
Abdominal Desmoid: Course, Severe Outcomes, and Unique Genetic Background in a Large Local Series

 , ,
, , 
Abstract
Simple Summary
Abstract
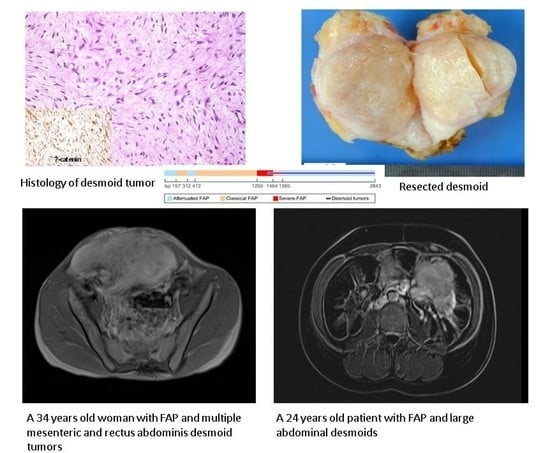
1. Introduction
2. Methods
3. Statistical Analysis
4. Results
4.1. Patients and Desmoid Characteristics
4.2. Genetic Findings
4.3. Desmoid Related Morbidity
4.4. Surgical and Medical Treatment
4.5. Oncologic Outcomes and Mortality
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alman, B.; Attia, S.; Baumgarten, C.; Benson, C.; Blay, J.-Y.; Bonvalot, S.; Breuing, J.; Cardona, K.; Casali, P.G.; van Coevorden, F.; et al. The management of desmoid tumours: A joint global consensus-based guideline approach for adult and paediatric patients. Eur. J. Cancer 2020, 127, 96–107. [Google Scholar] [CrossRef]
- Reitamo, J.J.; Hayry, P.; Nykyri, E.; Saxen, E. The desmoid tumor. I. Incidence, sex-, age- and anatomical distribution in the Finnish population. Am. J. Clin. Pathol. 1982, 77, 665–673. [Google Scholar] [CrossRef]
- Fallen, T.; Wilson, M.; Morlan, B.; Lindor, N.M. Desmoid tumors—A characterization of patients seen at Mayo Clinic 1976–1999. Fam. Cancer 2006, 5, 191–194. [Google Scholar] [CrossRef]
- Xhaja, X.; Church, J. Small bowel obstruction in patients with familial adenomatous polyposis related desmoid disease. Colorectal Dis. 2013, 15, 1489–1492. [Google Scholar] [CrossRef]
- Soravia, C.; Berk, T.; McLeod, R.S.; Cohen, Z. Desmoid disease in patients with familial adenomatous polyposis. Dis. Colon Rectum 2000, 43, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhou, Y.; Li, Q.; Tong, H.; Lu, W. Intestinal perforation during chemotherapeutic treatment of intra-abdominal desmoid tumor in patients with Gardner’s syndrome: Report of two cases. World J. Surg. Oncol. 2016, 14, 178. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Azam, B. Case report of an intra-abdominal desmoid tumour presenting with bowel perforation. Mcgill J. Med. 2007, 10, 90–92. [Google Scholar] [PubMed]
- Tsiaousis, P.; Christopoulous, P.; Iordanidou, I.; Atmatzidis, S.; Panagiotopoulou, K.; Psaraleksis, K.; Atmatzidis, K. An intra-abdominal desmoid tumor presenting with bowel perforation. A case report. Surg. Chron. 2008, 13, 327–331. [Google Scholar]
- van Broekhoven, D.L.M.; Grünhagen, D.J.; den Bakker, M.A.; van Dalen, T.; Verhoef, C. Time Trends in the Incidence and Treatment of Extra-Abdominal and Abdominal Aggressive Fibromatosis: A Population-Based Study. Ann. Surg. Oncol. 2015, 22, 2817–2823. [Google Scholar] [CrossRef] [PubMed]
- Koskenvuo, L.; Ristimaki, A.; Lepisto, A. Comparison of sporadic and FAP-associated desmoid-type fibromatoses. J. Surg. Oncol. 2017, 116, 716–721. [Google Scholar] [CrossRef]
- Koskenvuo, L.; Peltomaki, P.; Renkonen-Sinisalo, L.; Gylling, A.; Nieminen, T.T.; Ristimaki, A.; Lepisto, A. Desmoid tumor patients carry an elevated risk of familial adenomatous polyposis. J. Surg. Oncol. 2016, 113, 209–212. [Google Scholar] [CrossRef]
- Fiore, M.; Coppola, S.; Cannell, A.J.; Colombo, C.; Bertagnolli, M.M.; George, S.; Le Cesne, A.; Gladdy, R.A.; Casali, P.G.; Swallow, C.J.; et al. Desmoid-type fibromatosis and pregnancy: A multi-institutional analysis of recurrence and obstetric risk. Ann. Surg. 2014, 259, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Schlemmer, M. Desmoid tumors and deep fibromatoses. Hematol. Oncol. Clin. N. Am. 2005, 19, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Tekkis, P.P.; Gibbons, D.C.; Phillips, R.K.; Clark, S.K. Risk factors predicting desmoid occurrence in patients with familial adenomatous polyposis: A meta-analysis. Colorectal Dis. 2011, 13, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Burtenshaw, S.M.; Cannell, A.J.; McAlister, E.D.; Siddique, S.; Kandel, R.; Blackstein, M.E.; Swallow, C.J.; Gladdy, R.A. Toward Observation as First-line Management in Abdominal Desmoid Tumors. Ann. Surg. Oncol. 2016, 23, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Bonvalot, S.; Ternes, N.; Fiore, M.; Bitsakou, G.; Colombo, C.; Honore, C.; Marrari, A.; Le Cesne, A.; Perrone, F.; Dunant, A.; et al. Spontaneous regression of primary abdominal wall desmoid tumors: More common than previously thought. Ann. Surg. Oncol. 2013, 20, 4096–4102. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.K.; Neale, K.F.; Landgrebe, J.C.; Phillips, R.K. Desmoid tumours complicating familial adenomatous polyposis. Br. J. Surg. 1999, 86, 1185–1189. [Google Scholar] [CrossRef]
- Rodriguez-Bigas, M.A.; Mahoney, M.C.; Karakousis, C.P.; Petrelli, N.J. Desmoid tumors in patients with familial adenomatous polyposis. Cancer 1994, 74, 1270–1274. [Google Scholar] [CrossRef]
- Fiore, M.; Colombo, C.; Radaelli, S.; Callegaro, D.; Palassini, E.; Barisella, M.; Morosi, C.; Baldi, G.G.; Stacchiotti, S.; Casali, P.G.; et al. Hormonal manipulation with toremifene in sporadic desmoid-type fibromatosis. Eur. J. Cancer 2015, 51, 2800–2807. [Google Scholar] [CrossRef]
- Chugh, R.; Wathen, J.K.; Patel, S.R.; Maki, R.G.; Meyers, P.A.; Schuetze, S.M.; Priebat, D.A.; Thomas, D.G.; Jacobson, J.A.; Samuels, B.L.; et al. Efficacy of Imatinib in Aggressive Fibromatosis: Results of a Phase II Multicenter Sarcoma Alliance for Research through Collaboration (SARC) Trial. Clin. Cancer Res. 2010, 16, 4884–4891. [Google Scholar] [CrossRef]
- Gounder, M.M.; Mahoney, M.R.; Van Tine, B.A.; Ravi, V.; Attia, S.; Deshpande, H.A.; Gupta, A.A.; Milhem, M.M.; Conry, R.M.; Movva, S.; et al. Sorafenib for Advanced and Refractory Desmoid Tumors. N. Engl. J. Med. 2018, 379, 2417–2428. [Google Scholar] [CrossRef]
- Constantinidou, A.; Jones, R.L.; Scurr, M.; Al-Muderis, O.; Judson, I. Advanced aggressive fibromatosis: Effective palliation with chemotherapy. Acta Oncol. 2011, 50, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Shindle, M.K.; Khanna, A.J.; McCarthy, E.F.; O’Neill, P.J.; Sponseller, P.D. Desmoid Tumor of the Spinal Canal Causing Scoliosis and Paralysis. Spine 2002, 27, E304–E307. [Google Scholar] [CrossRef] [PubMed]
- Puvanesarajah, V.; Lina, I.A.; Liauw, J.A.; Hsu, W.; Burger, P.C.; Witham, T.F. Desmoid Tumor Formation following Posterior Spinal Instrumentation Placement. Evid. Based. Spine. Care. J. 2013, 4, 137–142. [Google Scholar] [CrossRef][Green Version]
- Huang, K.; Fu, H.; Shi, Y.-Q.; Zhou, Y.; Du, C.-Y. Prognostic factors for extra-abdominal and abdominal wall desmoids: A 20-year experience at a single institution. J. Surg. Oncol. 2009, 100, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Ballo, M.T.; Zagars, G.K.; Pollack, A.; Pisters, P.W.; Pollack, R.A. Desmoid tumor: Prognostic factors and outcome after surgery, radiation therapy, or combined surgery and radiation therapy. J. Clin. Oncol. 1999, 17, 158–167. [Google Scholar] [CrossRef]
- Salas, S.; Dufresne, A.; Bui, B.; Blay, J.-Y.; Terrier, P.; Ranchere-Vince, D.; Bonvalot, S.; Stoeckle, E.; Guillou, L.; Le Cesne, A.; et al. Prognostic Factors Influencing Progression-Free Survival Determined From a Series of Sporadic Desmoid Tumors: A Wait-and-See Policy According to Tumor Presentation. J. Clin. Oncol. 2011, 29, 3553–3558. [Google Scholar] [CrossRef] [PubMed]
- Shido, Y.; Nishida, Y.; Nakashima, H.; Katagiri, H.; Sugiura, H.; Yamada, Y.; Ishiguro, N. Surgical treatment for local control of extremity and trunk desmoid tumors. Arch. Orthop. Trauma Surg. 2009, 129, 929–933. [Google Scholar] [CrossRef]
- Quintini, C.; Ward, G.; Shatnawei, A.; Xhaja, X.; Hashimoto, K.; Steiger, E.; Hammel, J.; Diago Uso, T.; Burke, C.A.; Church, J.M. Mortality of intra-abdominal desmoid tumors in patients with familial adenomatous polyposis: A single center review of 154 patients. Ann. Surg. 2012, 255, 511–516. [Google Scholar] [CrossRef]
- Palassini, E.; Frezza, A.M.; Mariani, L.; Lalli, L.; Colombo, C.; Fiore, M.; Messina, A.; Casale, A.; Morosi, C.; Collini, P.; et al. Long-term Efficacy of Methotrexate Plus Vinblastine/Vinorelbine in a Large Series of Patients Affected by Desmoid-Type Fibromatosis. Cancer J. 2017, 23, 86–91. [Google Scholar] [CrossRef]
- Crombé, A.; Kind, M.; Ray-Coquard, I.; Isambert, N.; Chevreau, C.; André, T.; Lebbe, C.; Cesne, A.L.; Bompas, E.; Piperno-Neumann, S.; et al. Progressive Desmoid Tumor: Radiomics Compared With Conventional Response Criteria for Predicting Progression During Systemic Therapy-A Multicenter Study by the French Sarcoma Group. AJR. Am. J. Roentgenol. 2020, 215, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Quast, D.R.; Schneider, R.; Burdzik, E.; Hoppe, S.; Möslein, G. Long-term outcome of sporadic and FAP-associated desmoid tumors treated with high-dose selective estrogen receptor modulators and sulindac: A single-center long-term observational study in 134 patients. Fam. Cancer 2016, 15, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Skapek, S.X.; Anderson, J.R.; Hill, D.A.; Henry, D.; Spunt, S.L.; Meyer, W.; Kao, S.; Hoffer, F.A.; Grier, H.E.; Hawkins, D.S.; et al. Safety and efficacy of high-dose tamoxifen and sulindac for desmoid tumor in children: Results of a Children’s Oncology Group (COG) Phase II Study. Pediatr. Blood Cancer 2013, 60, 1108–1112. [Google Scholar] [CrossRef]
- Azzarelli, A.; Gronchi, A.; Bertulli, R.; Tesoro Tess, J.D.; Baratti, D.; Pennacchioli, E.; Dileo, P.; Rasponi, A.; Ferrari, A.; Pilotti, S.; et al. Low-dose chemotherapy with methotrexate and vinblastine for patients with advanced aggressive fibromatosis. Cancer 2001, 92, 1259–1264. [Google Scholar] [CrossRef]
- Penel, N.; Le Cesne, A.; Bui, B.N.; Perol, D.; Brain, E.G.; Ray-Coquard, I.; Guillemet, C.; Chevreau, C.; Cupissol, D.; Chabaud, S.; et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): An FNCLCC/French Sarcoma Group phase II trial with a long-term follow-up. Ann. Oncol. 2011, 22, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Kasper, B.; Gruenwald, V.; Reichardt, P.; Bauer, S.; Rauch, G.; Limprecht, R.; Sommer, M.; Dimitrakopoulou-Strauss, A.; Pilz, L.; Haller, F.; et al. Imatinib induces sustained progression arrest in RECIST progressive desmoid tumours: Final results of a phase II study of the German Interdisciplinary Sarcoma Group (GISG). Eur. J. Cancer 2017, 76, 60–67. [Google Scholar] [CrossRef]
- Manoukian, S.; Peissel, B.; Frigerio, S.; Lecis, D.; Bartkova, J.; Roversi, G.; Radice, P.; Bartek, J.; Delia, D. Two new CHEK2 germ-line variants detected in breast cancer/sarcoma families negative for BRCA1, BRCA2, and TP53 gene mutations. Breast Cancer Res. Treat. 2011, 130, 207–215. [Google Scholar] [CrossRef]
- Malkin, D. Li-fraumeni syndrome. Genes Cancer 2011, 2, 475–484. [Google Scholar] [CrossRef]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef]
- Hanenberg, H.; Andreassen, P.R. PALB2 (partner and localizer of BRCA2). Atlas Genet. Cytogenet. Oncol. Haematol. 2018, 22, 484–490. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Parameter | FAP (n = 46) | Non-FAP (n = 16) | All (n = 62) | p Value |
|---|---|---|---|---|
| Sex—male (%) | 21 (45.6) | 9 (56.2) | 30 (48.3) | 0.46 |
| Median age at desmoid diagnosis (IQR) | 31 (23–38) | 43.5 (33–63.25) | 34 (23–44) | 0.007 |
| Median follow up, months (IQR) | 83.1 (53.9–170.9) | 33.9 (21.9–61.5) | 72.4 (37.1–151.9) | 0.004 |
| Median number of desmoids per patient (IQR) | 2 (1–3) | 1 (1–2.2) | 2 (1–3) | 0.26 |
| Median desmoid size at diagnosis, cm (IQR) | 4.7 (3.5–7.6) | 8 (4.2–13) | 5.5 (3.5–8.8) | 0.08 |
| Median maximal desmoid size, cm (IQR) | 7.9 (4.5–10.7) | 8 (5.4–14) | 8 (4.7–11.5) | 0.76 |
| Desmoid location | ||||
| Abdominal wall (%) | 4 (8.7) | 1 (6.25) | 5 (8.1) | 0.13 |
| Mesentery (%) | 27 (58.7) | 14 (87.5) | 41 (66.1) | 0.6 |
| Both (%) | 15 (32.6) | 1 (6.25) | 16 (25.8) | 0.08 |
| Adverse events, patients (%) | 31 (67.3) | 10 (62.5) | 41 (66.1) | 0.72 |
| Bowel perforation (%) | 5 (10.8) | 2 (12.5) | 7 (11.3) | >0.999 |
| Bowel obstruction (%) | 16 (34.7) | 3 (18.7) | 19 (30.6) | 0.34 |
| Small bowel resection (%) | 13 (28.2) | 6 (37.5) | 19 (30.6) | 0.53 |
| Ureter obstruction (%) | 7 (15.2) | 1 (6.25) | 8 (12.9) | 0.66 |
| Ischemic colitis (%) | 2 (4.3) | 0 | 2 (3.2) | >0.999 |
| GI bleeding (%) | 2 (4.3) | 0 | 2 (3.2) | 0.56 |
| Need for TPN (%) | 7 (15.2) | 2 (12.5) | 9 (14.5) | >0.999 |
| Abscess formation (%) | 1 (2.7) | 1 (6.25) | 2 (3.2) | >0.999 |
| Spinal cord compression (%) | 1 (2.1) | 1 (6.25) | 2 (3.2) | 0.45 |
| Surgery before appearance of desmoid (%) | 42 (91.3) | 1 (6.25) | 43 (69.3) | <0.001 |
| Pregnancy before appearance of desmoid (% of females) | 6 (24) | 4 (57.1) | 10 (31.2) | 0.26 |
| Death (%) | 2 (4.3) | 0 | 2 (3.2) | >0.999 |
| Patient Number | Sex | Age at Diagnosis | Desmoid Location | Complications | Other Malignancies | Genetic NGS Panels Performed | Genes with Pathogenic Variants | Genes with VUS |
|---|---|---|---|---|---|---|---|---|
| 1 | Male | 67 | Mesenteric | None | None | Yes | CHEK2 | APC |
| 2 | Male | 29 | Mesenteric | None | Germ cell tumor | Yes | ERCC5 BLM | TP53 |
| 3 | Male | 54 | Mesenteric | Bowel perforation and liver abscess | None | Yes | - | BLM |
| 4 | Female | 20 | Mesenteric | None | None | Yes | - | MSH2 |
| 5 | Male | 69 | Mesenteric | Bowel obstruction and resection | RCC | Yes | - | CHEK2 |
| 6 | Female | 38 | Mesenteric | None | None | Yes | MSH6 | - |
| 7 | Male | 45 | Mesenteric | Bowel obstruction and need for TPN after resection | None | Yes | PALB2 | - |
| 8 | Male | 30 | Mesenteric | Bowel and ureter obstruction | RCC | Yes * | - | - |
| 9 | Female | 37 | Mesenteric and abdominal wall | Bowel perforation and abscess formation | None | No | - | - |
| 10 | Male | 62 | Abdominal wall | None | None | No | - | - |
| 11 | Male | 68 | Mesenteric | Small bowel resection | None | No | - | - |
| 12 | Female | 34 | Mesenteric | None | None | No | - | - |
| 13 | Female | 81 | Mesenteric | None | RCC | No | - | - |
| 14 | Male | 59 | Mesenteric | Need for TPN after resection | None | No | - | - |
| 15 | Female | 42 | Mesenteric | Small Bowel resection | None | No | - | - |
| 16 | Female | 9 | Mesenteric | Sciatic nerve involvement | None | No | - | - |
| Therapy | Patients Treated as 1st Line (%) | Patients Treated as 2nd Line (%) | Patients Treated as 3rd Line (%) | Patients Treated as 4th Line (%) | Treatment Initiation, Months from Diagnosis, Median (IQR) | Treatment Duration, Months from Initiation, Median (IQR) | Two Year Progression Free Survival (%) |
|---|---|---|---|---|---|---|---|
| COX 2 inhibitors | 29 (46.7) | 0 | 1 (1.6) | 0 | 4.1 (0–33.9) | 32.2 (6.6–92.9) | 57.7 |
| Hormonal | 33 (53.2) | 1 (1.6) | 1 (1.6) | 0 | 9.1 (0–48.7) | 44.1 (7.1–103.7) | 67.8 |
| Chemotherapy | 8 (12.9) | 7 (11.2) | 3 (4.8) | 0 | 16.8 (5.1–49.5) | 17.7 (7.9–23) | 38.4 |
| Imatinib | 1 (1.6) | 4 (6.4) | 2 (3.2) | 0 | 25.6 (7.7–60.8) | 7.7 (6.5–20.3) | 28.5 |
| Sorafenib | 1 (1.6) | 6 (9.6) | 1 (1.6) | 1 (1.6) | 26.3 (13.1–34.9) | 6.8 (3.6–11.2) | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ophir, G.; Sivan, S.; Hana, S.; Guy, R.; Nathan, G.; Naomi, F.I.; Joseph, K.; Ido, W.; Ofer, M.; Yael, G.; et al. Abdominal Desmoid: Course, Severe Outcomes, and Unique Genetic Background in a Large Local Series. Cancers 2021, 13, 3673. https://doi.org/10.3390/cancers13153673
Ophir G, Sivan S, Hana S, Guy R, Nathan G, Naomi FI, Joseph K, Ido W, Ofer M, Yael G, et al. Abdominal Desmoid: Course, Severe Outcomes, and Unique Genetic Background in a Large Local Series. Cancers. 2021; 13(15):3673. https://doi.org/10.3390/cancers13153673
Chicago/Turabian StyleOphir, Gilad, Shamai Sivan, Strul Hana, Rosner Guy, Gluck Nathan, Fliss Isakov Naomi, Klausner Joseph, Wolf Ido, Merimsky Ofer, Goldberg Yael, and et al. 2021. "Abdominal Desmoid: Course, Severe Outcomes, and Unique Genetic Background in a Large Local Series" Cancers 13, no. 15: 3673. https://doi.org/10.3390/cancers13153673
APA StyleOphir, G., Sivan, S., Hana, S., Guy, R., Nathan, G., Naomi, F. I., Joseph, K., Ido, W., Ofer, M., Yael, G., Zohar, L., Alona, Z., & Revital, K. (2021). Abdominal Desmoid: Course, Severe Outcomes, and Unique Genetic Background in a Large Local Series. Cancers, 13(15), 3673. https://doi.org/10.3390/cancers13153673