
Identification of an RNA-Binding-Protein-Based Prognostic Model for Ewing Sarcoma


Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. ES Data Processing
2.2. Identification of Differentially Expressed RBPs
2.3. Protein–Protein Interaction (PPI) Network Construction and Functional Enrichment Analyses
2.4. Prognosis-Related RBPs and the Interaction with TFs
2.5. Establishment and Validation of an RBP-Associated Prognostic Risk Model (RPRM)
2.6. Assessment of Gene Expression Level and Prognostic Significance in RPRM
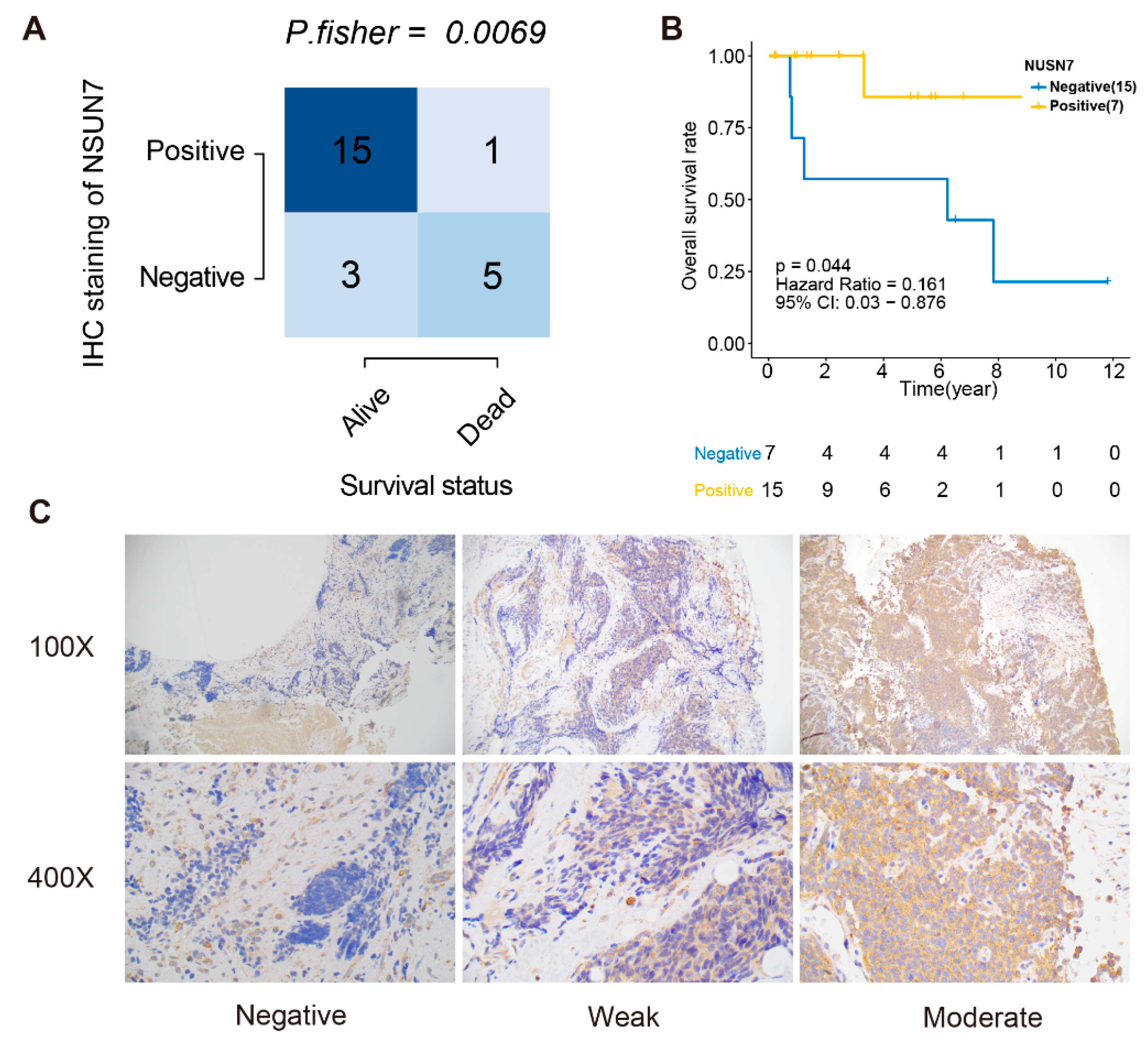
2.7. Immunohistochemistry
- Negative (“Most of the tumor cells were completely negative”): >75% completely negative, <25% with very weak cytoplasmic staining);
- Weak (“Cases exhibited a diffuse weak staining with small areas with negative or stronger immunoreactivity”): >75% of tumor cells exhibited weak immunoreactivity); and
- Moderate (“Cases showed stronger cytoplasmic immunoreactivity in most of the tumor cells, but smaller areas with either weaker or negative immunoreactivity were sometimes observed”): >75% of the tumor cells showed moderate cytoplasmic reactivity.
2.8. Statistical Analysis
3. Results
3.1. Data Preprocessing of the ES Dataset
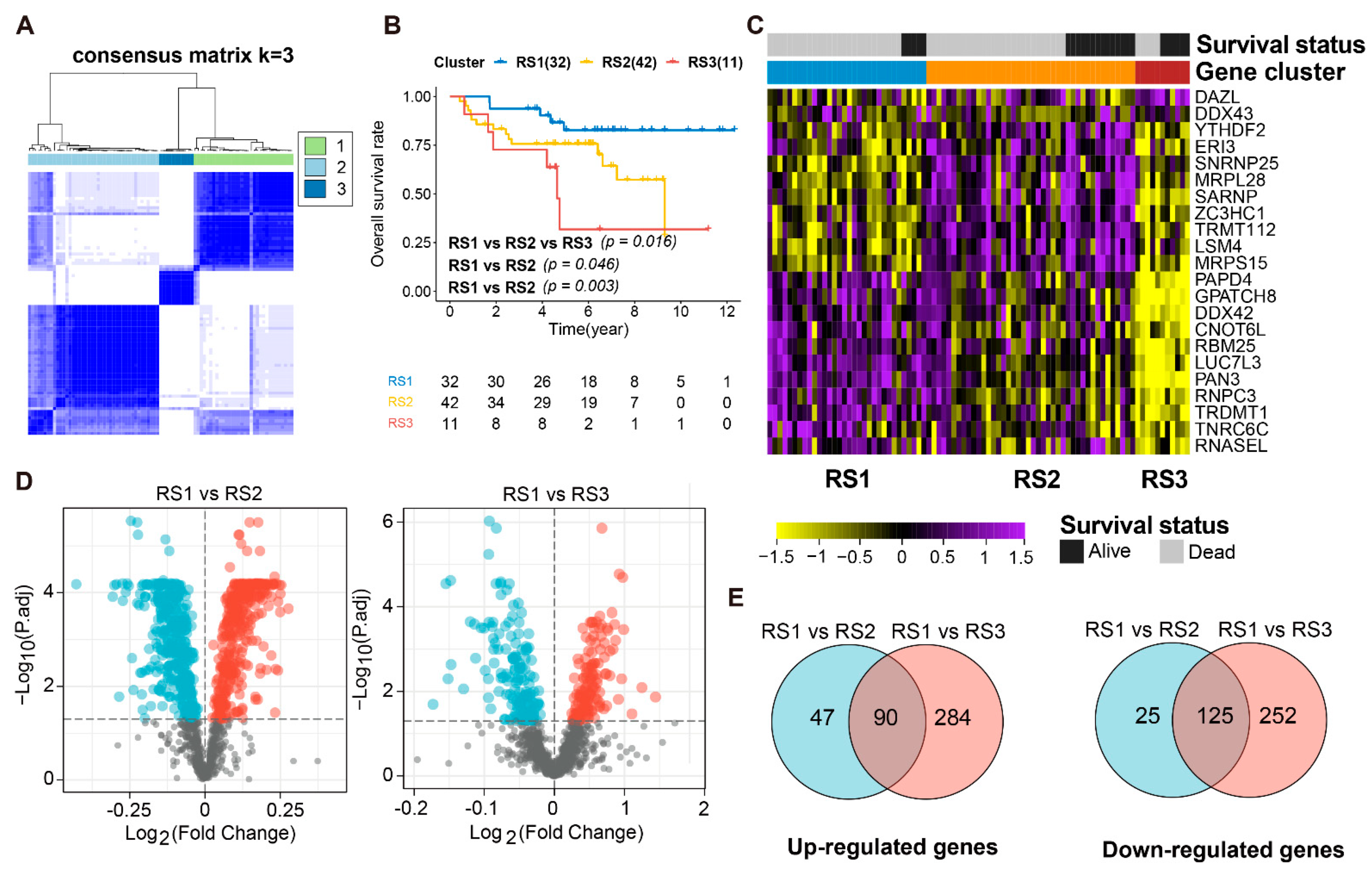
3.2. Identification of DERBPs in ES Patients and Transcriptional Subtypes
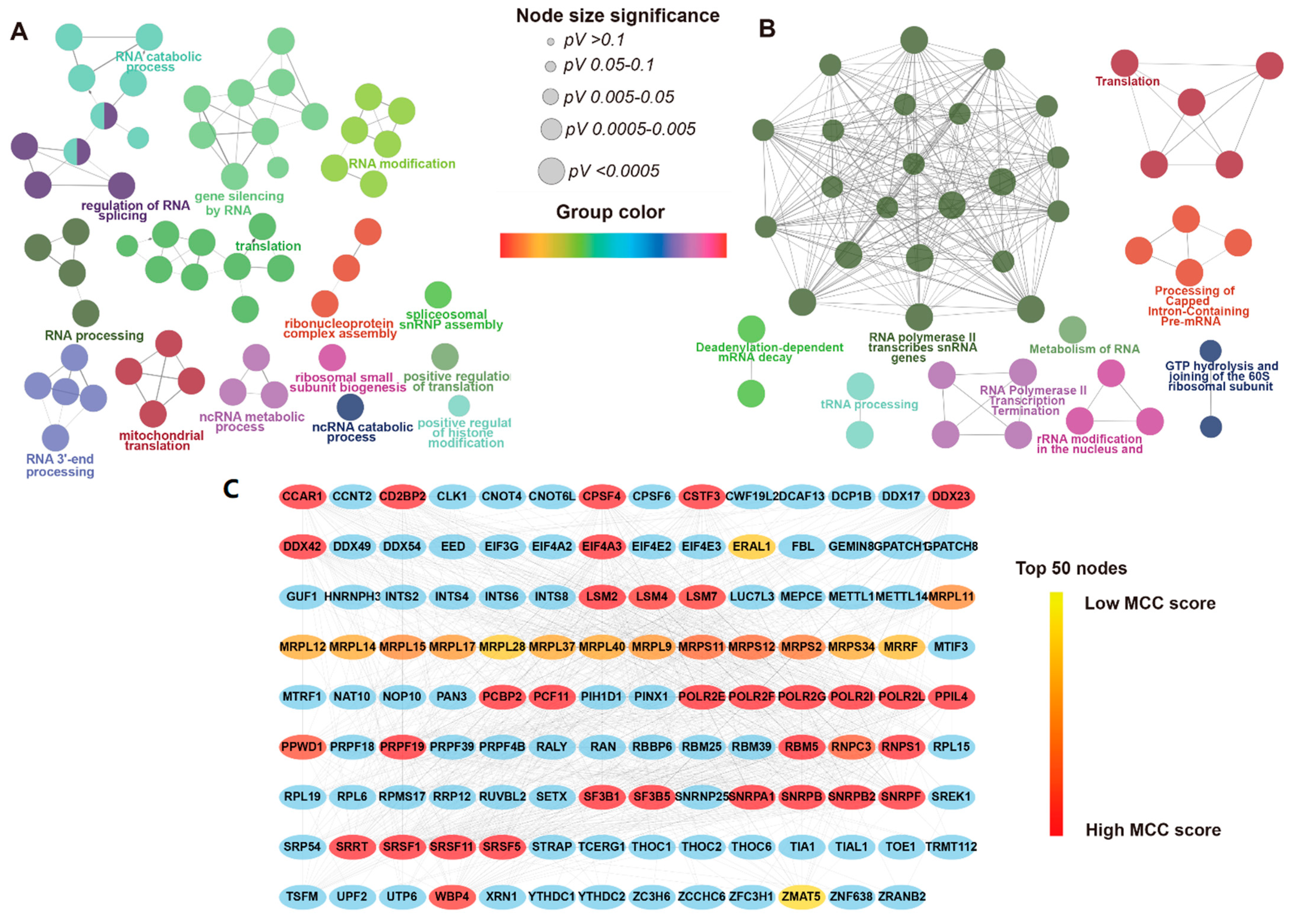
3.3. Functional Enrichment Analysis and PPI Network of DERBPs
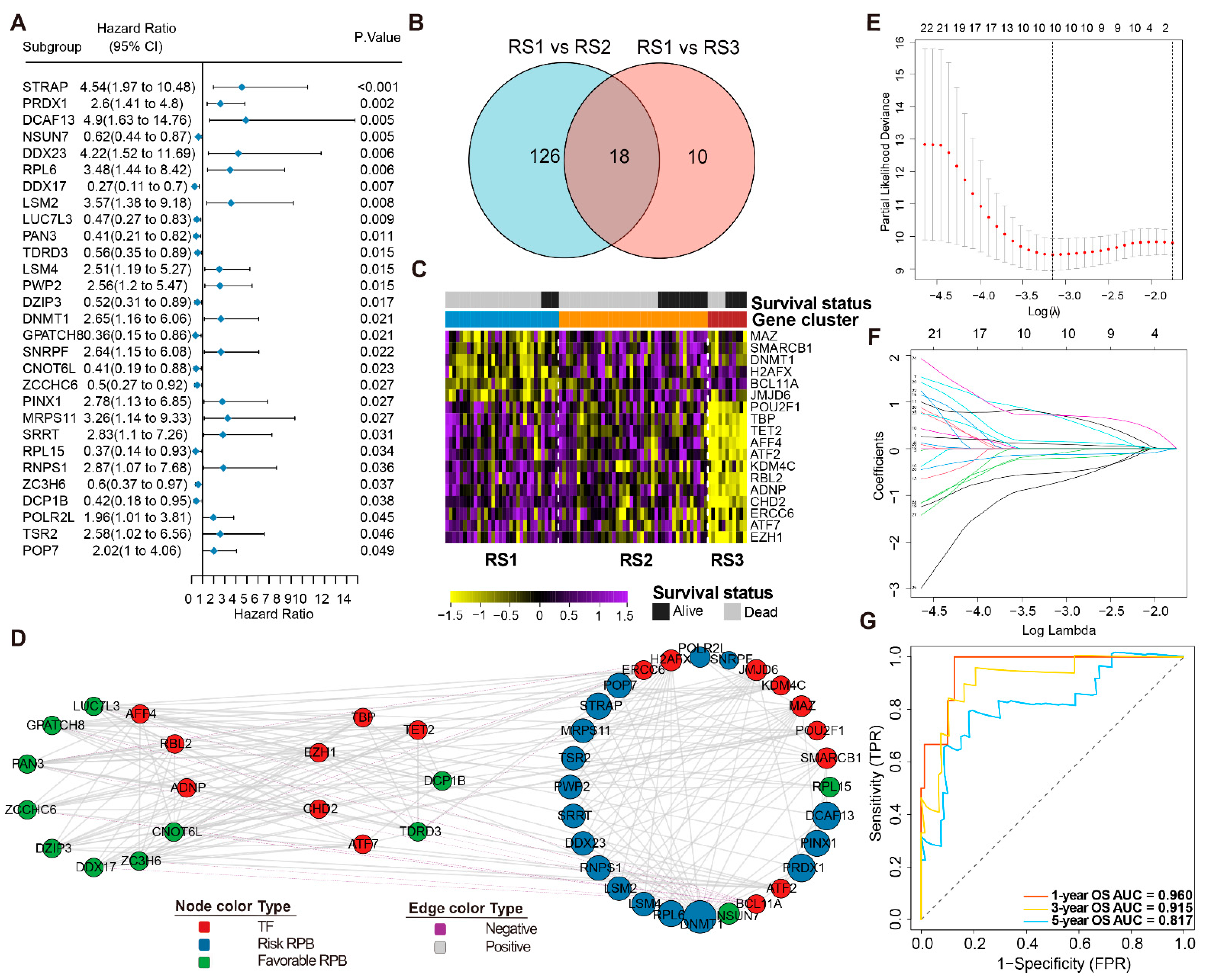
3.4. Prognosis-Related RPBs and the Regulatory Network
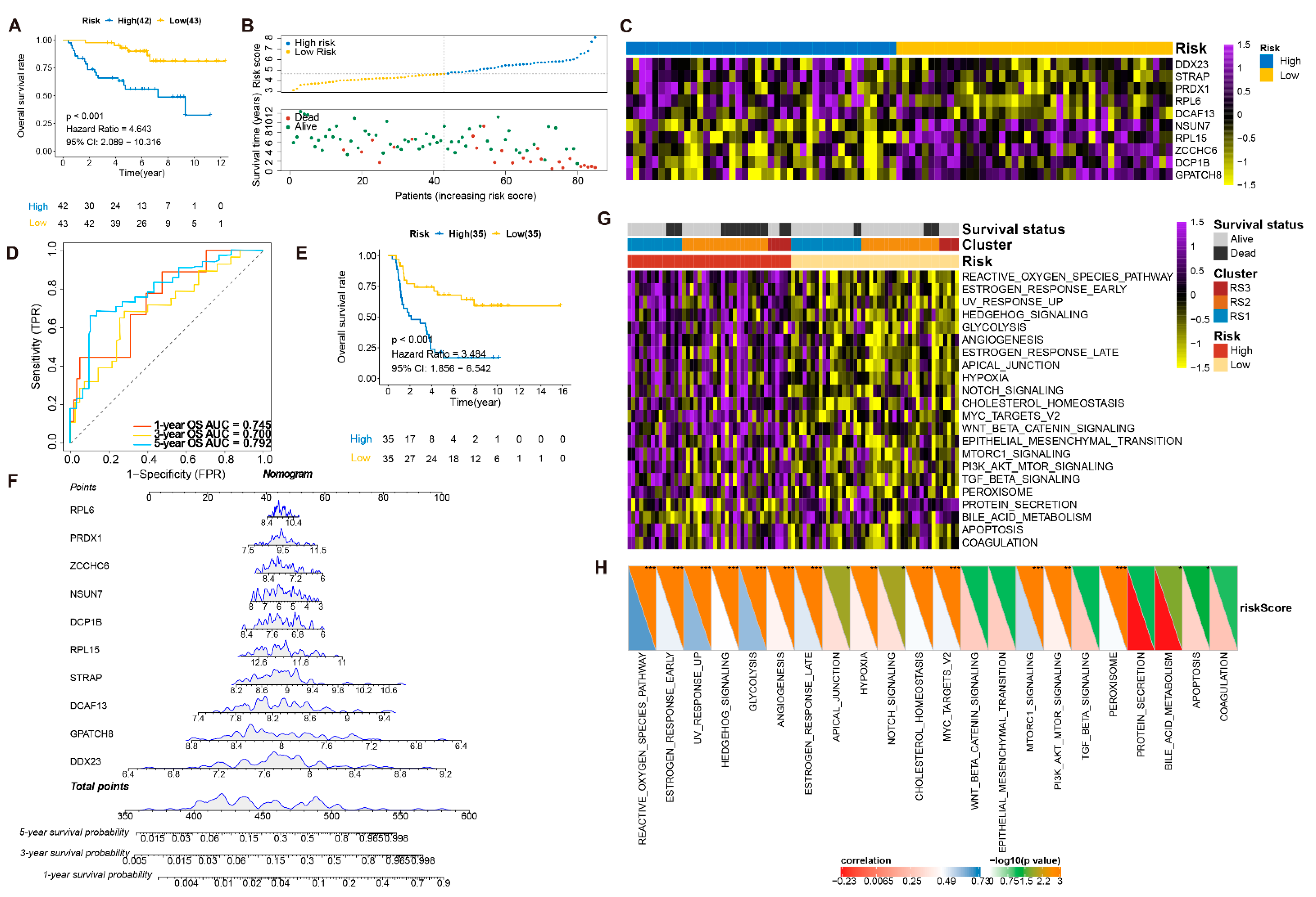
3.5. Construction and Validation of the RPBs-Associated Prognostic Risk Model (RPRM)
3.6. Validation of the Prognostic Value and Expression of the RBPs Involved in RPRM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arndt, C.A.; Rose, P.S.; Folpe, A.L.; Laack, N.N. Common musculoskeletal tumors of childhood and adolescence. Mayo Clin. Proc. 2012, 87, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Esiashvili, N.; Goodman, M.; Marcus, R.B., Jr. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J. Pediatr. Hematol. Oncol. 2008, 30, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.L.; Lessnick, S.L.; Jones, K.B.; Gouw, L.G.; Cummings, J.E.; Cannon-Albright, L.; Schiffman, J.D. Is There a Predisposition Gene for Ewing’s Sarcoma? J. Oncol. 2010, 2010, 397632. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.H.; Lessnick, S.L.; Lopez-Terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T. A second Ewing’s sarcoma translocation, t (21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Brohl, A.S.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014, 10, e1004475. [Google Scholar] [CrossRef]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef]
- Shukla, N.; Schiffman, J.; Reed, D.; Davis, I.J.; Womer, R.B.; Lessnick, S.L.; Lawlor, E.R.; The COG Ewing Sarcoma Biology Committee. Biomarkers in Ewing Sarcoma: The Promise and Challenge of Personalized Medicine. A Report from the Children’s Oncology Group. Front. Oncol. 2013, 3, 141. [Google Scholar] [CrossRef]
- Huang, H.Y.; Illei, P.B.; Zhao, Z.; Mazumdar, M.; Huvos, A.G.; Healey, J.H.; Wexler, L.H.; Gorlick, R.; Meyers, P.; Ladanyi, M. Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion: A highly lethal subset associated with poor chemoresponse. J. Clin. Oncol. 2005, 23, 548–558. [Google Scholar] [CrossRef]
- Kovar, H.; Auinger, A.; Jug, G.; Aryee, D.; Zoubek, A.; Salzer-Kuntschik, M.; Gadner, H. Narrow spectrum of infrequent p53 mutations and absence of MDM2 amplification in Ewing tumours. Oncogene 1993, 8, 2683–2690. [Google Scholar]
- Whelan, J.; Le Deley, M.C.; Dirksen, U.; Le Teuff, G.; Brennan, B.; Gaspar, N.; Hawkins, D.S.; Amler, S.; Bauer, S.; Bielack, S.; et al. High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared With Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E.W.I.N.G.99 and Ewing-2008. J. Clin. Oncol. 2018, 36, 3110. [Google Scholar] [CrossRef]
- Lawlor, E.R.; Sorensen, P.H. Twenty Years on: What Do We Really Know about Ewing Sarcoma and What Is the Path Forward? Crit. Rev. Oncog. 2015, 20, 155–171. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Kuwano, Y.; Nishikawa, T.; Masuda, K.; Rokutan, K. RNA Binding Proteins and Genome Integrity. Int. J. Mol. Sci. 2017, 18, 1341. [Google Scholar] [CrossRef]
- Martin, K.C.; Ephrussi, A. mRNA localization: Gene expression in the spatial dimension. Cell 2009, 136, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Proudfoot, N.J. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 2009, 136, 688–700. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Baltz, A.G.; Munschauer, M.; Schwanhausser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Araya, N.; Hirota, K.; Shimamoto, Y.; Miyagishi, M.; Yoshida, E.; Ishida, J.; Kaneko, S.; Kaneko, M.; Nakajima, T.; Fukamizu, A. Cooperative interaction of EWS with CREB-binding protein selectively activates hepatocyte nuclear factor 4-mediated transcription. J. Biol. Chem. 2003, 278, 5427–5432. [Google Scholar] [CrossRef] [PubMed]
- Rossow, K.L.; Janknecht, R. The Ewing’s sarcoma gene product functions as a transcriptional activator. Cancer Res. 2001, 61, 2690–2695. [Google Scholar]
- Chen, Y.; Hesla, A.C.; Lin, Y.; Ghaderi, M.; Liu, M.; Yang, C.; Zhang, Y.; Tsagkozis, P.; Larsson, O.; Haglund, F. Transcriptome profiling of Ewing sarcomas—Treatment resistance pathways and IGF-dependency. Mol. Oncol. 2020, 14, 1101–1117. [Google Scholar] [CrossRef]
- Mancarella, C.; Pasello, M.; Ventura, S.; Grilli, A.; Calzolari, L.; Toracchio, L.; Lollini, P.L.; Donati, D.M.; Picci, P.; Ferrari, S.; et al. Insulin-Like Growth Factor 2 mRNA-Binding Protein 3 is a Novel Post-Transcriptional Regulator of Ewing Sarcoma Malignancy. Clin. Cancer Res. 2018, 24, 3704–3716. [Google Scholar] [CrossRef]
- Volchenboum, S.L.; Andrade, J.; Huang, L.; Barkauskas, D.A.; Krailo, M.; Womer, R.B.; Ranft, A.; Potratz, J.; Dirksen, U.; Triche, T.J.; et al. Gene Expression Profiling of Ewing Sarcoma Tumors Reveals the Prognostic Importance of Tumor-Stromal Interactions: A Report from the Children’s Oncology Group. J. Pathol. Clin. Res. 2015, 1, 83–94. [Google Scholar] [CrossRef]
- Savola, S.; Klami, A.; Myllykangas, S.; Manara, C.; Scotlandi, K.; Picci, P.; Knuutila, S.; Vakkila, J. High Expression of Complement Component 5 (C5) at Tumor Site Associates with Superior Survival in Ewing’s Sarcoma Family of Tumour Patients. ISRN Oncol. 2011, 2011, 168712. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Veron, A.S.; Tirode, F.; Pierron, G.; Reynaud, S.; Kovar, H.; Oberlin, O.; Lapouble, E.; Ballet, S.; Lucchesi, C.; et al. Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat. Genet. 2012, 44, 323–327. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Meyer, C.A.; Zheng, R.; Qin, Q.; Wu, Q.; Jiang, P.; Li, B.; Shi, X.; Wang, B.; Fan, J.; et al. Cistrome Cancer: A Web Resource for Integrative Gene Regulation Modeling in Cancer. Cancer Res. 2017, 77, e19–e22. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, J.; Chen, G.; Li, M.; Wu, F.X.; Pan, Y. ClusterViz: A Cytoscape APP for Cluster Analysis of Biological Network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 815–822. [Google Scholar] [CrossRef]
- Goeman, J.J. L1 penalized estimation in the Cox proportional hazards model. Biom. J. 2010, 52, 70–84. [Google Scholar] [PubMed]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Biswas, B.; Bakhshi, S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J. Orthop. 2016, 7, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.; Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J. Stat. Softw. 2011, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gerds, T.A.; Kattan, M.W.; Schumacher, M.; Yu, C. Estimating a time-dependent concordance index for survival prediction models with covariate dependent censoring. Stat. Med. 2013, 32, 2173–2184. [Google Scholar] [CrossRef]
- Vickers, A.J.; Cronin, A.M.; Elkin, E.B.; Gonen, M. Extensions to decision curve analysis, a novel method for evaluating diagnostic tests, prediction models and molecular markers. BMC Med. Inform. Decis. Mak. 2008, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.; Wallden, B.; Schaper, C.; Ferree, S.; Liu, S.; Gao, D.; Barry, G.; Dowidar, N.; Maysuria, M.; Storhoff, J. Analytical validation of the PAM50-based Prosigna Breast Cancer Prognostic Gene Signature Assay and nCounter Analysis System using formalin-fixed paraffin-embedded breast tumor specimens. BMC Cancer 2014, 14, 177. [Google Scholar] [CrossRef] [PubMed]
- Stahl, D.; Gentles, A.J.; Thiele, R.; Gutgemann, I. Prognostic profiling of the immune cell microenvironment in Ewing s Sarcoma Family of Tumors. Oncoimmunology 2019, 8, e1674113. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, Q.; Wang, H.; Wang, Z.; Zuo, D.; Cai, Z.; Hua, Y. Prognostic significance of serum lactate dehydrogenase levels in Ewing’s sarcoma: A meta-analysis. Mol. Clin. Oncol. 2016, 5, 832–838. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO ID | No. of ES Cases Included | Platform | Age | Sex | Outcomes | |||
|---|---|---|---|---|---|---|---|---|
| <18 | ≥18 | Male | Female | Dead | Alive | |||
| GSE63155 | 46 | HuEx1.0 (GPL5175) | 41 | 5 | 27 | 19 | 14 | 32 |
| GSE63166 | 39 | HuEx1.0 (GPL5175) | 31 | 8 | 19 | 20 | 11 | 28 |
| GSE17679 | 32 | U133Plus2.0 (GPL570) | 16 | 16 | 22 | 10 | 20 | 12 |
| GSE34620 | 38 | U133Plus2.0 (GPL570) | 27 | 11 | 20 | 18 | 21 | 17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Su, H.; Su, Y.; Zhang, Y.; Lin, Y.; Haglund, F. Identification of an RNA-Binding-Protein-Based Prognostic Model for Ewing Sarcoma. Cancers 2021, 13, 3736. https://doi.org/10.3390/cancers13153736
Chen Y, Su H, Su Y, Zhang Y, Lin Y, Haglund F. Identification of an RNA-Binding-Protein-Based Prognostic Model for Ewing Sarcoma. Cancers. 2021; 13(15):3736. https://doi.org/10.3390/cancers13153736
Chicago/Turabian StyleChen, Yi, Huafang Su, Yanhong Su, Yifan Zhang, Yingbo Lin, and Felix Haglund. 2021. "Identification of an RNA-Binding-Protein-Based Prognostic Model for Ewing Sarcoma" Cancers 13, no. 15: 3736. https://doi.org/10.3390/cancers13153736
APA StyleChen, Y., Su, H., Su, Y., Zhang, Y., Lin, Y., & Haglund, F. (2021). Identification of an RNA-Binding-Protein-Based Prognostic Model for Ewing Sarcoma. Cancers, 13(15), 3736. https://doi.org/10.3390/cancers13153736