
Cell-Main Spectra Profile Screening Technique in Simulation of Circulating Tumour Cells Using MALDI-TOF Mass Spectrometry

 ,
,
Abstract
:Simple Summary
Abstract
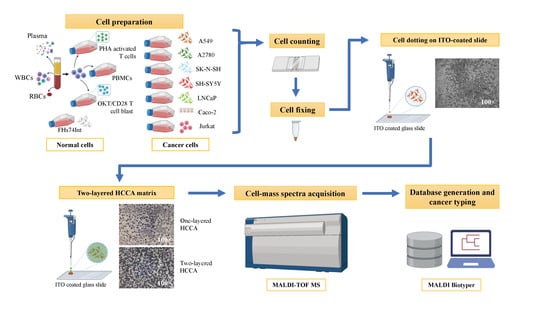
1. Introduction
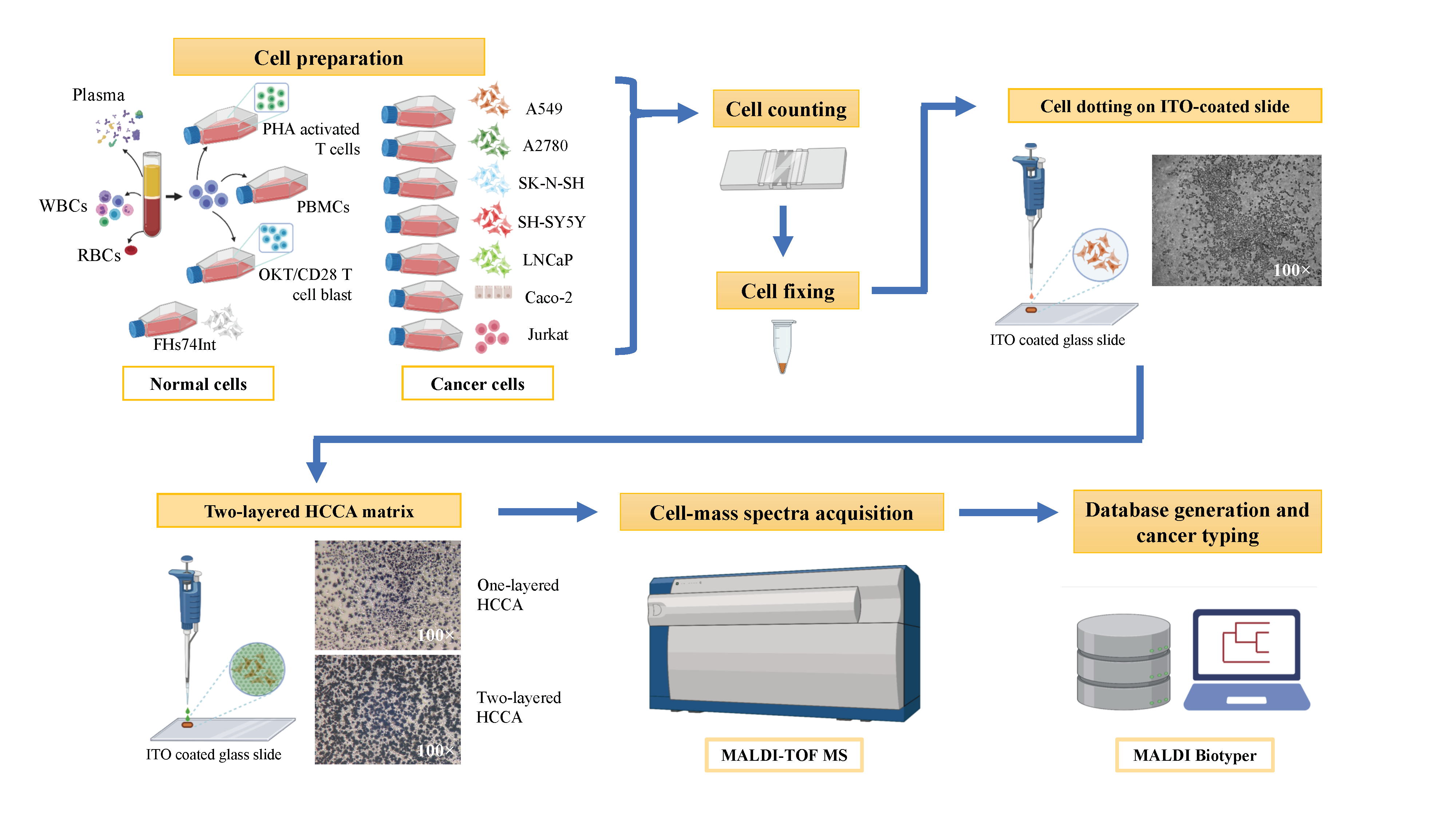
2. Materials and Methods
2.1. Culture of Immortalised Cell Lines
2.2. Generation of Activated T-Cells and OKT3/CD28 Blasts
2.3. Plasma and Red Blood Cell Preparation
2.4. hMX RBC Lysis and WBC Depletion
2.5. Recovery Rates of Cancer Cells during hMX Lysis and Depletion
2.6. Spiked Cancer Cell/WBC Sample Preparation
2.7. Cell Dotting on ITO Coated Slide
2.8. MALDI-TOF MS
2.9. Spectrum Analysis and Database Creation
3. Results
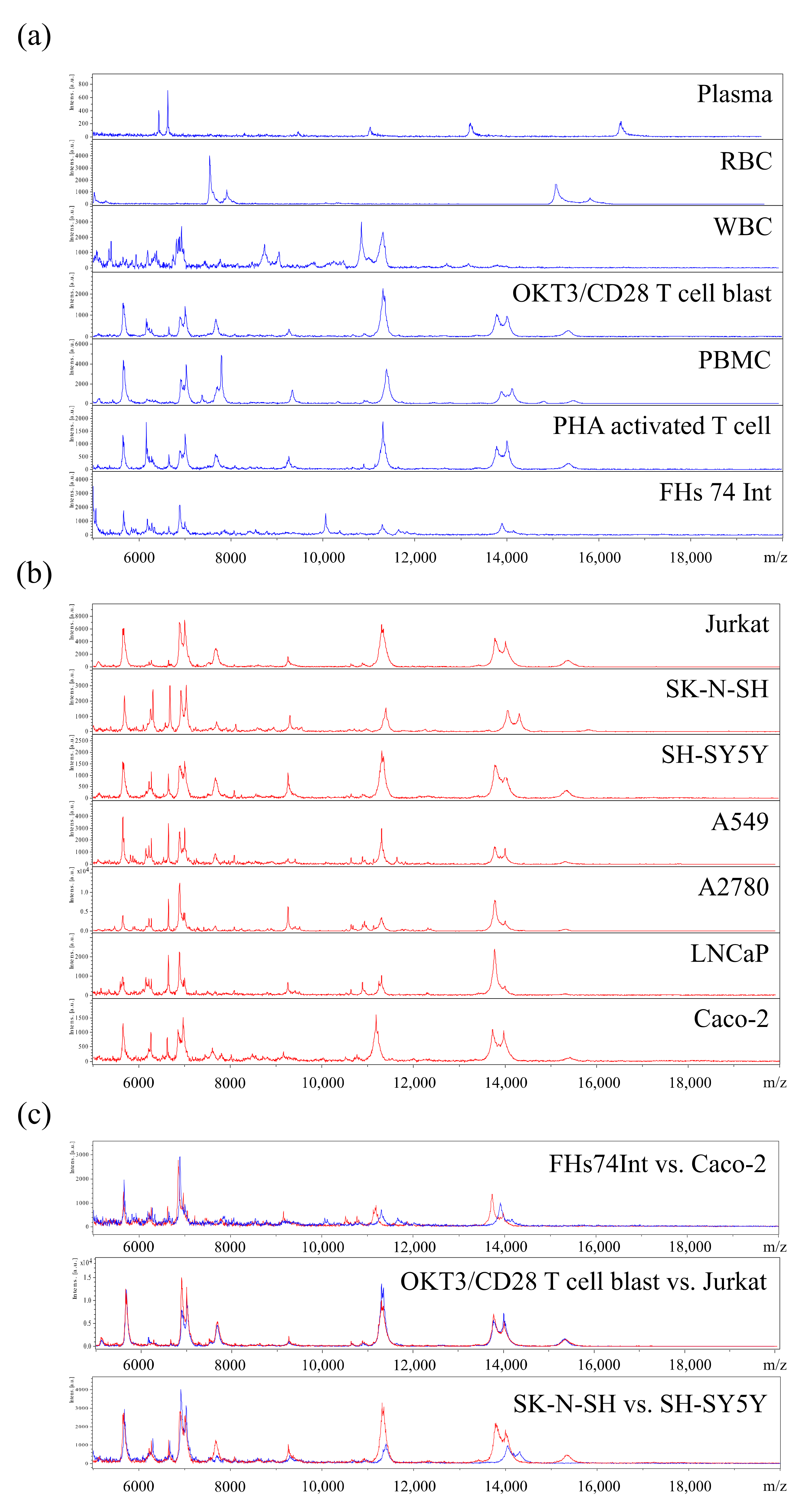
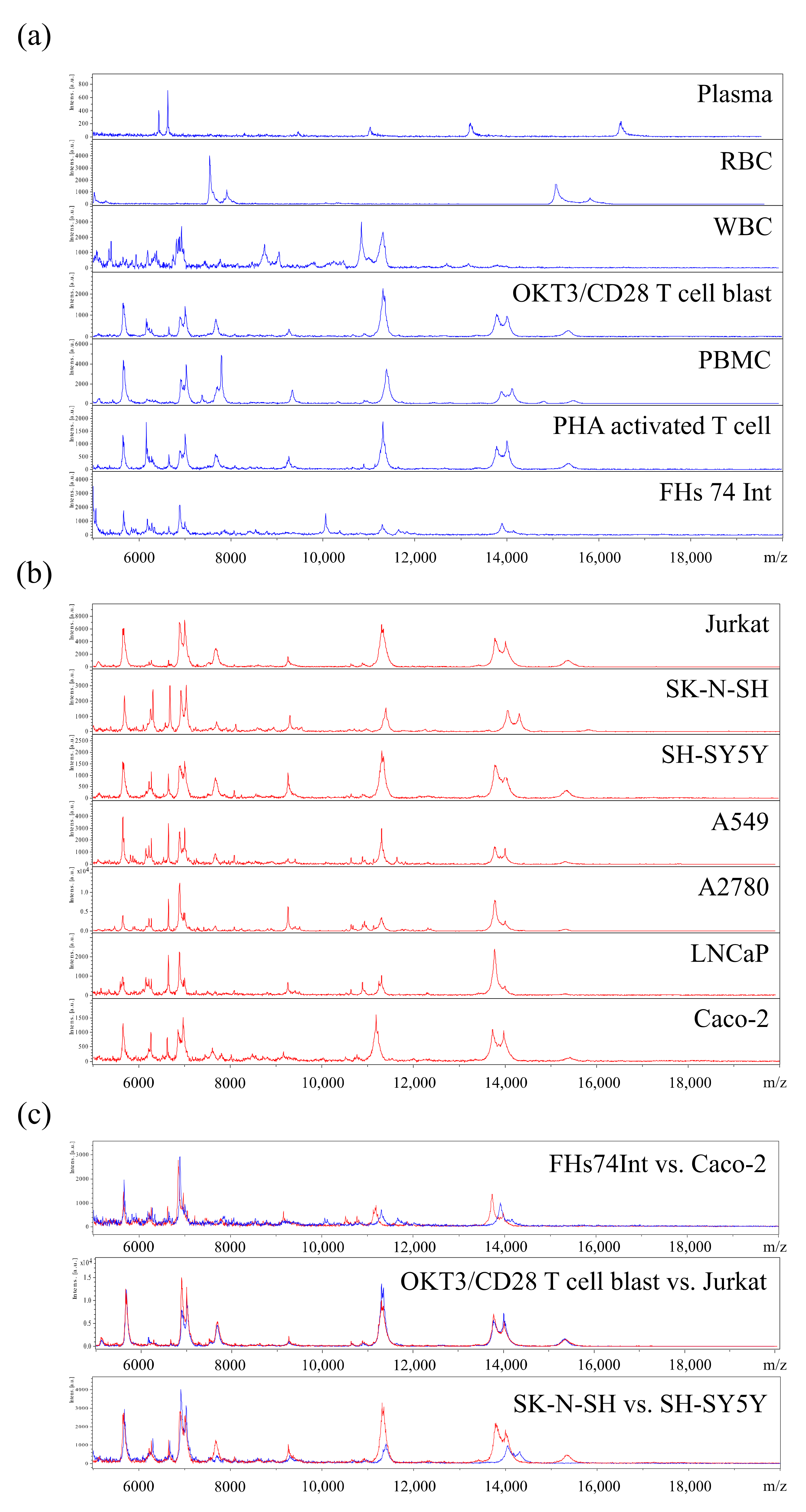
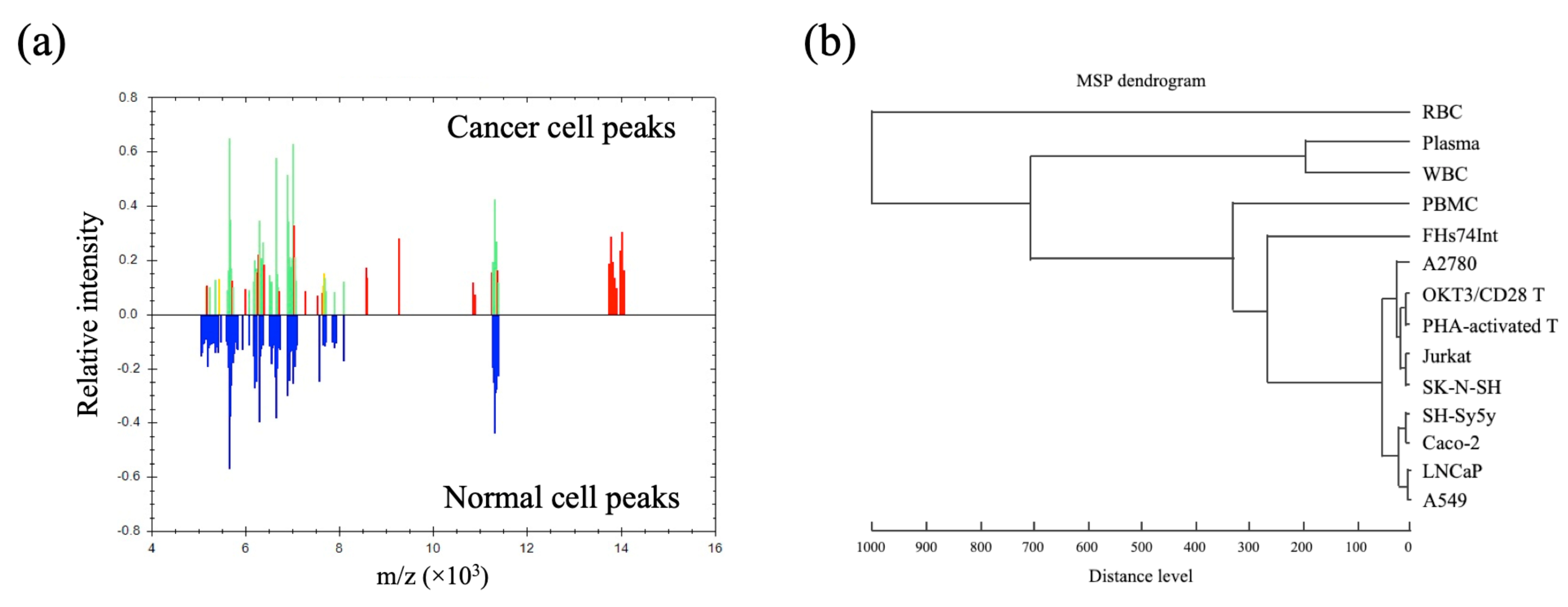
3.1. Profiling of MSP for Various Cell Types Using MALDI-TOF MS
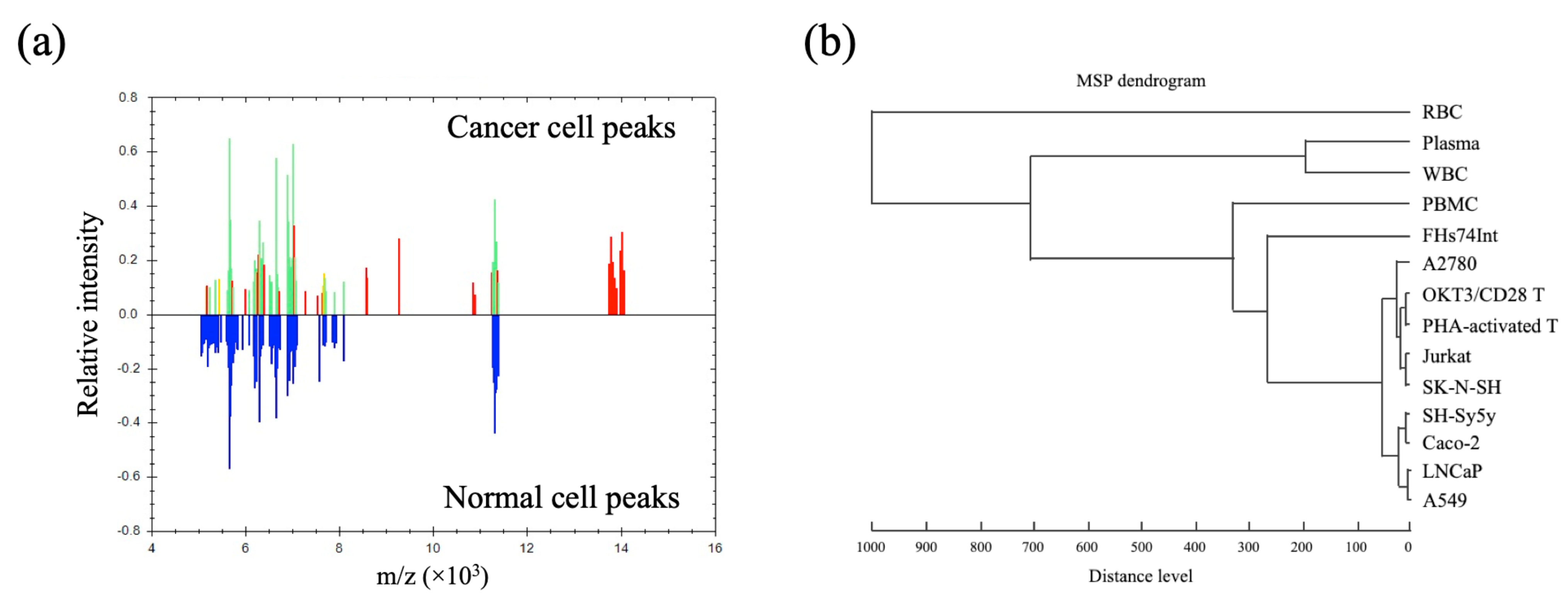
3.2. Cell-MSP Database Generation
3.3. Single Cancer Cell Typing
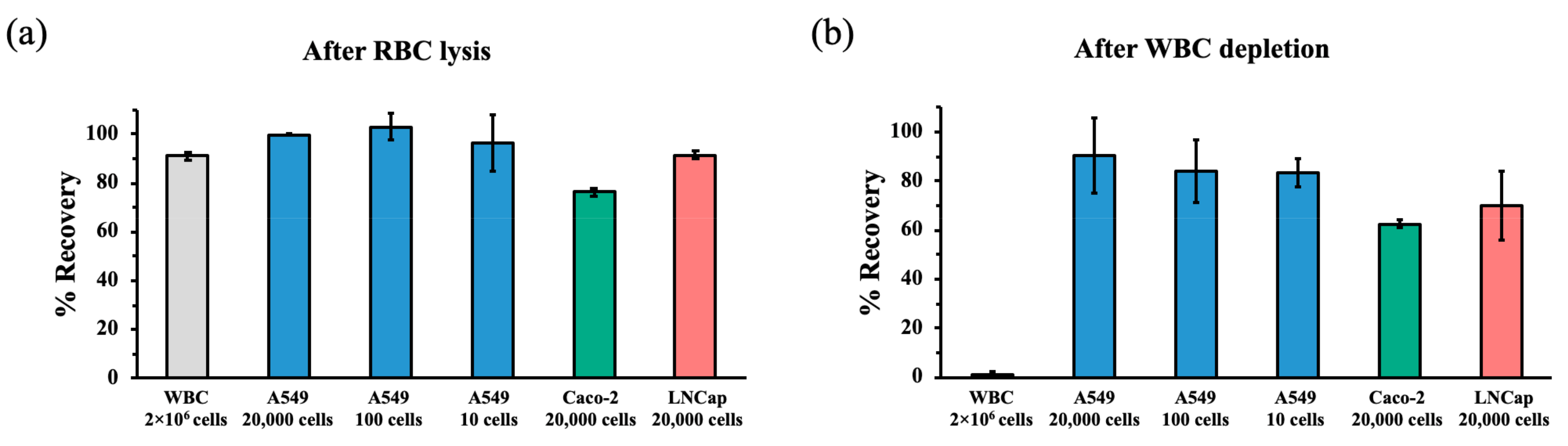
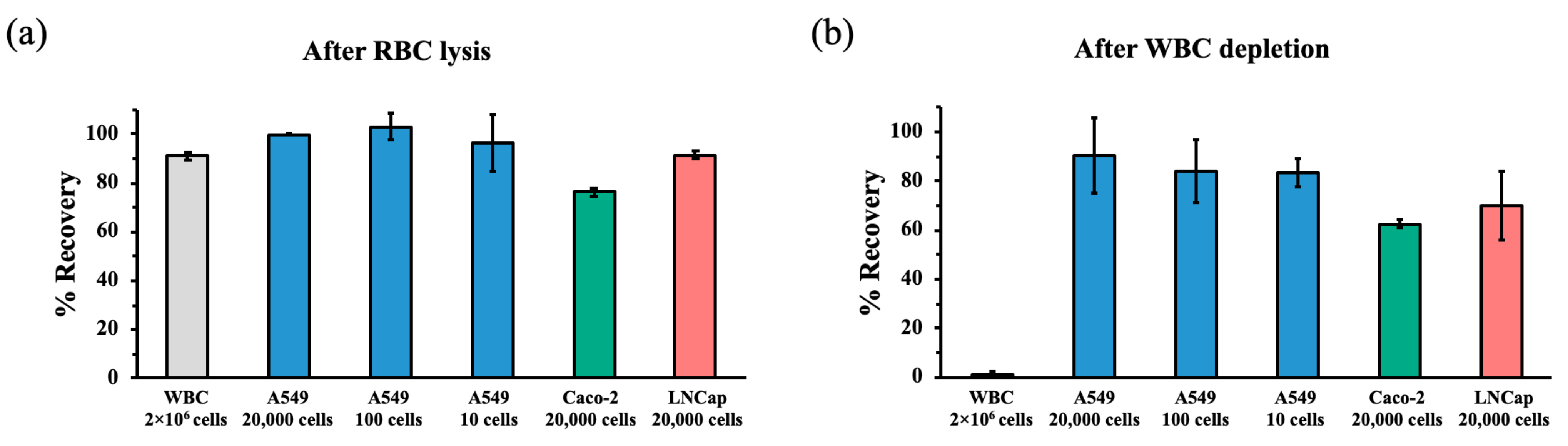
3.4. Recovery Rates of Cancer Cells after RBC Lysis and WBC Depletion during hMX Separation
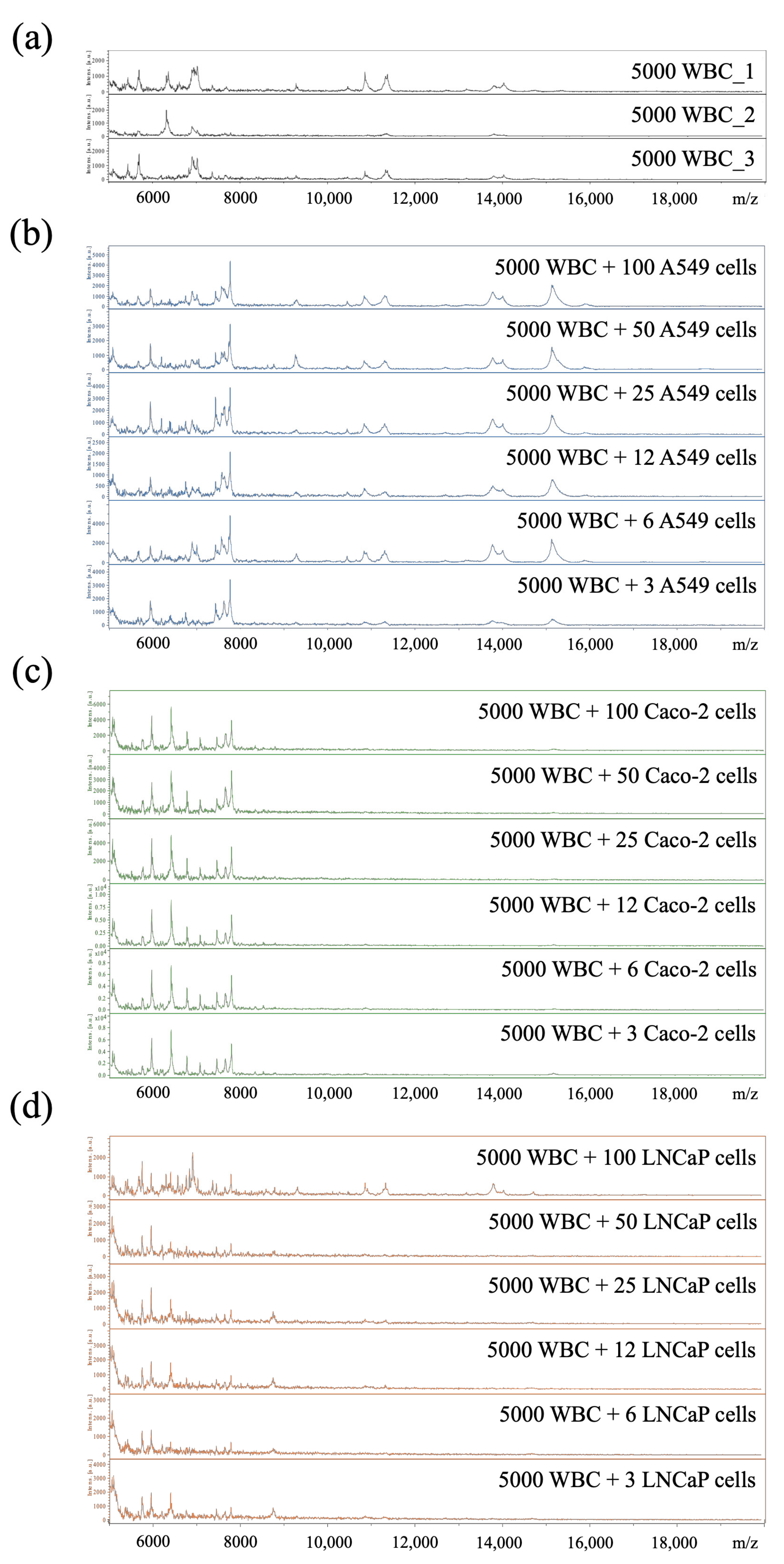
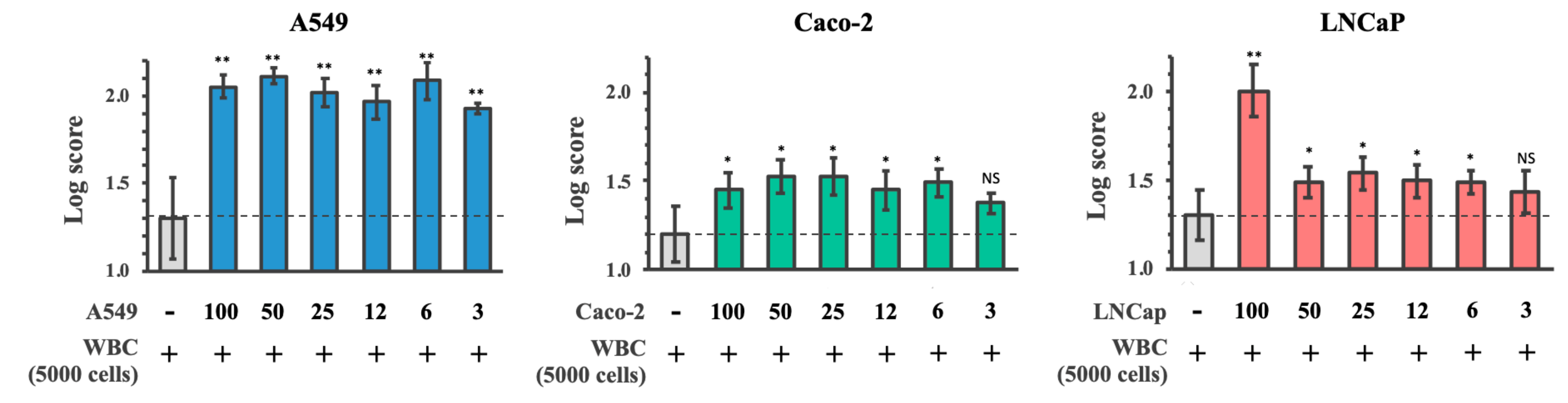
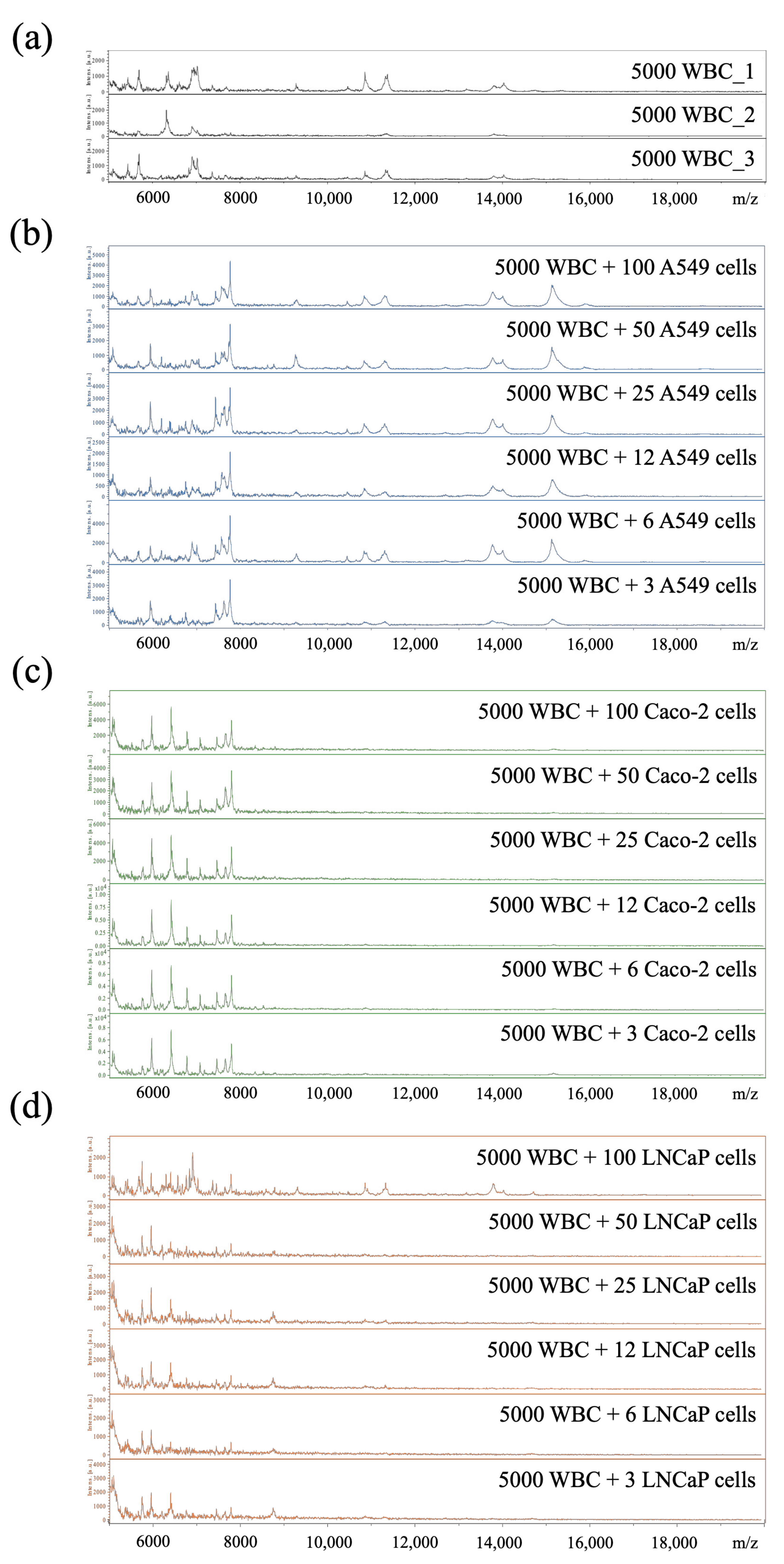
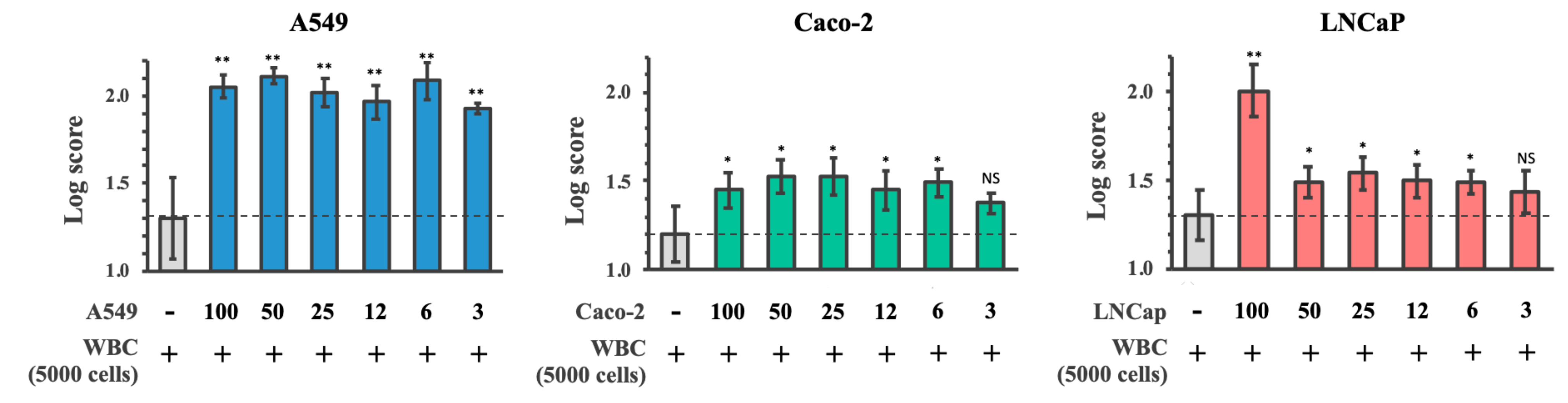
3.5. Limit of Detection of Cancer Cells Spiked into WBC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhakdi, S.C.P.; Suriyaphol, P.; Thaicharoen, P.; Grote, S.T.K.; Komoltri, C.; Chaiyaprasithi, B.; Charnkaew, K. Accuracy of Tumour-Associated Circulating Endothelial Cells as a Screening Biomarker for Clinically Significant Prostate Cancer. Cancers 2019, 11, 1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, A.C. Tumor Endothelial Cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.P.; Gires, O.; Wang, D.D.; Li, L.; Wang, H. Comprehensive in Situ Co-Detection of Aneuploid Circulating Endothelial and Tumor Cells. Sci. Rep. 2017, 7, 9789. [Google Scholar] [CrossRef] [Green Version]
- Cima, I.; Kong, S.L.; Sengupta, D.; Tan, I.B.; Phyo, W.M.; Lee, D.; Hu, M.; Iliescu, C.; Alexander, I.; Goh, W.L.; et al. Tumor-Derived Circulating Endothelial Cell Clusters in Colorectal Cancer. Sci. Transl. Med. 2016, 8, 345ra89. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Dicker, D.T.; Matthew, E.; El-Deiry, W.S.; Alpaugh, R.K. Circulating Tumor Cells: Silent Predictors of Metastasis. F1000Research 2017, 6, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J.; Obenauf, A.C. Metastatic Colonization by Circulating Tumour Cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, J.C.; Niederacher, D.; Topp, S.A.; Honisch, E.; Schumacher, S.; Schmitz, N.; Zacarias Fohrding, L.; Vay, C.; Hoffmann, I.; Kasprowicz, N.S.; et al. Diagnostic Leukapheresis Enables Reliable Detection of Circulating Tumor Cells of Nonmetastatic Cancer Patients. Proc. Natl. Acad. Sci. USA 2013, 110, 16580–16585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamrita, B.; Chahed, K.; Kabbage, M.; Guillier, C.L.; Trimeche, M.; Chaieb, A.; Chouchane, L. Identification of Tumor Antigens That Elicit a Humoral Immune Response in Breast Cancer Patients’ Sera by Serological Proteome Analysis (Serpa). Clin. Chim. Acta 2008, 393, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Tamminga, M.; Andree, K.C.; Hiltermann, T.J.N.; Jayat, M.; Schuuring, E.; van den Bos, H.; Spierings, D.C.J.; Lansdorp, P.M.; Timens, W.; Terstappen, L.W.M.N.; et al. Detection of Circulating Tumor Cells in the Diagnostic Leukapheresis Product of Non-Small-Cell Lung Cancer Patients Comparing Cellsearch® and Iset. Cancers 2020, 12, 896. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.L.; Zhu, P.; Makarova, O.V.; Martin, S.S.; Charpentier, M.; Chumsri, S.; Li, S.; Amstutz, P.; Tang, C.M. The systematic study of circulating tumor cell isolation using lithographic microfilters. RSC. Adv. 2014, 9, 4334–4342. [Google Scholar] [CrossRef]
- Vona, G.; Sabile, A.; Louha, M.; Sitruk, V.; Romana, S.; Schütze, K.; Capron, F.; Franco, D.; Pazzagli, M.; Vekemans, M.; et al. Isolation by size of epithelial tumor cells: A new method for the immunomorphological and molecular characterization of circulating tumor cells. Am. J. Pathol. 2000, 156, 57–63. [Google Scholar] [CrossRef]
- Weitz, J.; Kienle, P.; Lacroix, J.; Willeke, F.; Benner, A.; Lehnert, T.; Herfarth, C.; von Knebel Doeberitz, M. Dissemination of tumor cells in patients undergoing surgery for colorectal cancer. Clin. Cancer Res. 1998, 4, 343–348. [Google Scholar] [PubMed]
- Winer-Jones, J.P.; Vahidi, B.; Arquilevich, N.; Fang, C.; Ferguson, S.; Harkins, D.; Hill, C.; Klem, E.; Pagano, P.C.; Peasley, C.; et al. Circulating tumor cells: Clinically relevant molecular access based on a novel CTC flow cell. PLoS ONE 2014, 9, e86717. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Zhang, X.; Zhang, Q.; Yang, J.; Chen, Q.; Wang, J.; Li, X.; Chen, J.; Ma, T.; Li, G.; et al. Vimentin-positive circulating tumor cells as a biomarker for diagnosis and treatment monitoring in patients with pancreatic cancer. Cancer Lett. 2019, 452, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, A.; Kowalewska, M.; Góźdź, S. Current approaches for avoiding the limitations of circulating tumor cells detection methods-implications for diagnosis and treatment of patients with solid tumors. Transl. Res. 2017, 185, 58–84e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, N.; Meier-Stiegen, F.; Banys, M.; Neubauer, H.; Ruckhaeberle, E.; Fehm, T. Expression of stem cell and epithelial-mesenchymal transition markers in circulating tumor cells of breast cancer patients. BioMed Res. Int. 2014, 2014, 415721. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009, 69, 8536–8539. [Google Scholar] [CrossRef] [Green Version]
- Bhakdi, S.C.; Thaicharoen, P. Easy Employment and Crosstalk-Free Detection of Seven Fluorophores in a Widefield Fluorescence Microscope. Methods Protoc. 2018, 1, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coumans, F.A.; van Dalum, G.; Beck, M.; Terstappen, L.W. Filter characteristics influencing circulating tumor cell enrichment from whole blood. PLoS ONE 2013, 8, e61770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozkumur, E.; Shah, A.M.; Ciciliano, J.C.; Emmink, B.L.; Miyamoto, D.T.; Brachtel, E.; Yu, M.; Chen, P.I.; Morgan, B.; Trautwein, J.; et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci. Transl. Med. 2013, 5, 179ra47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, K.; Wei, W.; Jin, Y.; Wang, S.; Zhao, D.; Wang, S.; Gao, X.; Qiao, C.; Yue, H.; Ma, G.; et al. Biomimetic Immuno-Magnetosomes for High-Performance Enrichment of Circulating Tumor Cells. Adv. Mater. 2016, 28, 7929–7935. [Google Scholar] [CrossRef] [PubMed]
- Kowalewska, M.; Chechlińska, M.; Markowicz, S.; Kober, P.; Nowak, R. The relevance of RT-PCR detection of disseminated tumour cells is hampered by the expression of markers regarded as tumour-specific in activated lymphocytes. Eur. J. Cancer 2006, 42, 2671–2674. [Google Scholar] [CrossRef] [PubMed]
- Seng, P.; Drancourt, M.; Gouriet, F.; La Scola, B.; Fournier, P.E.; Rolain, J.M.; Raoult, D. Ongoing revolution in bacteriology: Routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin. Infect. Dis. 2009, 49, 543–551. [Google Scholar] [CrossRef]
- Lay, J.O., Jr.; Holland, R.D. Rapid identification of bacteria based on spectral patterns using MALDI-TOFMS. Methods Mol. Biol. 2000, 146, 461–487. [Google Scholar]
- Petersen, C.E.; Valentine, N.B.; Wahl, K.L. Characterization of microorganisms by MALDI mass spectrometry. Methods Mol. Biol. 2009, 492, 367–379. [Google Scholar]
- Sauget, M.; Valot, B.; Bertrand, X.; Hocquet, D. Can MALDI-TOF Mass Spectrometry Reasonably Type Bacteria? Trends Microbiol. 2017, 25, 447–455. [Google Scholar] [CrossRef]
- Ouedraogo, R.; Flaudrops, C.; Ben Amara, A.; Capo, C.; Raoult, D.; Mege, J.L. Global analysis of circulating immune cells by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. PLoS ONE 2010, 5, e13691. [Google Scholar] [CrossRef] [Green Version]
- Ouedraogo, R.; Textoris, J.; Gorvel, L.; Daumas, A.; Capo, C.; Mege, J.L. Analysis and Characterization of Immune Cells and Their Activation Status by Whole-Cell MALDI-TOF Mass Spectrometry. Methods Mol. Biol. 2019, 2024, 339–351. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Normal Condition | Cancers | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FHs74Int | PBMC | PHA-Activated T-Cell | Plasma | RBC | OKT/CD28 T-Cell Blast | WBC | A2780 | A549 | Caco-2 | Jurkat | LNCaP | SK-N-SH | SH-SY5Y | |
| FHs74Int | 2.1350 ± 0.2803 | 1.4915 ± 0.1994 | 1.6322 ± 0.3051 | 1.6348 ± 0.1322 | 0.6119 ± 0.3286 | 1.7119 ± 0.2587 | 1.8191 ± 0.0550 | 1.9049 ± 0.1933 | 2.0821 ± 0.1652 | 2.0012 ± 0.1727 | 1.7303 ± 0.169 | 2.0619 ± 0.1925 | 1.7640 ± 0.2299 | 1.8670 ± 0.2323 |
| PBMC | 1.7701 ± 0.1779 | 2.1496 ± 0.2651 | 1.4963 ± 0.2915 | 1.3458 ± 0.3576 | 0.5609 ± 0.4154 | 1.5627 ± 0.3147 | 1.6248 ± 0.1681 | 1.6007 ± 0.3079 | 1.5321 ± 0.3421 | 1.6986 ± 0.3045 | 1.7353 ± 0.3831 | 1.5169 ± 0.2545 | 1.5917 ± 0.3260 | 1.7939 ± 0.2621 |
| PHA-activated T-cell | 2.0488 ± 0.1909 | 1.6674 ± 0.1591 | 2.6313 ± 0.1089 | 0.7704 ± 0.1640 | 0.5730 ± 0.3678 | 2.5584 ± 0.1147 | 1.5338 ± 0.1197 | 2.1848 ± 0.1307 | 2.1576 ± 0.1746 | 2.1501 ± 0.1913 | 2.3600 ± 0.1887 | 2.1788 ± 0.0780 | 2.3289 ± 0.1192 | 2.3708 ± 0.0889 |
| Plasma | 1.3979 ± 0.2461 | 0.9940 ± 0.1887 | 0.7743 ± 0.2208 | 1.8753 ± 0.1970 | 0.4516 ± 0.3364 | 0.6832 ± 0.2934 | 1.7469 ± 0.1210 | 1.0028 ± 0.2665 | 1.1963 ± 0.3093 | 1.1208 ± 0.2618 | 0.9300 ± 0.2648 | 1.1220 ± 0.1533 | 0.8733 ± 0.2831 | 1.0638 ± 0.1367 |
| RBC | 0.5175 ± 0.6793 | 0.7217 ± 0.4881 | 0.2508 ± 0.2748 | 0.5910 ± 0.6671 | 1.9847 ± 0.3313 | 0.1100 ± 0.2578 | 0.7912 ± 0.6389 | 0.1550 ± 0.3025 | 0.5266 ± 0.5596 | 0.4926 ± 0.4792 | 0.3714 ± 0.2815 | 0.5056 ± 0.6176 | 0.2286 ± 0.3102 | 0.4845 ± 0.4842 |
| OKT/CD28 T-cell blast | 2.0774 ± 0.1380 | 1.6846 ± 0.0643 | 2.5799 ± 0.0458 | 0.8026 ± 0.2365 | 0.7324 ± 0.3286 | 2.7106 ± 0.0734 | 1.5184 ± 0.0869 | 2.2299 ± 0.0837 | 2.2066 ± 0.1191 | 2.1997 ± 0.1279 | 2.4063 ± 0.1366 | 2.2629 ± 0.0394 | 2.2910 ± 0.1400 | 2.4191 ± 0.0642 |
| WBC | 1.4917 ± 0.2092 | 1.1169 ± 0.3018 | 0.9202 ± 0.4080 | 1.6827 ± 0.2131 | 0.5963 ± 0.1684 | 0.8511 ± 0.2450 | 2.3397 ± 0.1838 | 1.1211 ± 0.2700 | 1.3692 ± 0.2490 | 1.2038 ± 0.1767 | 1.1408 ± 0.2247 | 1.3104 ± 0.2443 | 1.0331 ± 0.2621 | 1.1588 ± 0.2317 |
| A2780 | 2.1235 ± 0.1602 | 1.5215 ± 0.1751 | 2.0660 ± 0.2124 | 1.2680 ± 0.3177 | 0.3727 ± 0.3457 | 2.1136 ± 0.2224 | 1.6691 ± 0.0992 | 2.4944 ± 0.3423 | 2.2643 ± 0.1459 | 2.2756 ± 0.2128 | 2.2494 ± 0.2313 | 2.2714 ± 0.1548 | 2.2019 ± 0.2970 | 2.2298 ± 0.2702 |
| A549 | 2.1816 ± 0.2151 | 1.5275 ± 0.1524 | 1.8982 ± 0.2033 | 1.5506 ± 0.2227 | 0.3753 ± 0.3012 | 1.9355 ± 0.1950 | 1.6936 ± 0.1122 | 2.1782 ± 0.2156 | 2.4265 ± 0.1411 | 2.1329 ± 0.1696 | 2.0331 ± 0.2179 | 2.2404 ± 0.1166 | 2.0062 ± 0.1448 | 2.1106 ± 0.1536 |
| Caco-2 | 1.9453 ± 0.1441 | 1.5742 ± 0.2473 | 1.9038 ± 0.2818 | 1.3304 ± 0.2509 | 0.4539 ± 0.2892 | 1.9255 ± 0.3473 | 1.8031 ± 0.0916 | 1.9760 ± 0.2606 | 2.0722 ± 0.1635 | 2.3448 ± 0.1935 | 1.9798 ± 0.2957 | 2.0498 ± 0.1952 | 1.9230 ± 0.2567 | 1.9398 ± 0.3225 |
| Jurkat | 1.9232 ± 0.1248 | 1.7166 ± 0.1735 | 2.1508 ± 0.1942 | 1.0892 ± 0.3198 | 0.6737 ± 0.3454 | 2.1828 ± 0.1656 | 1.5729 ± 0.0996 | 2.1918 ± 0.1368 | 2.1099 ± 0.0933 | 2.2634 ± 0.1560 | 2.4694 ± 0.2012 | 2.1695 ± 0.1454 | 2.2253 ± 0.2057 | 2.2867 ± 0.1489 |
| LNCaP | 2.0905 ± 0.1492 | 1.4469 ± 0.1570 | 2.0563 ± 0.1790 | 1.3670 ± 0.3046 | 0.6243 ± 0.2958 | 2.1313 ± 0.1589 | 1.7233 ± 0.1455 | 2.2517 ± 0.1628 | 2.2276 ± 0.1262 | 2.1828 ± 0.1285 | 2.2080 ± 0.2369 | 2.5943 ± 0.1017 | 2.1270 ± 0.1728 | 2.1682 ± 0.1521 |
| SK-N-SH | 1.9944 ± 0.3021 | 1.7178 ± 0.2224 | 2.0928 ± 0.3945 | 1.1005 ± 0.2506 | 0.6191 ± 0.5308 | 2.1130 ± 0.4056 | 1.5143 ± 0.1422 | 2.1351 ± 0.3092 | 1.9520 ± 0.4005 | 2.1358 ± 0.2139 | 2.2001 ± 0.3488 | 1.9942 ± 0.4116 | 2.2406 ± 0.4680 | 2.2211 ± 0.2660 |
| SH-SY5Y | 2.0903 ± 0.1268 | 1.7604 ± 0.1654 | 2.2288 ± 0.1694 | 1.1884 ± 0.3393 | 0.7160 ± 0.5143 | 2.2949 ± 0.2257 | 1.6132 ± 0.1342 | 2.2423 ± 0.1941 | 2.1589 ± 0.1547 | 2.2678 ± 0.1251 | 2.3613 ± 0.2017 | 2.2732 ± 0.0793 | 2.2930 ± 0.1805 | 2.4820 ± 0.1413 |
| Sample | Cancers | ||||||
|---|---|---|---|---|---|---|---|
| A2780 | A549 | Caco-2 | Jurkat | LNCaP | SK-N-SH | SH-SY5Y | |
| WBC alone (5000 cells) | 1.0321 ± 0.2761 | 1.2988 ± 0.2297 | 1.2059 ± 0.1572 | 0.9033 ± 0.1858 | 1.3054 ± 0.1392 | 0.8873 ± 0.2569 | 1.1791 ± 0.1741 |
| WBC + A549 100 cells | 1.9630 ± 0.1058 | 2.0542 ±0.0675 | 1.9288 ± 0.0837 | 1.9156 ± 0.0648 | 2.0164 ± 0.1132 | 1.9640 ± 0.1252 | 1.9562 ± 0.1576 |
| WBC + A549 50 cells | 2.0150 ± 0.0131 | 2.1133 ±0.0446 | 2.0340 ± 0.0342 | 1.8923 ± 0.0985 | 2.0093 ± 0.0276 | 2.0413 ± 0.0614 | 2.0740 ± 0.0288 |
| WBC + A549 25 cells | 1.8237 ± 0.1525 | 2.0170 ±0.0832 | 1.8877 ± 0.1299 | 1.8937 ± 0.0535 | 1.9943 ± 0.0488 | 1.9313 ± 0.0803 | 1.9473 ± 0.1320 |
| WBC + A549 12 cells | 1.7277 ± 0.1466 | 1.9660 ±0.0957 | 1.7647 ± 0.1791 | 1.6070 ± 0.1327 | 1.8267 ± 0.1426 | 1.5607 ± 0.0715 | 1.7387 ± 0.1296 |
| WBC + A549 6 cells | 2.0620 ± 0.1580 | 2.0867 ±0.1100 | 1.9493 ± 0.1955 | 1.9973 ± 0.0304 | 2.0787 ± 0.1186 | 2.0723 ± 0.0509 | 2.1767 ± 0.0850 |
| WBC + A549 3 cells | 1.6043 ± 0.1917 | 1.9287 ±0.0325 | 1.6313 ± 0.1517 | 1.4933 ± 0.1420 | 1.6990 ± 0.1413 | 1.5620 ± 0.1274 | 1.6447 ± 0.0674 |
| WBC + Caco-2 100 cells | 0.9962 ± 0.2365 | 1.2082 ± 0.1990 | 1.4487 ±0.0973 | 0.9223 ± 0.1924 | 1.1708 ± 0.0903 | 0.9162 ± 0.2979 | 1.1570 ± 0.1816 |
| WBC + Caco-2 50 cells | 1.1342 ± 0.1317 | 1.2037 ± 0.0660 | 1.5260 ±0.0979 | 1.0262 ± 0.1944 | 1.3920 ± 0.1757 | 0.9752 ± 0.1857 | 1.2365 ± 0.0965 |
| WBC + Caco-2 25 cells | 1.1372 ± 0.2578 | 1.3704 ± 0.1549 | 1.5273 ±0.1048 | 1.1706 ± 0.2425 | 1.3114 ± 0.1364 | 1.0816 ± 0.2680 | 1.1946 ± 0.1729 |
| WBC + Caco-2 12 cells | 1.0020 ± 0.2398 | 1.1852 ± 0.3041 | 1.4497 ±0.1100 | 1.0293 ± 0.1586 | 1.2397 ± 0.1651 | 0.8930 ± 0.1938 | 1.1543 ± 0.1732 |
| WBC + Caco-2 6 cells | 1.2602 ± 0.1989 | 1.4042 ± 0.1298 | 1.4900 ±0.0757 | 1.2568 ± 0.2329 | 1.3110 ± 0.1401 | 0.9445 ± 0.1940 | 1.3058 ± 0.2022 |
| WBC + Caco-2 3 cells | 1.1419 ± 0.2372 | 1.2013 ± 0.1595 | 1.3747 ±0.0539 | 1.0453 ± 0.3087 | 1.1704 ± 0.1621 | 0.8650 ± 0.2197 | 1.2226 ± 0.1806 |
| WBC + LNCaP 100 cells | 1.9282 ± 0.1325 | 1.7682 ± 0.1501 | 1.7817 ± 0.2241 | 1.7393 ± 0.2260 | 2.0077 ±0.1448 | 1.7920 ± 0.2132 | 1.7323 ± 0.2172 |
| WBC + LNCaP 50 cells | 1.2056 ± 0.1210 | 1.4430 ± 0.0933 | 1.2372 ± 0.1781 | 0.9994 ± 0.0773 | 1.4907 ±0.0906 | 0.9514 ± 0.2849 | 1.2144 ± 0.1610 |
| WBC + LNCaP 25 cells | 1.1788 ± 0.1377 | 1.4313 ± 0.0577 | 1.2675 ± 0.2249 | 1.0938 ± 0.1024 | 1.5390 ±0.0897 | 0.8865 ± 0.1607 | 1.3278 ± 0.0426 |
| WBC + LNCaP 12 cells | 1.0170 ± 0.2486 | 1.2545 ± 0.1651 | 1.1175 ± 0.1503 | 1.0415 ± 0.1445 | 1.4963 ±0.0976 | 0.9748 ± 0.1699 | 1.1278 ± 0.2319 |
| WBC + LNCaP 6 cells | 1.1422 ± 0.1773 | 1.2156 ± 0.2179 | 1.1712 ± 0.0925 | 0.9804 ± 0.1444 | 1.4910 ±0.0646 | 1.0528 ± 0.1320 | 1.2680 ± 0.1429 |
| WBC + LNCaP 3 cells | 1.0447 ± 0.2740 | 1.4350 ±0.1640 | 1.1595 ± 0.1455 | 0.9908 ± 0.1840 | 1.4340 ± 0.1204 | 0.8315 ± 0.1774 | 1.1303 ± 0.2518 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiangjong, W.; Bhakdi, S.C.; Woramongkolchai, N.; Vanichapol, T.; Pongsakul, N.; Hongeng, S.; Chutipongtanate, S. Cell-Main Spectra Profile Screening Technique in Simulation of Circulating Tumour Cells Using MALDI-TOF Mass Spectrometry. Cancers 2021, 13, 3775. https://doi.org/10.3390/cancers13153775
Chiangjong W, Bhakdi SC, Woramongkolchai N, Vanichapol T, Pongsakul N, Hongeng S, Chutipongtanate S. Cell-Main Spectra Profile Screening Technique in Simulation of Circulating Tumour Cells Using MALDI-TOF Mass Spectrometry. Cancers. 2021; 13(15):3775. https://doi.org/10.3390/cancers13153775
Chicago/Turabian StyleChiangjong, Wararat, Sebastian Chakrit Bhakdi, Noppawan Woramongkolchai, Thitinee Vanichapol, Nutkridta Pongsakul, Suradej Hongeng, and Somchai Chutipongtanate. 2021. "Cell-Main Spectra Profile Screening Technique in Simulation of Circulating Tumour Cells Using MALDI-TOF Mass Spectrometry" Cancers 13, no. 15: 3775. https://doi.org/10.3390/cancers13153775
APA StyleChiangjong, W., Bhakdi, S. C., Woramongkolchai, N., Vanichapol, T., Pongsakul, N., Hongeng, S., & Chutipongtanate, S. (2021). Cell-Main Spectra Profile Screening Technique in Simulation of Circulating Tumour Cells Using MALDI-TOF Mass Spectrometry. Cancers, 13(15), 3775. https://doi.org/10.3390/cancers13153775