Are Recognized by TCRs on CD8+ T Cells
7.4. CAR-T Cells Promote T-Cell Expansion to Promote Anti-Tumor Function
8. MDM2–MDMX(MDM4)–p53 Axis
9. Future Perspectives
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvano, A.; Castiglia, M.; Rizzo, S.; Silvestris, N.; Brunetti, O.; Vaccaro, G.; Gristina, V.; Barraco, N.; Bono, M.; Guercio, G.; et al. Moving the Target on the Optimal Adjuvant Strategy for Resected Pancreatic Cancers: A Systematic Review with Meta-Analysis. Cancers 2020, 12, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Roessel, S.; van Veldhuisen, E.; Klompmaker, S.; Janssen, Q.P.; Abu Hilal, M.; Alseidi, A.; Balduzzi, A.; Balzano, G.; Bassi, C.; Berrevoet, F.; et al. Evaluation of Adjuvant Chemotherapy in Patients with Resected Pancreatic Cancer After Neoadjuvant FOLFIRINOX Treatment. JAMA Oncol. 2020, 6, 1733–1740. [Google Scholar] [CrossRef]
- Gamboa, A.C.; Rupji, M.; Switchenko, J.M.; Lee, R.M.; Turgeon, M.K.; Meyer, B.I.; Russell, M.C.; Cardona, K.; Kooby, D.A.; Maithel, S.K.; et al. Optimal timing and treatment strategy for pancreatic cancer. J. Surg. Oncol. 2020, 122, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, G.; Daye, D.; Wilson, N.M.; Noman, R.; Mahendra, A.M.; Doshi, M.H. Ablation in Pancreatic Cancer: Past, Present and Future. Cancers 2021, 13, 2511. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Ponz-Sarvise, M.; Tuveson, D.A.; Yu, K.H. Mouse Models of Pancreatic Ductal Adenocarcinoma. Hematol. Oncol. Clin. N. Am. 2015, 29, 609–617. [Google Scholar] [CrossRef]
- Kuo, K.K.; Kuo, C.J.; Chiu, C.Y.; Liang, S.S.; Huang, C.H.; Chi, S.W.; Tsai, K.B.; Chen, C.Y.; Hsi, E.; Cheng, K.H.; et al. Quantitative Proteomic Analysis of Differentially Expressed Protein Profiles Involved in Pancreatic Ductal Adenocarcinoma. Pancreas 2016, 45, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Zhen, D.B.; Rabe, K.G.; Gallinger, S.; Syngal, S.; Schwartz, A.G.; Goggins, M.G.; Hruban, R.H.; Cote, M.L.; McWilliams, R.R.; Roberts, N.J.; et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: A PACGENE study. Genet. Med. 2015, 17, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Axilbund, J.E.; Wiley, E.A. Genetic testing by cancer site: Pancreas. Cancer J. 2012, 18, 350–354. [Google Scholar] [CrossRef]
- Klein, A.P. Genetic susceptibility to pancreatic cancer. Mol. Carcinog. 2012, 51, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Hong, S.M.; Lim, P.; Kamiyama, H.; Khan, M.; Anders, R.A.; Goggins, M.; Hruban, R.H.; Eshleman, J.R. KRAS2 mutations in human pancreatic acinar-ductal metaplastic lesions are limited to those with PanIN: Implications for the human pancreatic cancer cell of origin. Mol. Cancer Res. 2009, 7, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Ollinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Habbe, N.; Shi, G.; Meguid, R.A.; Fendrich, V.; Esni, F.; Chen, H.; Feldmann, G.; Stoffers, D.A.; Konieczny, S.F.; Leach, S.D.; et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl. Acad. Sci. USA 2008, 105, 18913–18918. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, T.; Sunamura, M.; Horii, A. Molecular mechanisms of pancreatic carcinogenesis. Cancer Sci. 2006, 97, 1–7. [Google Scholar] [CrossRef]
- Furukawa, T.; Sunamura, M.; Motoi, F.; Matsuno, S.; Horii, A. Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am. J. Pathol. 2003, 162, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [Green Version]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [Green Version]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Hruban, R.H.; Kamiyama, M.; Borges, M.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Palmisano, E.; Brune, K.; Jaffee, E.M.; et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009, 324, 217. [Google Scholar] [CrossRef] [Green Version]
- Win, A.K.; Young, J.P.; Lindor, N.M.; Tucker, K.M.; Ahnen, D.J.; Young, G.P.; Buchanan, D.D.; Clendenning, M.; Giles, G.G.; Winship, I.; et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: A prospective cohort study. J. Clin. Oncol. 2012, 30, 958–964. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L.; Petersen, G.M.; Lerner-Ellis, J.; Holter, S.; Gallinger, S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015, 148, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffee, K.G.; Oberg, A.L.; McWilliams, R.R.; Majithia, N.; Allen, B.A.; Kidd, J.; Singh, N.; Hartman, A.R.; Wenstrup, R.J.; Petersen, G.M. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet. Med. 2018, 20, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, G.M.; Chaffee, K.G.; McWilliams, R.R.; Majithia, N.; Allen, B.; Kidd, J.; Singh, N.; Hartman, A.-R.; Oberg, A.L. Genetic heterogeneity and survival among pancreatic adenocarcinoma (PDAC) patients with positive family history. J. Clin. Oncol. 2016, 34, 4108. [Google Scholar] [CrossRef]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef]
- Hahn, S.A.; Greenhalf, B.; Ellis, I.; Sina-Frey, M.; Rieder, H.; Korte, B.; Gerdes, B.; Kress, R.; Ziegler, A.; Raeburn, J.A.; et al. BRCA2 germline mutations in familial pancreatic carcinoma. J. Natl. Cancer Inst. 2003, 95, 214–221. [Google Scholar] [CrossRef]
- Murphy, K.M.; Brune, K.A.; Griffin, C.; Sollenberger, J.E.; Petersen, G.M.; Bansal, R.; Hruban, R.H.; Kern, S.E. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: Deleterious BRCA2 mutations in 17%. Cancer Res. 2002, 62, 3789–3793. [Google Scholar] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Tuli, R.; Shiao, S.L.; Nissen, N.; Tighiouart, M.; Kim, S.; Osipov, A.; Bryant, M.; Ristow, L.; Placencio-Hickok, V.; Hoffman, D.; et al. A phase 1 study of veliparib, a PARP-1/2 inhibitor, with gemcitabine and radiotherapy in locally advanced pancreatic cancer. EBioMedicine 2019, 40, 375–381. [Google Scholar] [CrossRef] [Green Version]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy-the barrier or the path. J. Mol. Cell Biol. 2019, 11, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Zou, Z.Q.; Pirollo, K.; Blattner, W.; Chang, E.H. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 1990, 348, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 3010. [Google Scholar] [CrossRef]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Shetzer, Y.; Molchadsky, A.; Rotter, V. Oncogenic Mutant p53 Gain of Function Nourishes the Vicious Cycle of Tumor Development and Cancer Stem-Cell Formation. Cold Spring Harb. Perspect. Med. 2016, 6, a026203. [Google Scholar] [CrossRef]
- Synnott, N.C.; Murray, A.; McGowan, P.M.; Kiely, M.; Kiely, P.A.; O’Donovan, N.; O’Connor, D.P.; Gallagher, W.M.; Crown, J.; Duffy, M.J. Mutant p53: A novel target for the treatment of patients with triple-negative breast cancer? Int. J. Cancer 2017, 140, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Mello, S.S.; Attardi, L.D. Not all p53 gain-of-function mutants are created equal. Cell Death Differ. 2013, 20, 855–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, R.; Wang, W.; Dicker, D.T.; Rastinejad, F.; Lyssikatos, J.; el-Deiry, W.S. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol. Ther. 2002, 1, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rippin, T.M.; Bykov, V.J.; Freund, S.M.; Selivanova, G.; Wiman, K.G.; Fersht, A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zache, N.; Lambert, J.M.; Rokaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008, 2, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Moshfegh, A.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene 2010, 29, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinmann, L.; Wischhusen, J.; Demma, M.J.; Naumann, U.; Roth, P.; Dasmahapatra, B.; Weller, M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2008, 15, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Bromley, D.; Bauer, M.R.; Fersht, A.R.; Daggett, V. An in silico algorithm for identifying stabilizing pockets in proteins: Test case, the Y220C mutant of the p53 tumor suppressor protein. Protein Eng. Des. Sel. 2016, 29, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061. [Google Scholar] [CrossRef] [Green Version]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Burmakin, M.; Shi, Y.; Hedstrom, E.; Kogner, P.; Selivanova, G. Dual targeting of wild-type and mutant p53 by small molecule RITA results in the inhibition of N-Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin. Cancer Res. 2013, 19, 5092–5103. [Google Scholar] [CrossRef] [Green Version]
- Hiraki, M.; Hwang, S.Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef] [Green Version]
- Kung, A.L.; Zabludoff, S.D.; France, D.S.; Freedman, S.J.; Tanner, E.A.; Vieira, A.; Cornell-Kennon, S.; Lee, J.; Wang, B.; Wang, J.; et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 2004, 6, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staab, A.; Loeffler, J.; Said, H.M.; Diehlmann, D.; Katzer, A.; Beyer, M.; Fleischer, M.; Schwab, F.; Baier, K.; Einsele, H.; et al. Effects of HIF-1 inhibition by chetomin on hypoxia-related transcription and radiosensitivity in HT 1080 human fibrosarcoma cells. BMC Cancer 2007, 7, 213. [Google Scholar] [CrossRef] [Green Version]
- Kessler, J.; Hahnel, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. HIF-1alpha inhibition by siRNA or chetomin in human malignant glioma cells: Effects on hypoxic radioresistance and monitoring via CA9 expression. BMC Cancer 2010, 10, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, M.; Saxena, R.; Sinclair, E.; Fu, Y.; Jacobs, A.; Dyba, M.; Wang, X.; Cruz, I.; Berry, D.; Kallakury, B.; et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016, 23, 1615–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintus, S.S.; Ivanisenko, N.V.; Demenkov, P.S.; Ivanisenko, T.V.; Ramachandran, S.; Kolchanov, N.A.; Ivanisenko, V.A. The substitutions G245C and G245D in the Zn(2+)-binding pocket of the p53 protein result in differences of conformational flexibility of the DNA-binding domain. J. Biomol. Struct. Dyn. 2013, 31, 78–86. [Google Scholar] [CrossRef]
- Margalit, O.; Simon, A.J.; Yakubov, E.; Puca, R.; Yosepovich, A.; Avivi, C.; Jacob-Hirsch, J.; Gelernter, I.; Harmelin, A.; Barshack, I.; et al. Zinc supplementation augments in vivo antitumor effect of chemotherapy by restoring p53 function. Int. J. Cancer 2012, 131, E562–E568. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarado-Ortiz, E.; de la Cruz-Lopez, K.G.; Becerril-Rico, J.; Sarabia-Sanchez, M.A.; Ortiz-Sanchez, E.; Garcia-Carranca, A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2020, 8, 607670. [Google Scholar] [CrossRef] [PubMed]
- Escoll, M.; Gargini, R.; Cuadrado, A.; Anton, I.M.; Wandosell, F. Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 2017, 36, 3515–3527. [Google Scholar] [CrossRef]
- Hassan, Z.; Schneeweis, C.; Wirth, M.; Muller, S.; Geismann, C.; Neuss, T.; Steiger, K.; Kramer, O.H.; Schmid, R.M.; Rad, R.; et al. Important role of Nfkb2 in the Kras(G12D)-driven carcinogenesis in the pancreas. Pancreatology 2021. [Google Scholar] [CrossRef]
- Kim, M.P.; Li, X.; Deng, J.; Zhang, Y.; Dai, B.; Allton, K.L.; Hughes, T.G.; Siangco, C.; Augustine, J.J.; Kang, Y.; et al. Oncogenic KRAS recruits an expansive transcriptional network through mutant p53 to drive pancreatic cancer metastasis. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Capaci, V.; Bascetta, L.; Fantuz, M.; Beznoussenko, G.V.; Sommaggio, R.; Cancila, V.; Bisso, A.; Campaner, E.; Mironov, A.A.; Wisniewski, J.R.; et al. Mutant p53 induces Golgi tubulo-vesiculation driving a prometastatic secretome. Nat. Commun. 2020, 11, 3945. [Google Scholar] [CrossRef]
- Eliyahu, D.; Raz, A.; Gruss, P.; Givol, D.; Oren, M. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 1984, 312, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, J.R.; Rudge, K.; Currie, G.A. Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature 1984, 312, 651–654. [Google Scholar] [CrossRef]
- Parada, L.F.; Land, H.; Weinberg, R.A.; Wolf, D.; Rotter, V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 1984, 312, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction--a rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagosklonny, M.V.; Toretsky, J.; Neckers, L. Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene 1995, 11, 933–939. [Google Scholar]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Bi, J.; Li, Y.; Yang, S.; Zhang, Y.; Li, M.; Liu, H.; Li, Y.; McDonald, M.E.; Thiel, K.W.; et al. AZD1775 Increases Sensitivity to Olaparib and Gemcitabine in Cancer Cells with p53 Mutations. Cancers 2018, 10, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients with TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Hartman, S.J.; Bagby, S.M.; Yacob, B.W.; Simmons, D.M.; MacBeth, M.; Lieu, C.H.; Davis, S.L.; Leal, A.D.; Tentler, J.J.; Diamond, J.R.; et al. WEE1 Inhibition in Combination with Targeted Agents and Standard Chemotherapy in Preclinical Models of Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 642328. [Google Scholar] [CrossRef]
- Xiao, G.; Lundine, D.; Annor, G.K.; Canar, J.; Ellison, V.; Polotskaia, A.; Donabedian, P.L.; Reiner, T.; Khramtsova, G.F.; Olopade, O.I.; et al. Gain-of-Function Mutant p53 R273H Interacts with Replicating DNA and PARP1 in Breast Cancer. Cancer Res. 2020, 80, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Yang, J.; Adair, S.J.; Ozturk, H.; Kuscu, C.; Lee, K.Y.; Kane, W.J.; O’Hara, P.E.; Liu, D.; Demirlenk, Y.M.; et al. Targeted CRISPR screening identifies PRMT5 as synthetic lethality combinatorial target with gemcitabine in pancreatic cancer cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28068–28079. [Google Scholar] [CrossRef]
- Feng, M.; Xiong, G.; Cao, Z.; Yang, G.; Zheng, S.; Song, X.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017, 407, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Nummer, D.; Suri-Payer, E.; Schmitz-Winnenthal, H.; Bonertz, A.; Galindo, L.; Antolovich, D.; Koch, M.; Buchler, M.; Weitz, J.; Schirrmacher, V.; et al. Role of tumor endothelium in CD4+ CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J. Natl. Cancer Inst. 2007, 99, 1188–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Baine, M.J.; Meza, J.L.; Lin, C. Association of Immunotherapy with Survival Among Patients With Brain Metastases Whose Cancer Was Managed With Definitive Surgery of the Primary Tumor. JAMA Netw. Open 2020, 3, e2015444. [Google Scholar] [CrossRef]
- Cao, Z.; Kon, N.; Liu, Y.; Xu, W.; Wen, J.; Yao, H.; Zhang, M.; Wu, Z.; Yan, X.; Zhu, W.G.; et al. An unexpected role for p53 in regulating cancer cell-intrinsic PD-1 by acetylation. Sci. Adv. 2021, 7, eabf4148. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Lugli, E.; Kvistborg, P.; Galletti, G. Cancer neoantigens targeted by adoptive T cell transfer: Private no more. J. Clin. Investig. 2019, 129, 949–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlakis, E.; Stiewe, T. p53’s Extended Reach: The Mutant p53 Secretome. Biomolecules 2020, 10, 307. [Google Scholar] [CrossRef] [Green Version]
- Amelio, I.; Melino, G. Context is everything: Extrinsic signalling and gain-of-function p53 mutants. Cell Death Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Umut, Ö.; Gottschlich, A.; Endres, S.; Kobold, S. CAR T cell therapy in solid tumors: A short review. Memo Mag. Eur. Med. Oncol. 2021. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, Y.; Wu, Z.; Feng, K.; Tong, C.; Wang, Y.; Dai, H.; Shi, F.; Yang, Q.; Han, W. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy 2020, 22, 573–580. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Yan, Z.; Liao, X.; Li, Y.; Yang, J.; Wang, Z.G.; Zuo, Y.; Kawai, H.; Shadfan, M.; Ganapathy, S.; et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12001–12006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Zawacka-Pankau, J. The p53/MDM2/MDMX-targeted therapies-a clinical synopsis. Cell Death Dis. 2020, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Said, R.; Guibert, N.; Oxnard, G.R.; Tsimberidou, A.M. Circulating tumor DNA analysis in the era of precision oncology. Oncotarget 2020, 11, 188–211. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Incorvaia, L.; Del Re, M.; Malapelle, U.; Capoluongo, E.; Gristina, V.; Castiglia, M.; Danesi, R.; Fassan, M.; Giuffrè, G.; et al. The molecular profiling of solid tumors by liquid biopsy: A position paper of the AIOM-SIAPEC-IAP-SIBioC-SIC-SIF Italian Scientific Societies. ESMO Open 2021, 6, 100164. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial ID | Therapeutic Drug | Phase | Status | Condition | Primary Outcome |
|---|---|---|---|---|---|
| NCT03654716 | Drug: ALRN-6924 Drug: Cytarabine | Phase 1 | Recruiting | Solid Tumor | Percentage of patients with dose limiting toxicity by CTCAE V.5.0 for each dose level (Time Frame: 2 years) Percentage of patients with toxicity by CTCAE V.5.0 (Time Frame: 2 Years) |
| NCT02098967 | Drug: RO6839921 | Phase 1 | Completed | Neoplasms | Incidence of adverse events (Time Frame: Approximately 1 year) Incidence of dose-limiting toxicities (Time Frame: Approximately 1 year) |
| NCT01462175 | Drug: RO5503781 | Phase 1 | Completed | Neoplasms | Maximum Tolerated Dose (MTD) (Time Frame: Up to 28 days) Percentage of Participants With Dose Limiting Toxicities (DLTs) (Time Frame: Up to 28 days) Percentage of Participants With Adverse Events (AEs) and Serious Adverse Events (SAEs) (Time Frame: approximately 1.5 years) |
| NCT03362723 | Drug: Idasanutlin | Phase 1 | Completed | Solid Tumors | Area Under the Curve (AUC) of Idasanutlin ( Time Frame: Predose (within 2 h), 1, 2, 4, 6, and 10 h on Days 1, 8, 15, and 22; on Days 2, 3, 5, 9, 10, 12, 16, 17, 19, 23, 24, and 26; Day 29 (end of Cycle 1) or Cycle 2 Day 1 (for participants in the optional treatment extension phase) (cycle = 28 days)) Maximum Observed Plasma Concentration (Cmax) of Idasanutlin (Time Frame: Predose (within 2 h), 1, 2, 4, 6, and 10 h on Days 1, 8, 15, and 22; on Days 2, 3, 5, 9, 10, 12, 16, 17, 19, 23, 24, and 26; Day 29 (end of Cycle 1) or Cycle 2 Day 1 (for participants in the optional treatment extension phase) (cycle = 28 days)) |
| NCT03714958 | Drug: HDM201 Drug: Trametinib | Phase 1 | Recruiting | Advanced Cancer Metastatic Cancer | Dose Maximum Tolerated (Time Frame: During the first 2 cycles of treatment (1 cycle = 28 days)) |
| NCT02143635 | Drug: HDM201 Drug: ancillary treatment | Phase 1 | Completed | Advanced Solid and Hematological TP53wt Tumors | Incidence of dose limiting toxicities (DLTs) (Time Frame: up to 28 days) |
| NCT03449381 | Drug: BI 907828 | Phase 1 | Recruiting | Neoplasms | Phase Ia- Maximum tolerated dose (MTD) based on number of patients with dose limiting toxicities (DLTs) during first treatment cycle (Time Frame: Up to 28 days) Progression-free survival (Time Frame: Up to 24 months) Phase Ia—Number of patients with DLTs during first treatment cycle (21 days, Arm A; 28 days, Arm B) (Time Frame: Up to 28 days) Phase Ib—Number of patients with DLTs during the first treatment cycle (Time Frame: Up to 28 days) |
| NCT03964233 | Drug: BI 907828 Drug: BI 754091 Drug: BI 754111 | Phase 1 | Recruiting | Neoplasms | Phase Ia—maximum tolerated dose (MTD) of BI 907828 in combination with BI 754091 based on the number of patients with DLTs during the first treatment cycle (Time Frame: Up to 21 Days) Phase Ib—Objective response (OR) (Time Frame: Up to 24 months) |
| NCT01664000 | Drug: thioureidobutyronitrile | Phase 1 | Completed | Solid Tumors | Maximum Tolerated Dose (MTD) of Kevetrin (Time Frame: Up to 6 months) Dose Limiting Toxicities (DLT) of Kevetrin. (Time Frame: up to 4 weeks) |
| NCT03611868 | Drug: APG-115+Pembrolizumab | Phase 1 Phase 2 | Recruiting | Unresectable or Metastatic Melanoma or Advanced Solid Tumors P53 Mutation MDM2 Gene Mutation | Maximum Tolerated Dose (Time Frame: 21 days) Recommended Phase II Dose (Time Frame: 21 days) Overall Response Rate (Time Frame: Up to 12 months) |
| NCT02264613 | Drug: ALRN-6924 | Phase 1 Phase 2 | Completed | Solid Tumor | Evaluate the safety and tolerability of ALRN-6924 in adult patients with advanced solid tumors or lymphomas with wild-type (WT) TP53 who are refractory to or intolerant of standard therapy, or for whom no standard therapy exists—Phase 1 (Time Frame: From Day 1 of treatment until 30 days after the last cycle of treatment (each cycle is 28 days)) Evaluate the safety and tolerability of ALRN-6924 in adult patients with advanced solid tumors or lymphomas with wild-type (WT) TP53 who are refractory to or intolerant of standard therapy, or for whom no standard therapy exists—Phase 2 (Time Frame: From Day 1 of treatment until 30 days after the last cycle of treatment (each cycle is 28 days)) Determine the maximum tolerated dose (MTD)—Phase 1 (Time Frame: From the first dose until the end of the first cycle (each cycle is 28 days)) Determine Overall Response Rate—Phase 2 (Time Frame: From the first dose until the first documented date of progression or date of death from any cause, whichever comes first, assessed up to 100 months) |
| NCT04589845 | Drug: Entrectinib Drug: Alectinib Drug: Atezolizumab Drug: Ipatasertib Drug: Trastuzumab emtansine Drug: Idasanutlin Drug: Inavolisib Drug: Belvarafenib Drug: Pralsetinib | Phase 2 | Recruiting | Solid Tumor | All Cohorts: Independent Review Committee (IRC)-assessed objective response rate (ORR) based on confirmed objective response (OR) per Response Evaluation Criteria in Solid Tumors, Version 1.1 (RECIST v1.1) (Time Frame: Approximately up to 12 years) |
| Trial ID | Therapeutic Drug | Phase | Status | Condition | Primary Outcome |
|---|---|---|---|---|---|
| NCT04673448 | Biological: Dostarlimab Drug: Niraparib | Phase 1 | Not yet recruiting | Metastatic Pancreatic Carcinoma | Best objective response (Time Frame: 5 years) |
| NCT00892736 | Other: Laboratory Biomarker Analysis Other: Pharmacological Study Drug: Veliparib | Phase 1 | Completed | Pancreatic Carcinoma | MTD, DLT, recommended phase II dose of chronically dosed single-agent veliparib in patients with either a refractory BRCA 1/2- mutated solid cancer; platinum- refractory ovarian, fallopian tube, or primary peritoneal cancer; or basal-like breast cancer (Time Frame: 28 days) |
| NCT04644068 | Drug: AZD5305 Drug: Paclitaxel Drug: Carboplatin | Phase 1 | Recruiting | Pancreatic Cancer | The number of subjects with adverse events/serious adverse events (Time Frame: From time of Informed Consent to 28 days post last dose (approximately 1 year)) The number of subjects with dose-limiting toxicity (DLT), as defined in the protocol. (Time Frame: From first dose of study treatment until the end of Cycle 1. Approximately 35 days.) |
| NCT00576654 | Drug: Irinotecan Hydrochloride Other: Laboratory Biomarker Analysis Other: Pharmacological Study Drug: Veliparib | Phase 1 | Active, not recruiting | Stage III Pancreatic Cancer AJCC v6 and v7 | Optimal biologic dose (OBD) (Time Frame: Up to day 9 of course 1) Maximum administered dose of study drugs (Time Frame: Up to 21 days) Maximally tolerated dose (MTD) of study drugs (Time Frame: Up to 21 days) Recommended phase II dose (RP2D) of study drugs (Time Frame: Up to 21 days) |
| NCT00047307 | Drug: alvocidib Drug: gemcitabine hydrochloride Radiation: 3-dimensional conformal radiation therapy Other: laboratory biomarker analysis | Phase 1 | Completed | Adenocarcinoma of the Pancreas Recurrent Pancreatic Cancer Stage II Pancreatic Cancer Stage III Pancreatic Cancer Stage IV Pancreatic Cancer | Maximum tolerated dose of flavopiridol when administered biweekly in conjunction with radiation for patients with locally advanced pancreatic or extrahepatic bile duct cancer (Time Frame: 6 weeks) |
| NCT04764084 | Drug: Niraparib Drug: Anlotinib | Phase 1 | Not yet recruiting | Pancreatic Cancer | Dose limiting toxicity (DLT) and maximum tolerated dose (MTD) (Time Frame: 4 weeks) |
| NCT04182516 | Drug: NMS-03305293 | Phase 1 | Recruiting | Advanced/Metastatic Solid Tumors | Number of Participants with first-cycle dose limiting toxicity (Time Frame: Time interval between the date of the first dose administration in Cycle 1 (each cycle is 28 days) and the date of the first dose administration in Cycle 2 which is expected to be 28 days or up to 42 days in case of dose delay due to toxicity) |
| NCT00515866 | Drug: KU-0059436 (AZD2281)(PARP inhibitor) Drug: Gemcitabine | Phase 1 | Completed | Pancreatic Neoplasms | To establish the maximum tolerated dose (MTD) or a tolerable and effective dose of KU 0059436 in combination with gemcitabine (Time Frame: assessed at each visit) |
| NCT04503265 | Drug: AMXI-5001:Dose Escalation Phase I Drug: AMXI-5001:Dose Expansion Phase II | Phase 1 Phase 2 | Recruiting | Pancreatic Cancer | Determine the Maximum Tolerated Dose (MTD) (Time Frame: Approximately 12 months) |
| NCT01489865 | Drug: ABT-888 and mFOLFOX-6 | Phase 1 Phase 2 | Unknown | Metastatic Pancreatic Cancer | Dose limiting toxicities (Time Frame: 28 days) |
| NCT03337087 | Drug: Fluorouracil Other: Laboratory Biomarker Analysis Drug: Leucovorin Calcium Drug: Liposomal Irinotecan Drug: Rucaparib | Phase 1 Phase 2 | Recruiting | Metastatic Pancreatic Adenocarcinoma Stage IV Pancreatic Cancer AJCC v6 and v7 | Number of participants with dose limiting toxicities (Phase I) (Time Frame: Up to 28 days from start of treatment) Objective response (Phase Ib) (Time Frame: Baseline up to 3 years) Best response rate (Phase II) (Time Frame: At 32 weeks) |
| NCT04228601 | Drug: Fluzoparib Drug: Fluzoparib placebo Drug: mFOLFIRINOX | Phase 1 Phase 2 | Recruiting | Advanced Pancreatic Cancer | Number of Participants With a Dose Limited Toxicity (Time Frame: Within 28 Days after The First Dose) Maximum Tolerated Dose (Time Frame: Time Frame: Up to 8 months) Objective Response Rate (Time Frame: From Week 9 until documented disease progression or study discontinuation (approximately up to 24 months)) |
| NCT03404960 | Drug: Niraparib + Nivolumab Drug: Niraparib + Ipilimumab | Phase 1 Phase 2 | Recruiting | Pancreatic Adenocarcinoma | Progression-free survival (Time Frame: 6 months after initiation of study therapy) |
| NCT02042378 | Drug: Rucaparib | Phase 2 | Completed | Pancreatic Cancer Pancreatic Ductal Adenocarcinoma | Overall Response Rate (ORR) per RECIST v1.1 as assessed by the investigator (Time Frame: Screening, within 7 days prior to the start of every 3rd cycle of treatment, and Treatment Discontinuation Visit. Study to last for ~3 years.) |
| NCT03682289 | Drug: ATR Kinase Inhibitor AZD6738 Drug: Olaparib | Phase 2 | Recruiting | Metastatic Pancreatic Cancer Stage III Pancreatic Cancer Stage IV Pancreatic Cancer | Objective response rate (ORR) (Time Frame: Up to 2.5 years) Objective response rate (ORR) for other solid tumors (Time Frame: Up to 2.5 years) |
| NCT04493060 | Biological: Dostarlimab Drug: Niraparib | Phase 2 | Recruiting | Metastatic Pancreatic Ductal Adenocarcinoma Stage IV Pancreatic Cancer AJCC v8 | Disease control rate at 12 weeks (DCR12) (Time Frame: At 12 weeks) |
| NCT02498613 | Other: 18F-Fluoromisonidazole Drug: Cediranib Maleate Other: Laboratory Biomarker Analysis Drug: Olaparib Procedure: Positron Emission Tomography | Phase 2 | Recruiting | Metastatic Pancreatic Adenocarcinoma Pancreatic Ductal Adenocarcinoma Stage III Pancreatic Cancer AJCC v6 and v7 Stage IV Pancreatic Cancer AJCC v6 and v7 Unresectable Pancreatic Adenocarcinoma Unresectable Pancreatic Carcinoma | Objective response rate (Time Frame: Up to 4 weeks after completion of study treatment) |
| NCT04171700 | Drug: Rucaparib | Phase 2 | Recruiting | Solid Tumor | Best Overall Response Rate by Investigator (Time Frame: From first dose of study drug until disease progression (up to approximately 2 years)) |
| NCT04550494 | Procedure: Biopsy Drug: Talazoparib | Phase 2 | Recruiting | Advanced Pancreatic Carcinoma Metastatic Pancreatic Carcinoma Stage II Pancreatic Cancer AJCC v8 Stage IIA Pancreatic Cancer AJCC v8 Stage IIB Pancreatic Cancer AJCC v8 Stage III Pancreatic Cancer AJCC v8 Stage IV Pancreatic Cancer AJCC v8 | Percent of patients who demonstrate simultaneous Rad51 activation (Time Frame: At cycle 2 day 1) |
| NCT01286987 | Drug: Talazoparib | Phase 2 | Active, not recruiting | Metastatic Pancreatic Adenocarcinoma Pancreatic Ductal Adenocarcinoma Stage IV Pancreatic Cancer AJCC v6 and v7 | Objective response rate (defined as complete response or partial response) assessed using Response Evaluation Criteria in Solid Tumors 1.1 (Time Frame: At 24 weeks) |
| NCT03601923 | Drug: Niraparib | Phase 2 | Recruiting | Pancreatic Cancer | Progression Free Survival (Time Frame: 6 months) |
| NCT04409002 | Drug: Niraparib Drug: Dostarlimab Radiation: Radiation | Phase 2 | Recruiting | Pancreatic Cancer Metastatic Pancreatic Cancer | Disease control rate with RECIST 1.1 (Time Frame: 3 months up to 2 years) |
| NCT04548752 | Drug: Olaparib Biological: Pembrolizumab | Phase 2 | Recruiting | Metastatic Pancreatic Adenocarcinoma Stage IV Pancreatic Cancer AJCC v8 | Progression-free survival (PFS) (Time Frame: Up to 3 years) |
| NCT03140670 | Drug: RUCAPARIB | Phase 2 | Active, not recruiting | Pancreatic Cancer | Number of Adverse Events (Time Frame: 4 years) |
| NCT04858334 | Drug: Olaparib Drug: Placebo Administration | Phase 2 | Recruiting | Pancreatic Cancer | Improvement in relapse-free survival (RFS) (Time Frame: From randomization to first documentation of disease recurrence (primary tumor relapse) or death, assessed from 22 months to 44 months) |
| NCT02184195 | Drug: Olaparib Drug: Placebo | Phase 3 | Active, not recruiting | Germline BRCA1/2 Mutations and Metastatic Adenocarcinoma of the Pancreas | Progression-free Survival (PFS) by Blinded Independent Central Review (BICR) Using Modified Response Evaluation Criteria in Solid Tumours. This Study Used Modified RECIST Version (v) 1.1 (RECIST v1.1) (Time Frame: Up to 4 years) |
| Trial ID | Therapeutic Drug | Phase | Status | Condition | Primary Outcome |
|---|---|---|---|---|---|
| NCT00711997 | Biological: DTA-H19 | Phase 1 Phase 2 | Completed | Pancreatic Neoplasms | Maximal Tolerated Dose (MTD) & Dose Limiting Toxicity (DLT) of Intratumoral Injections of BC-819 (Time Frame: Week 4) |
| NCT00769483 | Drug: MK-0646 Drug: Gemcitabine Drug: Erlotinib | Phase 1 Phase 2 | Completed | Pancreatic Cancer Pancreatic Adenocarcinoma | MK-0646 Maximum Tolerable Dose (Time Frame: Up to 12 cycles) Progression Free Survival (Time Frame: From date of randomization until the date of first documented progression or date of death from any cause, whichever came first, assessed up to 100 months) |
| NCT03602079 | Drug: A166 | Phase 1 Phase 2 | Recruiting | Pancreatic Cancer | Phase I: Maximum Tolerated Dose (Time Frame: Minimum of 21 days from date of enrollment until the date of first documented progression or date of death from any cause, whichever came first, assessed up to 24 months) Phase II: Percentage of patients with an Objective Response Rate (ORR) (Complete Response (CR) + Partial Response (PR)) (Time Frame: From date of enrollment until the date of first documented progression or date of death from any cause, whichever came first, assessed up to 24 months) |
| NCT03190941 | Drug: Cyclophosphamide Drug: Fludarabine Biological: Anti-KRAS G12V mTCR PBL Drug: Aldesleukin | Phase 1 Phase 2 | Suspended (Administratively Suspended) | Pancreatic Cancer | Response rate (Time Frame: 6 weeks and 12 weeks following administration of the cell product, then every 3 months × 3, then every 6 months × 2 years, then per PI discretion) Frequency and severity of treatment-related adverse events (Time Frame: From time of cell infusion to two weeks after cell infusion) |
| NCT02705196 | Genetic: delolimogene mupadenorepvec Drug: gemcitabine Drug: nab-paclitaxel Biological: atezolizumab | Phase 1 Phase 2 | Recruiting | Pancreatic Cancer | Number of patient with dose-limiting toxicities (DLTs) as evaluated accordingly to CTCAE 4.0 (Time Frame: 9 months) |
| NCT03745326 | Drug: Cyclophosphamide Drug: Fludarabine Drug: Aldesleukin Biological: anti-KRAS G12D mTCR PBL | Phase 1 Phase 2 | Suspended ((suspension)) | Pancreatic Cancer | Frequency and severity of treatment-related adverse events (Time Frame: From time of cell infusion to two weeks after cell infusion) Response rate (Time Frame: 6 weeks and 12 weeks following administration of the cell product, then every 3 months × 3, then every 6 months × 2 years, then per PI discretion) |
| NCT01583686 | Drug: Fludarabine Biological: Anti-mesothelin chimeric T cell receptor (CAR) transduced peripheral blood lymphocytes (PBL) Drug: Cyclophosphamide Drug: Aldesleukin | Phase 1 Phase 2 | Terminated (Study terminated due to slow/insufficient accrual.) | Pancreatic Cancer | Number of Patients With Objective Tumor Regression (Time Frame: 3.5 mos.) Number of Participants With Serious and Non-serious Adverse Events Assessed by the Common Terminology Criteria in Adverse Events (CTCAE v4.0) (Time Frame: Date treatment consent signed to date off study, approximately 6 months and 17 days for Group A01, 16 months and 13 days for Group A02, 13 months and 3 days for Group A03, 10 months and 16 days for Group A04, and 11 months and 26 days for Group A05. ) |
| NCT03192462 | Biological: multiTAA specific T cells | Phase 1 Phase 2 | Active, not recruiting | Pancreatic Cancer | Number of patients with treatment related serious adverse events (Time Frame: 7 months) Number of patients who received 6 infusions of multiTAA-specific T cells (Time Frame: 6 months) |
| NCT02830724 | Drug: Cyclophosphamide Drug: Fludarabine Drug: Aldesleukin Biological: Anti-hCD70 CAR transduced PBL | Phase 1 Phase 2 | Suspended ((suspension)) | Pancreatic Cancer | Frequency and severity of treatment-related adverse events (Time Frame: From time of cell infusion to two weeks after cell infusion) Response rate (Time Frame: 6 weeks and 12 weeks following administration of the cell product, then every 3 months × 3, then every 6 months × 2 years, then per PI discretion) |
| NCT00255827 | Biological: HyperAcute-Pancreatic Cancer Vaccine | Phase 1 Phase 2 | Completed | Pancreatic Cancer | To assess the side effects, dose-limiting toxicity and maximum tolerated dose. (Time Frame: 6 months) |
| NCT04637698 | Biological: OH2 injection | Phase 1 Phase 2 | Recruiting | Pancreatic Cancer | The objective response rate of patients with pancreatic cancer receiving OH2 injection. (Time Frame: 2 years) |
| NCT00959946 | Drug: Bosutinib Drug: Capecitabine | Phase 1 Phase 2 | Terminated | Advanced Breast Cancer (Parts 1 and 2) Advanced Pancreatic Cancer (Part 1) Advanced Colorectal Cancer (Part 1) Advanced Cholangiocarcinoma (Part 1) Advanced Glioblastoma Multiforme (Part 1) | Maximum Tolerated Dose (MTD)—Part 1 (Time Frame: Part 1 Baseline up to Day 21) Percentage of Participants With Treatment-Emergent Adverse Events (AEs) or Serious Adverse Events (SAEs)—Part 1 (Time Frame: Part 1 Baseline up to 28 days after last dose of study treatment) |
| NCT04426669 | Drug: Cyclophosphamide Drug: Fludarabine Biological: Tumor-Infiltrating Lymphocytes (TIL) Drug: Aldesleukin | Phase 1 Phase 2 | Recruiting | Pancreatic Cancer | Maximum tolerated dose (MTD) (Time Frame: 28 Days Post IL-2) Preliminary efficacy of tumor reactive autologous lymphocytes with knockout of CISH gene in patients with refractory metastatic gastrointestinal epithelial cancers: changes in diameter (Time Frame: Every 4 Weeks for the first three months, then every 8 weeks thereafter, up to 2 years) Safety of tumor reactive autologous lymphocytes with knockout of the CISH gene—Incidence of Adverse Events (Time Frame: 2 Years or Disease Progression) |
| NCT04739046 | Theragene arm | Phase 2 | Recruiting | Pancreas Cancer | Objective Response Rate (Time Frame: 24 weeks) |
| NCT02340117 | Genetic: SGT-53 Drug: nab-paclitaxel Drug: Gemcitabine | Phase 2 | Recruiting | Metastatic Pancreatic Cancer | Progression free survival (PFS) at 5.5 months (Time Frame: 5.5 months) Objective response rate (ORR) (Time Frame: Up to 5 years) |
| NCT02806687 | Drug: Gene Therapy product CYL-02 Drug: Gemcitabine | Phase 2 | Recruiting | Pancreatic Adenocarcinoma | Progression-free survival (Time Frame: From date to randomization until the date of first documented progression or date of death, whichever came first, assessed up to 12 months) |
| NCT00868114 | Biological: KLH-pulsed autologous dendritic cell vaccine | Phase 2 | Terminated (Poor recruitment) | Metastatic Pancreatic Cancer | Overall Survival (Time Frame: Patients will be followed until death) |
| NCT00305760 | Drug: Cetuximab Biological: Pancreatic tumor vaccine Drug: Cyclophosphamide | Phase 2 | Completed | Pancreatic Cancer | Safety of Combining the Pancreatic Tumor Vaccine in Sequence With Cyclophosphamide and Erbitux. Safety is Defined as the Number of Treatment-related Grade 3 or 4 Adverse Events Observed in Greater Than 5% of the Patient Population (Time Frame: 7 months) |
| NCT00084383 | Biological: GVAX pancreatic cancer vaccine | Phase 2 | Completed | Pancreatic Cancer | Overall Survival (Time Frame: Participants were followed for the duration of the study, an average of 2 years)Disease-free Survival (Time Frame: Participants were followed for the duration of the study, an average of 2 years) |
| NCT00389610 | Biological: allogenic GM-CSF plasmid-transfected pancreatic tumor cell vaccine | Phase 2 | Active, not recruiting | Pancreatic Cancer | Safety as measured by local and systemic toxicities (Time Frame: Until progression) |
| NCT04548752 | Drug: Olaparib Biological: Pembrolizumab | Phase 2 | Recruiting | Metastatic Pancreatic Adenocarcinoma Stage IV Pancreatic Cancer AJCC v8 | Progression-free survival (PFS) (Time Frame: Up to 3 years) |
| NCT00305760 | Drug: Cetuximab Biological: Pancreatic tumor vaccine Drug: Cyclophosphamide | Phase 2 | Completed | Pancreatic Cancer | Safety of Combining the Pancreatic Tumor Vaccine in Sequence With Cyclophosphamide and Erbitux. Safety is Defined as the Number of Treatment-related Grade 3 or 4 Adverse Events Observed in Greater Than 5% of the Patient Population (Time Frame: 7 months) |
| NCT00084383 | Biological: GVAX pancreatic cancer vaccine | Phase 2 | Completed | Pancreatic Cancer | Overall Survival (Time Frame: Participants were followed for the duration of the study, an average of 2 years) Disease-free Survival (Time Frame: Participants were followed for the duration of the study, an average of 2 years) |
| NCT00389610 | Biological: allogenic GM-CSF plasmid-transfected pancreatic tumor cell vaccine | Phase 2 | Active, not recruiting | Pancreatic Cancer | Safety as measured by local and systemic toxicities (Time Frame: Until progression) |
| NCT04548752 | Drug: Olaparib Biological: Pembrolizumab | Phase 2 | Recruiting | Metastatic Pancreatic Adenocarcinoma Stage IV Pancreatic Cancer AJCC v8 | Progression-free survival (PFS) (Time Frame: Up to 3 years) |
| NCT01088789 | Biological: PANC 10.05 pcDNA-1/GM-Neo and PANC 6.03 pcDNA-1 neo vaccine. | Phase 2 | Recruiting | Pancreatic Cancer | Disease free overall survival. (Time Frame: Total of 13 years with 6 months between vaccines) |
| NCT04550494 | Procedure: Biopsy Drug: Talazoparib | Phase 2 | Recruiting | Advanced Pancreatic Carcinoma Metastatic Pancreatic Carcinoma Stage II Pancreatic Cancer AJCC v8 Stage IIA Pancreatic Cancer AJCC v8 Stage IIB Pancreatic Cancer AJCC v8 Stage III Pancreatic Cancer AJCC v8 Stage IV Pancreatic Cancer AJCC v8 | Percent of patients who demonstrate simultaneous Rad51 activation (Time Frame: At cycle 2 day 1) |
| NCT04383210 | Drug: Seribantumab | Phase 2 | Recruiting | Pancreatic Cancer | Objective Response Rate (Time Frame: Up to 12 months) |
| NCT04171700 | Drug: Rucaparib | Phase 2 | Recruiting | Solid Tumor | Best Overall Response Rate by Investigator (Time Frame: From first dose of study drug until disease progression (up to approximately 2 years)) |
| NCT02405585 | Drug: mFOLFIRINOX Biological: Algenpantucel-L Immunotherapy Radiation: SBRT Drug: Gemcitabine | Phase 2 | Terminated | Pancreatic Cancer Pancreatic Carcinoma Non-resectable | Progression Free Survival (Time Frame: 18 months (assuming enrollment period of 1 year)) |
| NCT00727441 | Biological: GVAX pancreatic cancer vaccine Drug: cyclophosphamide | Phase 2 | Completed | Pancreatic Cancer | Safety as Measured by Number of Participants With Treatment-related Grade 3 or 4 Local and Systemic Toxicity as Defined by NCI CTCAE v3.0 (Time Frame: 7 years) Amount of T-regulatory Cells (Tregs) and CD4+ and CD8+ Effector T Cells, After Neoadjuvant GVAX Pancreatic Cancer Vaccination. (Time Frame: Up to 8 years) Change in the Number and Function of Peripheral Mesothelin-specific CD8+ T Cells and CD4+, FoxP3+, and GITR+ Tregs (Time Frame: Up to 8 years) |
| NCT00051467 | Genetic: TNFerade | Phase 3 | Completed | Pancreatic Cancer | Not Provided |
| NCT01836432 | Drug: FOLFIRINOX Biological: Algenpantucel-L Immunotherapy Radiation: 5-FU Chemoradiation Drug: Gemcitabine Drug: Capecitabine Drug: Nab-Paclitaxel | Phase 3 | Terminated (Company decision) | Pancreatic Cancer Pancreatic Carcinoma Non-resectable Locally Advanced Malignant Neoplasm | Overall Survival (Time Frame: 13.5 months (assuming enrollment period of 1–2 years)) |
| NCT01286987 | Drug: Talazoparib | Phase 2 | Active, not recruiting | Metastatic Pancreatic Adenocarcinoma Pancreatic Ductal Adenocarcinoma Stage IV Pancreatic Cancer AJCC v6 and v7 | Objective response rate (defined as complete response or partial response) assessed using Response Evaluation Criteria in Solid Tumors 1.1 (Time Frame: At 24 weeks) |
| Trial ID | Therapeutic Drug | Phase | Status | Condition | Primary Outcome |
|---|---|---|---|---|---|
| NCT00603863 | Biological: IMMU-107 (hPAM4) | Phase 1 Phase 2 | Completed | Pancreatic Cancer | Safety will be evaluated based upon physical examinations, hematology, and chemistry laboratory testing as well as toxicity (Time Frame: Over 12 weeks) |
| NCT01631552 | Drug: Sacituzumab Govitecan (SG) | Phase 1 Phase 2 | Completed | Pancreatic Cancer | Percentage of Participants Experiencing Any Treatment Emergent Adverse Events and Serious Treatment Emergent Adverse Events (Time Frame: First dose date up to last dose (maximum duration: 55.2 months) plus 30 days Percentage of Participants Who Permanently Discontinued Sacituzumab Govitecan (SG) Due to Any Adverse Events, Excluding Adverse Events Leading to Death (Time Frame: First dose date up to last dose (maximum duration: 55.2 months)) Percentage of Participants Who Required Dose Interruption Due to Any Adverse Events (Time Frame: First dose date up to last dose (maximum duration: 55.2 months)) Objective Response Rate (ORR) by Independent Central Review (ICR) (Time Frame: Up to 74 months) Objective Response Rate by Local Assessment (Time Frame: Up to 74 months) |
| NCT01956812 | Drug: IMMU-107 Drug: placebo Drug: Gemcitabine | Phase 3 | Terminated (The DSMB conducted an interim analysis on overall survival, which showed that the treatment arm did not demonstrate a sufficient improvement in OS vs. placebo.) | Metastatic Pancreatic Cancer Pancreatic Cancer | overall survival (Time Frame: 24 months) |
| Mutant p35 | ||||||
|---|---|---|---|---|---|---|
| Metastasis | High Proliferation Rate | Resistance to Apoptosis/Drugs | Stem Cell | Chronic Inflammation | Genomic Instability | Metabolism |
| CDH1 | c-MYC | MDR1 | CD133 | sIL-1Ra | BRCA1 | SREBP |
| TFG-β | NF-Y | VDR | OCT3/4 | TLR3 | E2F | RhoA/ROCK |
| MAP2K1 | MAP2K3 | IGF2 | CD44 | NF-κB | RAD17 | ETS2 |
| SENP1/RAC1 | CDK1 | p63 | NANOG | HIF1α | SP1 | GLUT1 |
| TWIST1 | IGF1R | p71 | SOX2 | STAT3 | MRE11 | HK2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, K.-K.; Hsiao, P.-J.; Chang, W.-T.; Chuang, S.-C.; Yang, Y.-H.; Wuputra, K.; Ku, C.-C.; Pan, J.-B.; Li, C.-P.; Kato, K.; et al. Therapeutic Strategies Targeting Tumor Suppressor Genes in Pancreatic Cancer. Cancers 2021, 13, 3920. https://doi.org/10.3390/cancers13153920
Kuo K-K, Hsiao P-J, Chang W-T, Chuang S-C, Yang Y-H, Wuputra K, Ku C-C, Pan J-B, Li C-P, Kato K, et al. Therapeutic Strategies Targeting Tumor Suppressor Genes in Pancreatic Cancer. Cancers. 2021; 13(15):3920. https://doi.org/10.3390/cancers13153920
Chicago/Turabian StyleKuo, Kung-Kai, Pi-Jung Hsiao, Wen-Tsan Chang, Shih-Chang Chuang, Ya-Han Yang, Kenly Wuputra, Chia-Chen Ku, Jia-Bin Pan, Chia-Pei Li, Kohsuke Kato, and et al. 2021. "Therapeutic Strategies Targeting Tumor Suppressor Genes in Pancreatic Cancer" Cancers 13, no. 15: 3920. https://doi.org/10.3390/cancers13153920
APA StyleKuo, K.-K., Hsiao, P.-J., Chang, W.-T., Chuang, S.-C., Yang, Y.-H., Wuputra, K., Ku, C.-C., Pan, J.-B., Li, C.-P., Kato, K., Liu, C.-J., Wu, D.-C., & Yokoyama, K. K. (2021). Therapeutic Strategies Targeting Tumor Suppressor Genes in Pancreatic Cancer. Cancers, 13(15), 3920. https://doi.org/10.3390/cancers13153920