Simple Summary
In addition to cancer-related factors, anti-cancer chemotherapy treatment can drive life-threatening body wasting in a syndrome known as cachexia. Emerging evidence has described the impact of several key chemotherapeutic agents on skeletal muscle in particular, and the mechanisms are gradually being unravelled. Despite this evidence, there remains very little research regarding therapeutic strategies to protect muscle during anti-cancer treatment and current global grand challenges focused on deciphering the cachexia conundrum fail to consider this aspect—chemotherapy-induced myopathy remains very much on the dark side of the cachexia sphere. This review explores the impact and mechanisms of, and current investigative strategies to protect against, chemotherapy-induced myopathy to illuminate this serious issue.
Abstract
Cancer cachexia is a debilitating multi-factorial wasting syndrome characterised by severe skeletal muscle wasting and dysfunction (i.e., myopathy). In the oncology setting, cachexia arises from synergistic insults from both cancer–host interactions and chemotherapy-related toxicity. The majority of studies have surrounded the cancer–host interaction side of cancer cachexia, often overlooking the capability of chemotherapy to induce cachectic myopathy. Accumulating evidence in experimental models of cachexia suggests that some chemotherapeutic agents rapidly induce cachectic myopathy, although the underlying mechanisms responsible vary between agents. Importantly, we highlight the capacity of specific chemotherapeutic agents to induce cachectic myopathy, as not all chemotherapies have been evaluated for cachexia-inducing properties—alone or in clinically compatible regimens. Furthermore, we discuss the experimental evidence surrounding therapeutic strategies that have been evaluated in chemotherapy-induced cachexia models, with particular focus on exercise interventions and adjuvant therapeutic candidates targeted at the mitochondria.
1. Introduction
Chemotherapy constitutes a group of anti-neoplastic agents that were progressively discovered throughout the 19th century and became commonplace in oncological treatment as first-line or complementary therapeutic strategies for nearly all cancer types [1]. In general, chemotherapy targets cell cycle arrest through DNA-damage pathways that promote apoptotic cell death, although agents are heterogeneously stratified into different classes depending on their mode of action [2]. Independent of their different mechanisms, chemotherapies remain effective at inducing cancer cytotoxicity to abate the hyper-active neoplastic cell cycle [3]. Despite exerting anti-cancer efficacy, chemotherapy also elicits detrimental off-target side-effects to otherwise healthy cells due to their non-specific cytotoxicity [4]. Systemic toxicities are pervasive in the blood cell population as well as the central and peripheral nervous, gastrointestinal, cardiovascular, and integumentary systems [5,6,7,8]. Over the past decade, interest has developed concerning the non-specific impact of chemotherapy-induced toxicity on the skeletal muscular system [9,10,11]. Specifically, chemotherapeutic agents reduce body mass concurrent with skeletal muscle atrophy and dysfunction (referred to herein, as cachectic myopathy). These side-effects are clinically evident during patient deconditioning in the oncological setting [12], where weight loss and fatigue are two key debilitating events prominent in metabolic wasting syndrome, cachexia [13].
Cachexia is a multifactorial condition characterised by the loss of body mass and composition (highlighted by lean mass loss, with or without loss of fat mass) and progressive functional impairment [14]. The centrally afflicted organ in cachexia is skeletal muscle, which is driven by a multitude of factors including metabolic dysregulation; anorexia; systemic inflammation; and insulin resistance [15]. Skeletal muscle mass is an integral prognostic marker in cancer cachexia diagnosis—this is because increased adiposity de- sensitises the utility of body weight and body mass index (a crude indicator of body composition) during cachexia diagnosis [16,17]. Once instigated, cachectic myopathy propagates a vicious cycle involving increased risk of dose-related toxicities, which influences patient risk stratification and clinical decision making [18]. The result is compromised treatment efficacy (i.e., dose-reduction or treatment cessation) that increases the risk of morbidity and mortality [18]. Cachectic myopathy is the result of two synergistic insults from each of: (1) cancer–host interactions and (2) chemotherapy toxicity [15,19]. These effects can be acute, yet are most often life-long [20]. While the number of studies concerning cachexia is increasing exponentially, the proportion of these studies focusing on the impact of chemotherapy is far behind [9].
Overall, cancer cachexia represents a significant burden on patients and clinicians, with estimates suggesting that cachexia affects 50–80% of cancer patients and accounts for up to 20% of cancer deaths [21]. There are no current treatment options outside of standard nutrition interventions for body mass loss, which are largely unsuccessful [22,23]. Thus, cachexia is a significant unmet challenge in need of pre-clinical investigations to identify novel drug-targets and evaluate therapeutic interventions for clinical translation. Herein, we present a review of the literature surrounding experimental chemotherapy-induced cachexia, with particular focus on the skeletal muscle-specific side-effects and underlying mechanisms of myopathy. We also discuss the challenges associated with current experimental approaches used to investigate chemotherapy-induced cachexia and adjuvant strategies to protect against it.
2. Mechanisms of Chemotherapy-Induced Cachectic Myopathy
Chemotherapeutic agents work through different modes of action, which likely influences the degree to which they can induce cachectic myopathy. However, of the agents that do induce myopathy, there are some common underlying mechanisms [9]. It has been proposed that chemotherapy promotes systemic inflammation via the central nervous system, specifically through stimulation of the hypothalamus–pituitary–adrenal axis, to induce an adaptive illness response [24]. This response simultaneously induces the release of glucocorticoids and the production of pro-inflammatory cytokines (e.g., interleukin-1α and β, interleukin-6, and tumour necrosis factor-α (TNF-α)), which are both key events in the induction of skeletal muscle atrophy [25,26]. In particular, over-production of pro-inflammatory cytokines can directly induce skeletal muscle atrophy via engagement of membranous receptors and activation of a pro-catabolic transcription program [9,13]. Braun et al. proposes that instigation of the inflammatory milieu caused by chemotherapy increases activity of the gene, regulated in development and DNA damage response 1 (REDD1), which is associated with skeletal muscle atrophy [24]. REDD1 transcription regulates the adaptive stress response including the activation of stress-sensitive molecular targets, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and mitogen activated protein kinase (MAPK) [27,28]. These two targets share common signalling pathways during oxidant-induced stress [29]. In particular, MAPK activity promotes phosphorylation of the NF-κB subunit, p65, resulting in NF-κB activation and the induction of skeletal muscle atrophy [29]. This atrophic response is achieved primarily through the ubiquitin-proteasome system (UPS)-mediated transcription of classic atrogenes, the E3 ubiquitin ligases, MuRF-1, and Atrogin-1 [30,31]. Additionally, it has been demonstrated by our laboratory and others that several chemotherapeutic agents can promote reactive oxygen species (ROS) production in C2C12 myotubes, which is associated with impaired myotube morphometry [32,33,34,35]. These data suggest that stress-sensitive molecular targets could be common signalling pathways in chemotherapy-induced cachectic myopathy. Furthermore, excessive ROS production is associated with the onset of mitochondrial dysfunction, an event postulated to be the crucial trigger for the induction of skeletal muscle wasting by chemotherapy [36].
As well as through directly targeting differentiated skeletal muscle tissue, chemotherapeutic agents also target myoprogenitor activity (i.e., satellite cell replication), which impacts skeletal muscle growth, repair, and turnover (as reviewed by us previously [37]). Many chemotherapeutic agents arrest cell cycling as their principal mechanisms of action against rampant cancer cell proliferation, resulting in visible side-effects in high-turnover cells including hair, bone marrow, skin, and gastrointestinal epithelium. It has recently emerged that muscle satellite (stem) cells, which normally undergo rapid proliferation and differentiation in response to muscle damage (e.g., inflammation) and growth factors (e.g., growth hormone and androgens) are also impacted, contributing to the net loss of muscle mass observed during cancer-related cachexia [38,39,40].
The research to date has predominantly focused on three key chemotherapeutic agents to study the mechanisms that govern chemotherapy-induced cachectic myopathy: the anthracycline, doxorubicin (DOX); the platinum-based alkylating agent, cisplatin (cis-diamminedichloroplatinum(II) (CDDP)); and the anti-metabolite, 5-fluorouracil (5FU). As such, we will further explore the current understanding of the connection between these stress-signalling cascades and the induction of cachectic myopathy in the context of specific chemotherapeutic agents and/or regimens.
2.1. Doxorubicin (DOX)
DOX is a member of the anthracycline class of chemotherapies, and, as such, it elicits cytotoxicity via topoisomerase-II inhibition to induce DNA damage and cell cycle arrest [41]. Despite its potent anti-cancer efficacy, DOX is notorious for its cardiotoxic properties, which significantly limits its clinical utility [42]. Subsequently, there has been substantial interest in the effect of DOX on skeletal muscle health (summarised in Figure 1), particularly since skeletal muscle is an active compartment in the metabolism of DOX and its metabolites (e.g., DOX metabolite, doxorubicinol) is retained within skeletal muscle tissue for up to five days post-intravenous delivery [43,44]. The clinical implications of DOX administration are remarkable. Patients often present with debilitating fatigue, muscle weakness, and impaired ambulatory capacity [45,46]. At the cellular level, DOX treatment induces skeletal muscle dysfunction [47,48] characterised by reduced force production, impaired calcium (Ca2+) dynamics, and increased susceptibility to physiological fatigue [47,48,49]. Importantly, Gilliam et al. highlighted that these symptoms are dependent on the activity of pro-inflammatory cytokine, TNF-α [50]. This pro-inflammatory scenario has been shown to promote pyroptosis, a biological process of programmed cell death, which is characterised by the nucleotide binding oligmerisation domain (NOD), leucine rich repeat-containing proteins (NLR) family member, NRLP3 inflammasome formation, and activation of apoptotic caspases [51,52]. Furthermore, TNF-α mediates skeletal muscle contractile dysfunction through enhanced ROS production [53], although this is considered to be an additive effect to DOX-induced oxidative stress. DOX can directly stimulate ROS production through redox cycling at NADH dehydrogenase/Complex I of the mitochondrial electron transport chain [54]. As a result, ROS are produced, in particular superoxide anion radicals, via the reduction of DOX’s quinone moiety to an unstable semiquinone [54]. This redox cycling significantly elevates skeletal muscle hydrogen peroxide (H2O2) emission without perturbing homeostatic antioxidant buffering capacity [55,56,57] and alters bioenergetic efficiency by impinging on the functionality of respiratory complexes, leading to modifications that promote oxidative damage (e.g., lipid peroxidation) [56,58]. Recent findings from the Hulmi group suggest that DOX-induced skeletal muscle perturbations are predominately influenced by enhanced transcription of REDD1 as part of the transcriptional program regulated by oxidative stress sensitive tumour suppressor protein, p53, a master regulator of cellular homeostasis [59,60,61]. Interestingly, there are few data connecting the promotion of inflammatory cytokines and oxidative stress from DOX to increased NF-κB activity in skeletal muscle, which is a downstream target of REDD1 transcription. Supriya et al. showed that DOX potentiated NF-κB activity in the skeletal muscle of diabetic mice [62]. The authors suggested that several mechanisms downstream of REDD1 were likely contributing [62].
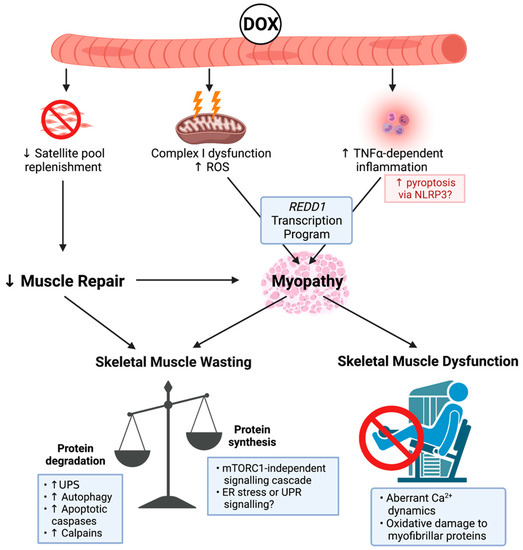
Figure 1.
Known mechanisms of doxorubicin (DOX)-induced cachectic myopathy. DOX promotes reactive oxygen species (ROS) production primarily via Complex I dysfunction, which induces mitochondrial dysfunction and tumour-necrosis factor-α (TNF-α)-dependent inflammation, which can promote pyroptosis via increased nucleotide binding oligmerisation domain, leucine rich repeat-containing protein 3 (NLRP3) inflammasome formation, and activation of apoptotic caspases. This stimulates a regulation in the development and DNA damage response 1 (REDD1) transcription program, which overarches DOX-induced cachectic myopathy. DOX also reduces the replenishment of the satellite cell pool, which contributes to cachectic myopathy through impaired muscle repair. Underlying skeletal muscle wasting, DOX increases protein degradation via the ubiquitin-proteasomal system (UPS), autophagy, apoptotic caspases, and the calcium (Ca2+)-dependent proteases, calpains, while also reducing protein synthesis in a mammalian target of rapamycin complex 1 (mTORC1)-independent manner. While the exact mechanism has not been fully elucidated, endoplasmic reticulum (ER) stress or the unfolded protein response (UPR) signalling may be contributing factors. DOX also alters Ca2+ dynamics and promotes oxidative damage to myofibrillar proteins causing skeletal muscle dysfunction. Created with biorender.com (accessed on 6 July 2021).
Enhanced oxidative damage from DOX administration induces modifications that increase the catabolism of myofibrillar proteins, in particular actin and myosin, two integral proteins of the contractile apparatus [33,63]. The pro-catabolic signalling-cascade potentiated by DOX can activate multiple proteolytic systems including the UPS, autophagy, apoptotic caspases, and Ca2+-dependent proteases (i.e., calpains) [56,63,64]. The pro-catabolic shift in skeletal muscle protein balance has led to the consensus that DOX can cause skeletal muscle mass loss [11]. While DOX mechanisms are largely centred around proteolysis, there is also a rationale to suggest DOX impairs anabolism. This is highlighted by a reduced rate of protein synthesis independent of the classical signalling of mechanistic target of rapamycin complex 1 (mTORC1) [59], a key mediator of protein synthesis (for extensive review see [65]). There is also some evidence that DOX administration can impair the regenerative capacity of muscle through inhibiting satellite cell proliferation [66]. These data implicate alternative pathways that regulate protein synthesis during DOX administration. One such pathway was the activation of the endoplasmic reticulum stress/unfolded protein response signalling cascade, which can negatively regulate protein synthesis [67]. However, there were divergent responses between different types of striated muscle (i.e., heart, diaphragm, and limb skeletal muscles) and the expression of markers involved in this signalling cascade, elicited by DOX [55,68]. Thus, further research is required to enrich the understanding of the processes regulating the skeletal muscle protein balance during DOX treatment.
2.2. Cisplatin (CDDP)
CDDP is a platinum-based alkylating agent that enhances DNA damage via the aquation of its chloride ligands to form a highly reactive mono-aquated complex. This complex can bind with DNA residues to develop CDDP-DNA adducts, which induce DNA crosslinking and cell cycle arrest [69]. CDDP administration is associated with several toxicities, notably nephrotoxicity and neurotoxicity [70,71]. However, it also has deleterious impact on skeletal muscle (critically reviewed in detail previously [10,72]). CDDP was originally considered a potent inducer of negative protein balance—predominantly through impacting caloric intake (i.e., food consumption), which drives protein degradation to reduce body and skeletal muscle mass [73] (Figure 2). Importantly, Sakai et al. demonstrated that CDDP can drive protein degradation independently of caloric intake (through using pair-fed controls), in part, via an Akt and forkhead box O (FoxO)-dependent signalling cascade that enhances the transcription of atrogenes MuRF-1 and Atrogin-1 [74] while synergistically activating other constituents of the UPS [75]. CDDP also promotes the accumulation of autophagosomes (an indicator of autophagy-lysosome system dysregulation) via a similar, although currently undefined, Akt/FoxO3a-dependent mechanism [34,76,77]. Furthermore, CDDP suppresses protein synthesis via a protein kinase B (Akt)-dependent mechanism, which leads to the de-phosphorylation of p70S6k1, a downstream target of mTORC1 [78]. Thus, muscle anabolism signalling also appears to be impacted by CDDP (Figure 2). Damreuer et al. demonstrated that CDDP-mediated atrogene transcription occurs in response to trans-activation of NF-κB, specifically through the heterodimerisation of key subunit proteins, p50 and p65 [79]. Enhanced proteolysis induced by CDDP underscores skeletal muscle wasting, and this was initially thought to be the driver of functional decline [80]. However, recent findings from Conte et al. demonstrate that CDDP also dysregulates Ca2+ ion homeostasis, which is necessary for optimal skeletal muscle function [81]. CDDP increases the intracellular concentration of Ca2+, compromises Ca2+ dynamics, and de-sensitises the excitability of action potentials, resulting in reduced force production [82]. CDDP-induced oxidative stress is also thought to contribute to a dysfunctional contractile apparatus, although the underlying mechanisms are presently unclear. Sirago et al. proposed that CDDP drives H2O2 production and oxidative stress based upon evidence of increased peroxiredoxin (PRX) sulphonylation in skeletal muscle [77,83], which is also central to CDDP’s anti-cancer properties [84]. This mechanism has fascinating potential for therapeutic intervention given that PRX suppression is a key stimulant for NF-κB trans-activation [85], a critical target for CDDP-induced myopathy [9].
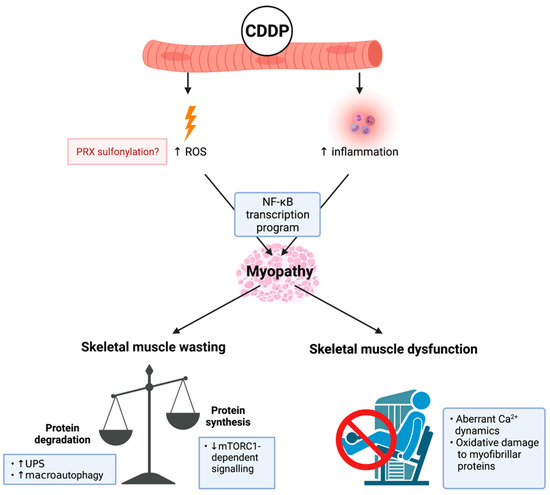
Figure 2.
Known mechanisms of cisplatin (cis-diamminedichloropaltinum(II) (CDDP))-induced cachectic myopathy. CDDP promotes reactive oxygen species (ROS) production potentially through: (1) increased peroxiredoxin (PRX) sulphonylation; and (2) inflammation induced by pro-inflammatory cytokine mediated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription program activation, which is a central mechanism of CDDP-induced cachectic myopathy. The underlying mechanism regulating CDDP-induced skeletal muscle wasting is increased protein degradation involving elevated ubiquitin-proteasomal system (UPS) activity and the promotion of macroautophagy. Additionally, there is evidence of reduced protein synthesis via mammalian target of rapamycin complex 1 (mTORC1)-dependent signalling cascades. CDDP induces skeletal muscle dysfunction through promoting aberrant calcium (Ca2+) dynamics and oxidative damage to myofibrillar proteins. Created with biorender.com (accessed on 6 July 2021).
While CDDP is the predominate platinum-based chemotherapeutic agent experimentally evaluated for its effect on skeletal muscle health, our laboratory and others have demonstrated that analogues, carboplatin [86,87] and oxaliplatin (OXA) [88,89,90] can also contribute to the induction of cachectic myopathy. A complicating factor in the research surrounding platinum-based agents is the reliance on solvent, dimethyl sulphoxide (DMSO), to prepare experimental drug solutions. Using DMSO to deliver the chemotherapeutic agents CDDP and carboplatin suppresses their relative cytotoxicity in cell culture, whereas the cytotoxicity elicited by OXA is not impacted [91,92]. This highlights the complexity of interpreting the experimental data surrounding platinum-based complexes in comparison to vehicle control groups that do not disclose information regarding DMSO utilisation, an all too common feature in the accumulated literature thus far [92].
2.3. 5-Fluorouracil (5FU)
5FU is a chemotherapeutic agent from the anti-metabolite class that elicits cytotoxicity via: (1) the misincorporation of nucleotides into RNA and DNA; and (2) the inhibition of the nucleotide enzyme, thymidylate synthase. This leads to both DNA and RNA damage and cell cycle arrest [93]. 5FU is primarily utilised against colorectal cancer as a backbone constituent of multi-agent regimens. However, it can elicit debilitating side-effects independently [93], emphasised by gastrointestinal toxicities such as mucositis and enteric neuropathy [94,95,96]. With respect to the side-effects of 5FU on skeletal muscle, there is mixed evidence as to whether it can independently drive the loss of mass and function [97,98,99]. Interestingly, VanderVeen et al. demonstrated that 5FU administration impairs the homeostatic coordination of skeletal muscle repair and remodelling through reducing M1-like macrophage abundance [98]. This suggests that 5FU dysregulates monocyte recruitment and drives a shift towards a pro-fibrotic skeletal muscle microenvironment [98] (Figure 3). The Bonetto group have extensively demonstrated that the 5FU-based combination regimen, FOLFIRI [5FU, leucovorin (LV), and irinotecan (IRI)], can drive cachectic myopathy, with several mechanisms explored [35,100,101,102,103]. In skeletal muscle, FOLFIRI: (1) promotes the phosphorylation of MAPK isoforms, p38 and ERK1/2; (2) increases serum ROS levels; (3) reduces mitochondrial number and size; and (4) downregulates the expression of protein markers indicative of mitochondrial maintenance and turnover (i.e., biogenesis, fission, and fusion) [35,101,102]. Despite FOLFIRI inducing cachectic myopathy, the 5FU-based combination regimen FOLFOX [5FU, LV, and OXA] has limited impact on skeletal muscle [35]. While these data may be accounted for by methodological specifics concerning the treatment timeline and/or dosages of the FOLFOX constituents, it is an interesting observation when considered in context of findings from our laboratory. We have demonstrated that IRI (constituent of the FOLFIRI regimen) monotherapy induces cachectic myopathy [104], which is characterised by reduced expression of dystrophin, a key structural protein that connects the sarcolemma to the actin cytoskeleton and maintains cytoskeletal integrity [104]. Similarly, we recently demonstrated that 5FU monotherapy also reduces dystrophin protein expression, in addition to desmin, an intermediate filament that provides stability to sarcomeres. However, these cytoskeletal protein changes are not associated with overt cachectic myopathy or loss of function [99]. These findings highlight that: (1) 5FU may prime skeletal muscle for myopathy by reducing the abundance of key cytoskeletal structural proteins; and (2) these events (i.e., loss of dystrophin and other cytoskeletal proteins) apparently precede alterations to skeletal muscle mass or function (Figure 3). Our work suggests that cytoskeletal proteins may have potential as early biomarkers for chemotherapy-induced cachectic myopathy, although this requires further investigation to be confirmed.
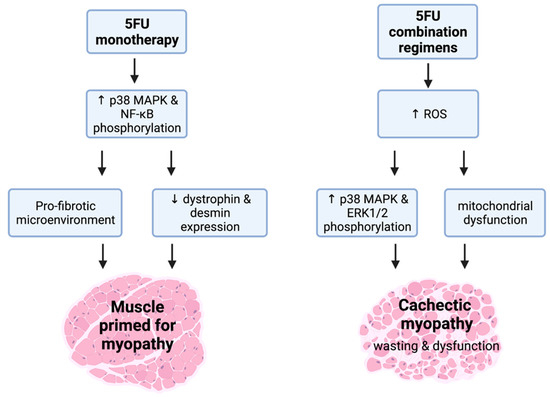
Figure 3.
(5FU)-related cachectic myopathy. 5FU monotherapy promotes the phosphorylation of atrophic regulators, p38 mitogen activated protein kinase (MAPK), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). These mechanistic targets are likely to be stimulated via signalling modulators including reactive oxygen species (ROS). 5FU does induce a pro-fibrotic skeletal muscle microenvironment and reduces the expression of the key cytoskeletal proteins, desmin and dystrophin, which suggests that 5FU primes muscle for cachectic myopathy. Interestingly, when additional chemotoxic insult to skeletal muscle occurs alongside 5FU such as in 5FU combination regimens, the induction of cachectic myopathy is observed. This is underscored by increased ROS production that stimulates the phosphorylation of p38 MAPK and ERK1/2 alongside mitochondrial dysfunction, leading to skeletal muscle wasting and dysfunction. Created with biorender.com (accessed on 6 July 2021).
2.4. Other Chemotherapeutic Agents
Several other chemotherapeutic agents have been investigated for their impact on skeletal muscle, albeit to a far lesser extent than DOX, CDDP, and 5FU. Distinct from platinum-based alkylating agents, chemotherapies arising from the alternative alkylating agent classes such as nitrosourea (i.e., cystemustine (CMN)) and nitrogen mustard (i.e., cyclophosphamide (CYP)) have also been investigated for their effect on skeletal muscle [73,105,106,107]. Interestingly, in cancer-free mice, CMN acutely reduces body mass, and, while post-chemotherapy catch-up growth is evident, skeletal muscle mass does not completely recover [106]. However, in tumour-bearing mice, the effect of CMN manifests differently. CMN treatment (10 days) reduces skeletal muscle mass, albeit, paradoxically, concurrent with enhanced protein synthesis and reduced proteasome-dependent proteolysis at the molecular level [105,106]. These data perhaps reflect an ongoing, yet unsuccessful, attempt to recover skeletal muscle mass in response to CMN treatment. Similar to CMN, CYP administration induces an acute loss of body mass followed by compensatory catch-up growth in cancer-free mice, but skeletal muscle mass loss is only mildly impacted [107]. However, CYP reduces ambulatory capacity and skeletal muscle adenosine triphosphate (ATP) production through impairing mitochondrial function [107]. CYP may also impact skeletal muscle through inducing neutropenia (CYP is used experimentally to generate rodent models of neutropenia) [108], possibly through impacting skeletal muscle repair and remodelling efficiency. A similar effect has been reported for 5FU [98].
Gemcitabine (GCB) is a chemotherapeutic agent from the anti-metabolite class, which has often been experimentally investigated as part of a combination regimen with CDDP consistent with its clinical utility for the treatment of metastatic cancers [109,110,111]. Given the extensive evidence supporting that CDDP treatment induces cachectic myopathy [10,72], it is currently unclear as to what extent (if at all) GCB contributes. Current evidence demonstrates that the CDDP and GCB combination potentiates tumour-induced skeletal muscle mass loss and proteolytic activity, while GCB alone has no impact [112,113,114]. This does not rule out the possibility that GCB exacerbates the effects of CDDP in skeletal muscle. Another chemotherapeutic agent from the anti-metabolite class is methotrexate (MTX), which is not only utilised in cancer, but in hyper-inflammatory conditions such as rheumatoid arthritis [115]. There are no data available surrounding the effect of MTX on skeletal muscle health in cancer-based models. However, in a mouse model of diabetes, MTX was shown to elicit benefits on skeletal muscle glucose metabolism [116,117]. These findings suggest that chemotherapeutic agents from the anti-metabolite class display a modest myotoxic profile compared to other chemotherapy classes.
Mitotic inhibitors are a class of chemotherapeutic agents that have been investigated for their effect on skeletal muscle, in particular taxanes and vinca alkaloids [118]. These drugs are known for their microtubule de-stabilising properties and potent neurotoxicity [119,120,121]. Docetaxel (DTX) is a taxane-based chemotherapy that demonstrably reduces skeletal muscle mass. This effect is evidenced by serum markers of malnutrition and inflammation, and a pro-catabolic transcriptional program, yet skeletal muscle contractile function is not affected [122,123]. Paclitaxel (PTX) is another taxane-based agent, albeit not extensively investigated with respect to skeletal muscle. Ramos et al. highlighted the capacity of PTX to alter skeletal muscle microtubule architecture observed through α-tubulin disorganisation [118]. Additionally, alterations to microtubules by PTX are suggested to be underpinned by impaired adenosine diphosphate (ADP)-dependent bioenergetics via the binding of tubulin to the mitochondrial ADP/ATP exchanger voltage-dependent anion channel (VDAC) [118]. Ramos et al. also studied the vinca alkaloid, vinblastine (VBL), and demonstrated a similar capacity to alter tubulin architecture. However, distinct from PTX, the connection with impaired ADP bioenergetics is seemingly independent of the interaction between tubulin and VDAC [118]. Rather, the mechanism may be reflective of the lesser-known role of VBL as an inhibitor of microtubule-associated proteins, 1A/1B light chain 3B (LC3B)-II, degradation. Thus, VBL is a likely suppressor of auto-lysosomal maturation and autophagic flux [124]. Further investigation of taxanes and vinca alkaloids with regard to skeletal muscle health is warranted, given the association with microtubule perturbations and induction of dystrophic phenotypes [125].
3. Therapeutic Strategies to Mitigate Chemotherapy-Induced Cachectic s Myopathy: An Update
Currently, cachexia represents a significant unmet challenge in cancer, with no treatment approved for clinical use. This is likely due to the complexity of the syndrome, especially at the skeletal muscle level. There are multiple contributing factors to the induction of myopathy during anti-cancer therapy. For example, muscle deconditioning due to hospitalisation-dependent factors such as prolonged periods of bed rest, reduced opportunity to undertake physical activity, and depressive mood/fatigue could contribute significantly to cachexia progression during chemotherapy administration [126]. Several investigations into therapeutic strategies to mitigate the debilitating effects of cancer cachexia are underway with multiple candidates showing promise. These include exercise and multi-target pharmaceutical/nutraceutical adjuvant interventions [127,128] (Figure 4). Herein, we summarise the current knowledge and provide insights surrounding these candidate strategies to better inform future investigations. In particular, this section will focus on therapeutic strategies that elicit protection against mitochondrial dysfunction, a key mechanistic event in the induction of cachectic myopathy [129,130]. Specifically, mitochondrial degeneration is suggested as an event preceding muscle wasting in cachexia [131], and thus is a prime therapeutic target for early intervention.
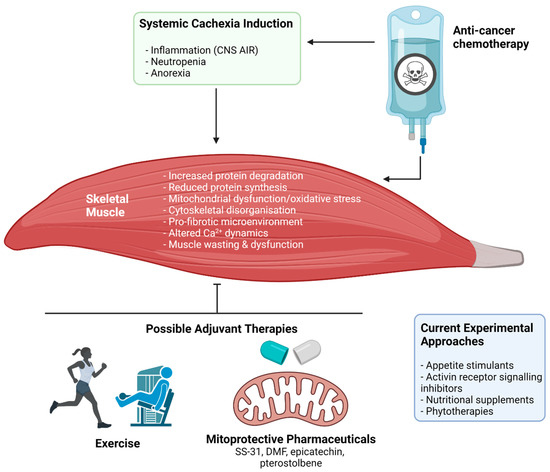
Figure 4.
The impact of anti-cancer chemotherapy treatment on cachectic myopathy and possible protective therapeutic interventions. Broadly, chemotherapeutic agents used in clinical cancer treatment can both directly and indirectly target skeletal muscle through induction or amplification of systemic cachexia. The result is the initiation of a wasting and dysfunction program within skeletal muscle, involving: increased muscle protein degradation, reduced protein synthesis, mitochondrial dysfunction and oxidative stress, cytoskeletal disorganisation and reduction of key cytoskeletal proteins that stabilise the muscle membrane, pro-fibrotic signalling within the extracellular matrix, and altered calcium (Ca2+) dynamics. The result is muscle wasting and dysfunction that leaves patients weak and fatigued, which affects their capacity to undertake activities of daily living and reduces quality of life. Several potential therapeutic approaches are currently being investigated to protect against or treat these symptoms including appetite stimulants, activin receptor signalling inhibitors, nutritional supplements, and phytotherapies. Novel therapeutic strategies could include exercise and mitoprotective compounds (e.g., SS-31, BGP-15, dimethyl fumarate (DMF), epicatechin, and pterostilbene). Abbreviations: CNS AIR: central nervous system acute illness response. Created with biorender.com (accessed on 6 July 2021).
3.1. Exercise Interventions
Exercise is a non-pharmacological and cost-effective strategy that is currently being investigated for therapeutic purposes against cachexia. The therapeutic potential of exercise in the cancer setting is multi-faceted, but includes both modulation of systemic inflammation and regulatory control of the redox balance [130], two inherently related mechanisms that drive cachexia [129]. These mechanisms are also prominent drivers of chemotherapy-induced cachectic myopathy. Thus, exercise interventions could prevent the atrophy and loss of function associated with anti-cancer chemotherapy treatment. As such, exercise has been investigated in experimental models exploring its efficacy to mitigate the impact of chemotherapy (particularly DOX) on skeletal muscle health (summarised in Table 1). In general, exercise has shown modest protective efficacy against the loss of skeletal muscle mass and functionality from chemotherapy administration [132]. Smuder proposes that this is linked to the capacity of exercise to promote endogenous antioxidant expression, protein chaperoning via heat shock protein-70 (HSP-70) to mitigate proteolytic activity, and increased multi-drug resistant proteins [133]. It is important to note that the majority of studies listed in Table 1 employed exercise as a pre-conditioning strategy prior to the administration of DOX, demonstrating the capacity of exercise to prime skeletal muscle to resist DOX-induced stress [63,64,134,135,136,137,138]. However, the clinical compatibility of this approach is questionable since cancer treatment would need to be delayed while muscles were conditioned using exercise programs. There is also a lack of data on the efficacy of pre-treatment exercise strategies for other chemotherapeutic agents aside from DOX. Jones and Alfano highlight a paucity of clinical studies investigating the utility of exercise interventions in the pre-treatment stage [139]. Rather, exercise interventions throughout the cancer survivorship continuum have predominantly been studied during and post-chemotherapy treatment, as per the adapted Physical Exercise Across the Cancer Experience (PEACE) framework [139]. To date, only four pre-clinical studies have investigated the efficacy of exercise during chemotherapy administration. These studies investigated different chemotherapeutic agents and/or regimens as well as diverse exercise modalities [78,140,141,142]. This makes it difficult to form a consensus sufficient to facilitate clinical exercise prescription based upon pre-clinical data. For example, some studies employed a maximal treadmill running test to exhaustion [141,143]. This is a low-cost alternative to incorporating the use of metabolic studies that enable the quantification of peak/maximum oxygen consumption (VO2 peak/max), which can be used to identify a relative exercise intensity [144]. Other studies have incorporated metabolic analyses enabling calculation of a relative VO2 peak/max [144]. However, given the lack of consistency across different rodent models, a knowledge gap remains in translating exercise intensity criteria between rodents and humans [145]. Additionally, there are no known experimental studies assessing the utility of exercise in the post-treatment recovery stage.

Table 1.
Experimental studies on the effect of exercise against chemotherapy-induced cachectic myopathy.
Thus far, animal experiments investigating the utility of exercise programs to resist chemotherapy-induced cachectic myopathy have not been without their pitfalls. There are numerous confounding variables highlighted in Table 1 including the selection of rodent species, strain, age, gender, and muscle type. These are all factors that can influence the protective efficacy of exercise in the context of chemotherapy-induced myopathy [145,146,147,148,149]. Additionally, tissue harvest time, relative to final chemotherapy dose, is a crucial consideration since transient events versus adaptive responses can be confused when making static measurements at a fixed timepoint. It is also imperative for experimental models to evaluate the feasibility and efficacy of resistance training during chemotherapy administration, since in vivo load-induced hypertrophy has emerged as having translational potential (for review see [150]). Animal studies to date have predominately focused on endurance training. In particular, it would be of great interest to determine whether chemotherapy-treated skeletal muscle could recover from the myotrauma induced by resistance training interventions. Indeed, Huang et al. [151] demonstrates that DOX can impair the inflammatory response necessary for skeletal muscle repair and remodelling following damaged caused by eccentric-exercise. Thus, further investigation is warranted.
Experimental animal models used to investigate the efficacy of exercise interventions during the administration of chemotherapy often utilise a treatment regimen that involves the metronomic delivery of chemotherapeutic agents spread out over an extended duration. This approach, while clinically compatible, results in milder skeletal muscle effects compared to the pre-conditioning studies, which utilise a single near-maximum tolerable dosage (MTD) bolus injection of a clinically-relevant accumulated human dose [152]. However, metronomic delivery of chemotherapy is a more compatible representation of the clinical scenario. This is especially true for DOX, which is clinically administered through slow-intravenous infusion or as repeated fractional doses cyclically over several weeks, to regulate plasma concentration of the drug. As such, severe cytotoxicity is prevented while anti-cancer efficacy is maintained [153]. The utility of metronomic delivery regimens in experimental models of chemotherapy cachexia still require a balance between retaining a clinically compatible cumulative dose and maintaining survival of the animals, so that exercise training interventions can be implemented. Going forward, future studies should refer to current gold standard models published in both de Lima et al. [143] and Ballaro et al. [89], which evaluate the effect of exercise during the synergistic-induction of cachectic myopathy from both chemotherapy and cancer-related factors. If possible, a chemotherapy control group should be included alongside a cancer control group when models are being optimised. This would help inform decision making regarding choice of exercise modality and secondary targets outside of mass and function preservation (e.g., mitigating systemic inflammation or insulin resistance) [50,154,155]. Current models need to be improved to enrich the clinical interpretability of the findings, thus promoting wider translational capability.
Current clinical studies indicate that aerobic exercise training can reduce fatigue and anxiety/depressive moods to improve participation in physical activity, while resistance exercise training can improve lean mass and muscular strength [156,157,158]. Interestingly, there is only weak evidence that either exercise modality can preserve skeletal muscle mass or cross-sectional area (CSA) [156,159,160]. This may reflect difficulties associated with measuring muscle mass in humans [161] or the barriers associated with incorporating muscle biopsies in clinical trials such as high costs and low patient recruitment due to the invasiveness of sampling. Nevertheless, these data highlight inconsistency with data derived from chemotherapy treated, cancer-free mouse models, which demonstrate protective efficacy from exercise (see Table 1) [78,142]. Perhaps even more interesting, and consistent with the clinical data, is that exercise did not alter skeletal muscle mass or CSA in two tumour-burdened mouse models treated with chemotherapy [89,143]. Thus, tumour factors appear to be most influential in dictating whether skeletal muscle can be modulated by exercise. While mass may not necessarily be modifiable by exercise therapy, de Lima et al. importantly demonstrated that exercise training improved skeletal muscle recovery after the cessation of chemotherapy, a paradigm yet to be explored in clinical studies [143]. Considering that current exercise interventions only elicit modest protective efficacy against chemotherapy and cancer-induced cachectic myopathy at best, there is an emphasis to explore multi-modal therapeutic strategies that involve exercise prescription programs alongside pharmacological interventions for synergistic benefits. Furthermore, given that some cancer patients may have a reduced opportunity to undertake exercise training due to hospitalisation-related deconditioning, multi-targeted pharmacological and/or supplementary interventions strategies that mimic specific aspects of exercise should be identified and explored for their therapeutic efficacy.
3.2. Adjuvant Therapies
Complementary to exercise strategies, several adjuvant candidates have been evaluated for their protective potential to combat chemotherapy-induced cachectic myopathy. Drug candidates focus on different biochemical targets that are involved in diverse molecular and physiological aspects that characterise the cachectic myopathy phenotype [40]. These candidates arise from a range of different therapeutic classes including activin receptor signalling inhibitors [59,60,100,103,165], appetite stimulants [77,80,82,166,167,168,169,170], nutritional supplements [171,172,173,174,175,176], and phytotherapies [112,113,177,178,179,180,181,182,183,184,185]. Activin receptor signalling inhibitors have shown strong pre-clinical efficacy to mitigate cancer and chemotherapy-induced cachexia through preserving skeletal muscle mass and function [186]. However, there has been limited success in the clinical translation of this pharmacological target, with a Phase II trial conducted by Novartis Pharmaceuticals (NCT01433263) demonstrating that activin receptor antibody, BYM338 (Bimagrumab), improved lean mass and muscle volume, but contributed to a net loss of body mass [187].
Appetite stimulants, in particular ghrelin receptor agonists, have shown promise in the pre-clinical setting to normalise food intake and muscle mass/CSA of chemotherapy-treated mice compared to healthy counterparts [80,82,170]. These findings translated clinically as observed through the ROMANA 1 & 2 trials (NCT01387269 & NCT01387282), where anamorelin (a ghrelin receptor agonist) increased body and lean mass of advanced cancer patients. However, it was not approved for clinical use because it failed to improve grip strength—a primary endpoint of the trial [188].
Nutritional supplements including essential amino acids and fatty acids have shown the capacity to protect against cancer and chemotherapy-induced cachexia, highlighting translational potential in single supplement interventions [189]. However, nutritional supplements are only a complementary piece of the cachexia puzzle as their clinical utility is primarily dependent on the anabolic potential of the individual [190]. Additionally, nutrition-related guidelines in cancer are based mostly on expert consensus, rarely on clinical trial evidence, highlighting a greater need to investigate multi-combination nutritional supplement interventions at the clinical level [191]. Similarly, compounds from the phytotherapies class have typically not been clinically evaluated for their therapeutic efficacy to mitigate cachectic myopathy given their rare utility in Western medicine [192]. This aspect should be re-considered based on encouraging pre-clinical data—specifically, the reduction of atrogene transcription, which underscores skeletal muscle wasting and holds strong promise as a therapeutic target against cachexia [193].
An emerging therapeutic class of particular interest to our laboratory group, and others, is the utility of mitoprotective compounds to combat skeletal muscle oxidative stress. Enhanced ROS production is a common underlying mechanism associated with multiple chemotherapies [32,33] and is a key contributing factor to mitochondrial dysfunction, a tenet of cachectic myopathy [36,37]. To date, mitoprotective compounds have not been evaluated in clinical trials. However, multiple mitoprotective agents have been experimentally evaluated for their potential protective efficacy against chemotherapy-induced cachectic myopathy, which will be discussed herein.
One of the first mitoprotective agents investigated for its therapeutic efficacy alongside chemotherapy was SS-31, a cardiolipin-targeting peptide, which preserves mitochondrial cristae structure and promotes oxidative phosphorylation [194]. SS-31 was shown to attenuate DOX-induced activity of multiple proteolytic systems (e.g., UPS, apoptosis, and calpains) in various muscle types, which prevented skeletal muscle atrophy [56,68]. Further, SS-31 normalises DOX-induced ROS emission in C2C12 myotube cultures and rodent muscle, with the latter observed as an acute event [33,56]. However, Ballaro et al. demonstrated that SS-31 administration yielded a modest mitoprotection, and subsequently, limited therapeutic efficacy in a more clinically compatible model of cachectic myopathy. In this study, mice were injected with C26 colorectal adenocarcinoma cells and metronomically dosed with OXA and 5FU combination chemotherapy over five weeks [90]. These data highlight that SS-31 may be more efficacious at a preventative stage of cachexia (i.e., pre-cachexia) when mitochondria are less damaged [131]. Alternatively, SS-31 may elicit specific mitoprotection against the anthracycline DOX, which induces mitochondrial stress differently to platinum-based alkylating agents, albeit this would need to be confirmed in future studies of DOX utilising more clinically compatible models.
Another adjuvant mitoprotective agent is BGP-15, a hydroximic acid derivative nicotinic acid-amidoxime small molecule that can preserve skeletal muscle metabolic homeostasis and mitochondrial quality control processes [195,196]. In particular, BGP-15 has been touted as an inhibitor of poly-(ADP-ribose) polymerase-1 (PARP-1) and co-inducer of HSP-70 [197,198], which are mechanisms associated with improved mitochondrial content, function, and oxidative capacity [199,200]. Furthermore, BGP-15 has also shown pleiotropic capacity to elicit mitoprotection independent of these mechanisms [201], a likely explanation of its therapeutic potential in a range of myopathies [202,203,204,205]. Our laboratory group has evaluated the therapeutic utility of BGP-15 alongside multiple chemotherapies including OXA, IRI, and 5FU with mixed efficacy [88,99,104]. BGP-15 was protective against OXA-induced lean mass loss, while also normalising ROS generation and mitochondrial viability [88]. However, alongside IRI, BGP-15 paradoxically rescued (partially) body, lean, and skeletal muscle mass in addition to muscle contractile function, while exacerbating the IRI-induced muscle protein synthesis inhibition and reduced expression of the cytoskeletal proteins, dystrophin, and β-dystroglycan [104]. The recovery of muscle mass and function may have been due to BGP-15′s enhancement of ATP production and mitochondrial density. Indeed, the latter was also enhanced when BGP-15 was delivered alongside 5FU and was associated with improved mitochondrial fusion. Furthermore, BGP-15 suppressed the 5FU-induced phosphorylation of NF-κB and MAPK isoforms [99], two likely mechanisms involved in the induction of cachectic myopathy. Given the heterogeneity of our findings, further investigations are required with a focus on clinically compatible combination chemotherapy regimens before BGP-15 can be considered a viable therapeutic candidate to protect against cachectic myopathy.
Future studies investigating adjuvant mitoprotective candidates should consider agents that can synergistically target chemotherapy driven oxidative damage, alongside other signalling pathways involved in the induction of cachectic myopathy. One potential compound is the nuclear factor erythroid 2-related factor 2 (Nrf2) transcriptional activator, dimethyl fumarate (DMF). DMF is a methyl ester of fumaric acid purported to upregulate cytoprotective response genes, and suppress NF-κB signalling, which imparts an anti-oxidative and -inflammatory effect [206] Another Nrf2 activator with therapeutic potential in this setting could be pterostilbene [207], a dimethoxylated analogue of resveratrol, a compound with demonstrated efficacy against DOX-mediated skeletal and cardiac myopathy [208,209]. Like resveratrol, pterostilbene can prolong lifespan, mitigate oxidative stress and normalise dysregulated autophagy in experimental models [210,211]. However, pterostilbene has greater bioavailability and a longer half-life compared to resveratrol [212], suggesting greater translational potential. Finally, epicatechin is another potentially viable mitoprotective candidate, which activates Nrf2 to inhibit oxidative stress [213]. Interestingly, epicatechin has been shown to restore the expression of dystrophin and other key cytoskeletal proteins in models of myopathy such as Becker muscular dystrophy and diabetes [214,215]. This highlights the protective potential of epicatechin against the chemotherapy-induced reduction of dystrophin expression shown by our group [99,104], and in cancer-induced cachectic myopathy in which dystrophin is also reduced [216]. Importantly, the adjuvant candidates proposed are not associated with cancer growth, and thus are unlikely to impact the anti-cancer efficacy of chemotherapy treatment [206,217,218].
Other compounds have also been evaluated for their therapeutic efficacy against chemotherapy-induced cachectic myopathy, whereby mitoprotection is elicited as a secondary effect to the central mechanism. Sodium nitrate (SN) supplementation is one of these strategies. SN potentiates the nitrate/nitrite/nitric oxide (NO) pathway to increase endogenous NO production as the central mechanism [219]. Mitoprotection occurs concurrently via an alternate pathway [220]. SN is cardioprotective during DOX treatment in mice, with the preservation of left ventricular function dependent on the mitigation of oxidative stress and mitochondrial Complex I dysfunction [221,222]. However, our evaluation of SN supplementation alongside DOX administration failed to elicit a protective effect against cachectic myopathy [223]. Interestingly, the metabolic cytoprotectant, metformin, has also been investigated alongside DOX and demonstrated no protective benefit against chemotherapy-induced cachectic myopathy. These drug candidates both promote adenosine monophosphate-activated protein kinase (AMPK) signalling to preserve cellular energy status [220,224]. Thus, they may have greater utility as a therapeutic strategy to mitigate aberrant skeletal muscle glucose uptake during chemotherapy treatment.
4. Future Directions and Conclusions
Chemotherapy is an under-appreciated contributing factor in the induction of cachectic myopathy. To date, research has predominately contextualised cancer cachexia as governed by tumour-related factors without considering the other side of the cachexia sphere—chemotherapy [9]. This is problematic with respect to the clinical compatibility of this paradigm, as the large majority of cancer patients typically receive chemotherapy as part of their treatment strategy. This highlights the need for experimental studies to reflect the synergistic insult from cancer and chemotherapy in the induction of cachectic myopathy [15]. Models that allow the exploration of both factors in combination, and, using the spectra of chemotherapeutic agents, multi-therapy regimens, and cancer sub-types, are required. Future investigations in this area also need to consider the impact of novel chemotherapeutic agents in clinical trials across all cancers. For example, the therapeutic utility of multi-kinase inhibitors is becoming increasingly prevalent [225] and they demonstrably have cachexia-inducing properties [226,227]. Additionally, drug compounds that are utilised to mitigate the side-effects of clinical chemotherapy treatment such as the well-described gastrointestinal toxicities elicited by dexamethasone [228] need to be stratified for their capacity to potentiate chemotherapy-induced cachectic myopathy [229,230].
The utility of a cachexia scoring system within investigative animal research such as the animal cachexia score (ACASCO) [231] is warranted to justify a relative cachectic burden concerning chemotherapy- and cancer-induced myopathy. This will assist with the clinical interpretation of experimental findings, and where they fit within the cachexia diagnostic continuum in human patients, which incorporates three key stages: pre-cachexia, cachexia, and refractory cachexia [14]. Specific to chemotherapy-induced cachectic myopathy, a modified scoring system should also be considered to incorporate biomarkers of oxidative stress and skeletal muscle damage, which are currently being developed for other severe skeletal myopathies that share similar features with cachectic myopathy (i.e., dystrophin loss and oxidative stress) as highlighted by Grounds et al. [232].
Despite emerging as a burgeoning sub-field of skeletal muscle wasting conditions, chemotherapy-induced myopathy is, to date, a pariah with respect to research effort and directed funding in contrast to cancer-induced myopathy [9]—comparatively, it remains on the ‘dark side’ of the cachexia sphere. Illuminating the mechanisms involved and the physiological repercussions as well as actively pursuing protective therapeutics will enrich clinical decision making, patient outcomes, and the quality of cancer survivorship.
Author Contributions
Conceptualisation, D.G.C., C.A.G., and E.R.; Writing—original draft preparation, D.G.C.; Writing—review and editing, E.R. and C.A.G.; Supervision, E.R. and C.A.G.; Project administration, E.R.; Funding acquisition, E.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a program establishment grant (62962602) from the Institute for Sport, Exercise, and Active Living and the Centre for Chronic Disease, Victoria University (now the Institute for Health and Sport, Victoria University).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors wish to thank Cara Timpani for proof-reading the manuscript.
Conflicts of Interest
E.R. is a consultant to Santhera Pharmaceuticals and Epirium Bio. The other authors declare no conflict of interest.
References
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- Reuvers, T.G.A.; Kanaar, R.; Nonnekens, J. DNA Damage-Inducing Anticancer Therapies: From Global to Precision Damage. Cancers 2020, 12, 2098. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg Robert, A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chen, Y.; Jungsuwadee, P.; Vore, M.; Butterfield, D.A.; St Clair, D.K. Collateral damage in cancer chemotherapy: Oxidative stress in nontargeted tissues. Mol. Interv. 2007, 7, 147–156. [Google Scholar] [CrossRef]
- Love, R.R.; Leventhal, H.; Easterling, D.V.; Nerenz, D.R. Side effects and emotional distress during cancer chemotherapy. Cancer 1989, 63, 604–612. [Google Scholar] [CrossRef]
- Florescu, M.; Cinteza, M.; Vinereanu, D. Chemotherapy-induced Cardiotoxicity. Maedica 2013, 8, 59–67. [Google Scholar]
- Giglio, P.; Gilbert, M.R. Neurologic complications of cancer and its treatment. Curr. Oncol. Rep. 2010, 12, 50–59. [Google Scholar] [CrossRef]
- Maxwell, M.B.; Maher, K.E. Chemotherapy-induced myelosuppression. Semin. Oncol. Nurs. 1992, 8, 113–123. [Google Scholar] [CrossRef]
- Coletti, D. Chemotherapy-induced muscle wasting: An update. Eur. J. Transl. Myol. 2018, 28, 7587. [Google Scholar] [CrossRef]
- Conte, E.; Bresciani, E.; Rizzi, L.; Cappellari, O.; De Luca, A.; Torsello, A.; Liantonio, A. Cisplatin-Induced Skeletal Muscle Dysfunction: Mechanisms and Counteracting Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 1242. [Google Scholar] [CrossRef]
- Hiensch, A.E.; Bolam, K.A.; Mijwel, S.; Jeneson, J.A.L.; Huitema, A.D.R.; Kranenburg, O.; van der Wall, E.; Rundqvist, H.; Wengstrom, Y.; May, A.M. Doxorubicin-induced skeletal muscle atrophy: Elucidating the underlying molecular pathways. Acta Physiol. 2019, e13400. [Google Scholar] [CrossRef]
- Gilliam, L.A.; St Clair, D.K. Chemotherapy-induced weakness and fatigue in skeletal muscle: The role of oxidative stress. Antioxid. Redox Signal. 2011, 15, 2543–2563. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet. Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Berardi, E.; Madaro, L.; Lozanoska-Ochser, B.; Adamo, S.; Thorrez, L.; Bouche, M.; Coletti, D. A Pound of Flesh: What Cachexia Is and What It Is Not. Diagnostics 2021, 11, 116. [Google Scholar] [CrossRef]
- Caan, B.J.; Meyerhardt, J.A.; Kroenke, C.H.; Alexeeff, S.; Xiao, J.; Weltzien, E.; Feliciano, E.C.; Castillo, A.L.; Quesenberry, C.P.; Kwan, M.L.; et al. Explaining the Obesity Paradox: The Association between Body Composition and Colorectal Cancer Survival (C-SCANS Study). Cancer Epidemiol. Biomark. Prev. 2017, 26, 1008–1015. [Google Scholar] [CrossRef]
- Martin, L.; Birdsell, L.; Macdonald, N.; Reiman, T.; Clandinin, M.T.; McCargar, L.J.; Murphy, R.; Ghosh, S.; Sawyer, M.B.; Baracos, V.E. Cancer cachexia in the age of obesity: Skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J. Clin. Oncol. 2013, 31, 1539–1547. [Google Scholar] [CrossRef]
- Bozzetti, F. Forcing the vicious circle: Sarcopenia increases toxicity, decreases response to chemotherapy and worsens with chemotherapy. Ann. Oncol. 2017, 28, 2107–2118. [Google Scholar] [CrossRef]
- Rausch, V.; Sala, V.; Penna, F.; Porporato, P.E.; Ghigo, A. Understanding the common mechanisms of heart and skeletal muscle wasting in cancer cachexia. Oncogenesis 2021, 10, 1. [Google Scholar] [CrossRef]
- Scheede-Bergdahl, C.; Jagoe, R.T. After the chemotherapy: Potential mechanisms for chemotherapy-induced delayed skeletal muscle dysfunction in survivors of acute lymphoblastic leukaemia in childhood. Front. Pharmacol. 2013, 4, 49. [Google Scholar] [CrossRef]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Thomas, D.R. Distinguishing starvation from cachexia. Clin. Geriatr Med. 2002, 18, 883–891. [Google Scholar] [CrossRef]
- Argilés, J.M.; López-Soriano, F.J.; Stemmler, B.; Busquets, S. Novel targeted therapies for cancer cachexia. Biochem. J. 2017, 474, 2663–2678. [Google Scholar] [CrossRef]
- Braun, T.P.; Szumowski, M.; Levasseur, P.R.; Grossberg, A.J.; Zhu, X.; Agarwal, A.; Marks, D.L. Muscle atrophy in response to cytotoxic chemotherapy is dependent on intact glucocorticoid signaling in skeletal muscle. PLoS ONE 2014, 9, e106489. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.P.; Grossberg, A.J.; Krasnow, S.M.; Levasseur, P.R.; Szumowski, M.; Zhu, X.X.; Maxson, J.E.; Knoll, J.G.; Barnes, A.P.; Marks, D.L. Cancer- and endotoxin-induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J. 2013, 27, 3572–3582. [Google Scholar] [CrossRef]
- Braun, T.P.; Zhu, X.; Szumowski, M.; Scott, G.D.; Grossberg, A.J.; Levasseur, P.R.; Graham, K.; Khan, S.; Damaraju, S.; Colmers, W.F.; et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic-pituitary-adrenal axis. J. Exp. Med. 2011, 208, 2449–2463. [Google Scholar] [CrossRef]
- Pastor, F.; Dumas, K.; Barthélémy, M.-A.; Regazzetti, C.; Druelle, N.; Peraldi, P.; Cormont, M.; Tanti, J.-F.; Giorgetti-Peraldi, S. Implication of REDD1 in the activation of inflammatory pathways. Sci. Rep. 2017, 7, 7023. [Google Scholar] [CrossRef]
- Britto, F.A.; Dumas, K.; Giorgetti-Peraldi, S.; Ollendorff, V.; Favier, F.B. Is REDD1 a metabolic double agent? Lessons from physiology and pathology. Am. J. Physiol.-Cell Physiol. 2020, 319, C807–C824. [Google Scholar] [CrossRef] [PubMed]
- Kefaloyianni, E.; Gaitanaki, C.; Beis, I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 2006, 18, 2238–2251. [Google Scholar] [CrossRef]
- Webster, J.M.; Kempen, L.J.A.P.; Hardy, R.S.; Langen, R.C.J. Inflammation and Skeletal Muscle Wasting During Cachexia. Front. Physiol. 2020, 11, 597675. [Google Scholar] [CrossRef]
- Cai, D.; Frantz, J.D.; Tawa, N.E., Jr.; Melendez, P.A.; Oh, B.C.; Lidov, H.G.; Hasselgren, P.O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cheregi, B.D.; Sorensen, J.C.; Nurgali, K.; Hayes, A. Chemotherapeutic agents induce mitochondrial superoxide production and toxicity but do not alter respiration in skeletal muscle in vitro. Mitochondrion 2018, 42, 33–49. [Google Scholar] [CrossRef]
- Gilliam, L.A.; Moylan, J.S.; Patterson, E.W.; Smith, J.D.; Wilson, A.S.; Rabbani, Z.; Reid, M.B. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 2012, 302, C195–C202. [Google Scholar] [CrossRef] [PubMed]
- Fanzani, A.; Zanola, A.; Rovetta, F.; Rossi, S.; Aleo, M.F. Cisplatin triggers atrophy of skeletal C2C12 myotubes via impairment of Akt signalling pathway and subsequent increment activity of proteasome and autophagy systems. Toxicol. Appl. Pharmacol. 2011, 250, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Barreto, R.; Waning, D.L.; Gao, H.; Liu, Y.; Zimmers, T.A.; Bonetto, A. Chemotherapy-related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 2016, 7, 43442–43460. [Google Scholar] [CrossRef]
- Hyatt, H.W.; Powers, S.K. Mitochondrial Dysfunction Is a Common Denominator Linking Skeletal Muscle Wasting Due to Disease, Aging, and Prolonged Inactivity. Antioxidants 2021, 10, 588. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, J.C.; Cheregi, B.D.; Timpani, C.A.; Nurgali, K.; Hayes, A.; Rybalka, E. Mitochondria: Inadvertent targets in chemotherapy-induced skeletal muscle toxicity and wasting? Cancer Chemother Pharm. 2016, 78, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Mehl, K.A.; Davis, J.M.; Berger, F.G.; Carson, J.A. Myofiber degeneration/regeneration is induced in the cachectic ApcMin/+ mouse. J. Appl. Physiol. 2005, 99, 2379–2387. [Google Scholar] [CrossRef] [PubMed]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef]
- Martin, A.; Freyssenet, D. Phenotypic features of cancer cachexia-related loss of skeletal muscle mass and function: Lessons from human and animal studies. J. Cachexia Sarcopenia Muscle 2021, 12, 252–273. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Hayward, R.; Hydock, D.; Gibson, N.; Greufe, S.; Bredahl, E.; Parry, T. Tissue retention of doxorubicin and its effects on cardiac, smooth, and skeletal muscle function. J. Physiol. Biochem. 2013, 69, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Fabris, S.; MacLean, D.A. Skeletal Muscle an Active Compartment in the Sequestering and Metabolism of Doxorubicin Chemotherapy. PLoS ONE 2015, 10, e0139070. [Google Scholar] [CrossRef] [PubMed]
- Fairclough, D.L.; Fetting, J.H.; Cella, D.; Wonson, W.; Moinpour, C.M. Quality of life and quality adjusted survival for breast cancer patients receiving adjuvant therapy. Eastern Cooperative Oncology Group (ECOG). Qual. Life Res. 1999, 8, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.L.; Winters-Stone, K.; Gallucci, B. Exercise effects on bone mineral density in women with breast cancer receiving adjuvant chemotherapy. Oncol. Nurs. Forum 2007, 34, 627–633. [Google Scholar] [CrossRef] [PubMed]
- van Norren, K.; van Helvoort, A.; Argiles, J.M.; van Tuijl, S.; Arts, K.; Gorselink, M.; Laviano, A.; Kegler, D.; Haagsman, H.P.; van der Beek, E.M. Direct effects of doxorubicin on skeletal muscle contribute to fatigue. Br. J. Cancer 2009, 100, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Hydock, D.S.; Lien, C.Y.; Jensen, B.T.; Schneider, C.M.; Hayward, R. Characterization of the effect of in vivo doxorubicin treatment on skeletal muscle function in the rat. Anticancer Res. 2011, 31, 2023–2028. [Google Scholar]
- Zorzato, F.; Salviati, G.; Facchinetti, T.; Volpe, P. Doxorubicin induces calcium release from terminal cisternae of skeletal muscle. A study on isolated sarcoplasmic reticulum and chemically skinned fibers. J. Biol. Chem. 1985, 260, 7349–7355. [Google Scholar] [CrossRef]
- Gilliam, L.A.A.; Ferreira, L.F.; Bruton, J.D.; Moylan, J.S.; Westerblad, H.; St Clair, D.K.; Reid, M.B. Doxorubicin acts through tumor necrosis factor receptor subtype 1 to cause dysfunction of murine skeletal muscle. J. Appl. Physiol. 2009, 107, 1935–1942. [Google Scholar] [CrossRef]
- Dessouki, F.B.A.; Kukreja, R.C.; Singla, D.K. Stem Cell-Derived Exosomes Ameliorate Doxorubicin-Induced Muscle Toxicity through Counteracting Pyroptosis. Pharmaceuticals 2020, 13, 450. [Google Scholar] [CrossRef]
- Dessouki, F.B.; Kukreja, R.C.; Singla, D.K. Doxorubicin-Induced Muscle Toxicity: A Novel Mechanism Involving Inflammation-Mediated Pyroptosis in Soleus Muscle. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Hardin, B.J.; Campbell, K.S.; Smith, J.D.; Arbogast, S.; Smith, J.; Moylan, J.S.; Reid, M.B. TNF-α acts via TNFR1 and muscle-derived oxidants to depress myofibrillar force in murine skeletal muscle. J. Appl. Physiol. 2008, 104, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, L.A.A.; Fisher-Wellman, K.H.; Lin, C.T.; Maples, J.M.; Cathey, B.L.; Neufer, P.D. The anticancer agent doxorubicin disrupts mitochondrial energy metabolism and redox balance in skeletal muscle. Free Radic. Biol. Med. 2013, 65, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Doerr, V.; Montalvo, R.N.; Kwon, O.S.; Talbert, E.E.; Hain, B.A.; Houston, F.E.; Smuder, A.J. Prevention of Doxorubicin-Induced Autophagy Attenuates Oxidative Stress and Skeletal Muscle Dysfunction. Antioxidants 2020, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Kwon, O.S.; Smuder, A.J.; Wiggs, M.P.; Sollanek, K.J.; Christou, D.D.; Yoo, J.K.; Hwang, M.H.; Szeto, H.H.; Kavazis, A.N.; et al. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. Physiol. 2015, 593, 2017–2036. [Google Scholar] [CrossRef]
- Gilliam, L.A.; Lark, D.S.; Reese, L.R.; Torres, M.J.; Ryan, T.E.; Lin, C.T.; Cathey, B.L.; Neufer, P.D. Targeted overexpression of mitochondrial catalase protects against cancer chemotherapy-induced skeletal muscle dysfunction. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E293–E301. [Google Scholar] [CrossRef]
- Tarpey, M.D.; Amorese, A.J.; Balestrieri, N.P.; Fisher-Wellman, K.H.; Spangenburg, E.E. Doxorubicin causes lesions in the electron transport system of skeletal muscle mitochondria that are associated with a loss of contractile function. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, T.A.; Degerman, J.; Rasanen, M.; Poikonen, A.R.; Koskinen, S.; Mervaala, E.; Pasternack, A.; Ritvos, O.; Kivela, R.; Hulmi, J.J. Systemic blockade of ACVR2B ligands prevents chemotherapy-induced muscle wasting by restoring muscle protein synthesis without affecting oxidative capacity or atrogenes. Sci. Rep. 2016, 6, 32695. [Google Scholar] [CrossRef]
- Hulmi, J.J.; Nissinen, T.A.; Rasanen, M.; Degerman, J.; Lautaoja, J.H.; Hemanthakumar, K.A.; Backman, J.T.; Ritvos, O.; Silvennoinen, M.; Kivela, R. Prevention of chemotherapy-induced cachexia by ACVR2B ligand blocking has different effects on heart and skeletal muscle. J. Cachexia Sarcopenia Muscle 2018, 9, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Beyfuss, K.; Hood, D.A. A systematic review of p53 regulation of oxidative stress in skeletal muscle. Redox Rep. 2018, 23, 100–117. [Google Scholar] [CrossRef]
- Supriya, R.; Tam, B.T.; Pei, X.M.; Lai, C.W.; Chan, L.W.; Yung, B.Y.; Siu, P.M. Doxorubicin Induces Inflammatory Modulation and Metabolic Dysregulation in Diabetic Skeletal Muscle. Front. Physiol. 2016, 7. [Google Scholar] [CrossRef]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise protects against doxorubicin-induced oxidative stress and proteolysis in skeletal muscle. J. Appl. Physiol. 2011, 110, 935–942. [Google Scholar] [CrossRef]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise protects against doxorubicin-induced markers of autophagy signaling in skeletal muscle. J. Appl. Physiol. 2011, 111, 1190–1198. [Google Scholar] [CrossRef]
- Goodman, C.A. Role of mTORC1 in mechanically induced increases in translation and skeletal muscle mass. J. Appl. Physiol. 2019, 127, 581–590. [Google Scholar] [CrossRef] [PubMed]
- D’Lugos, A.C.; Fry, C.S.; Ormsby, J.C.; Sweeney, K.R.; Brightwell, C.R.; Hale, T.M.; Gonzales, R.J.; Angadi, S.S.; Carroll, C.C.; Dickinson, J.M. Chronic doxorubicin administration impacts satellite cell and capillary abundance in a muscle-specific manner. Physiol. Rep. 2019, 7, e14052. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, K.R.; McMillan, J.D.; Kumar, A. Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease. J. Cell Physiol. 2018, 233, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Montalvo, R.N.; Doerr, V.; Min, K.; Szeto, H.H.; Smuder, A.J. Doxorubicin-induced oxidative stress differentially regulates proteolytic signaling in cardiac and skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R227–R233. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Ali, B.A.; Zulli, A.; Apostolopoulos, V. Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers 2021, 13, 1572. [Google Scholar] [CrossRef]
- Kanat, O.; Ertas, H.; Caner, B. Platinum-induced neurotoxicity: A review of possible mechanisms. World J. Clin. Oncol. 2017, 8, 329–335. [Google Scholar] [CrossRef]
- Moreira-Pais, A.; Ferreira, R.; Gil da Costa, R. Platinum-induced muscle wasting in cancer chemotherapy: Mechanisms and potential targets for therapeutic intervention. Life Sci. 2018, 208, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Le Bricon, T.; Gugins, S.; Cynober, L.; Baracos, V.E. Negative impact of cancer chemotherapy on protein metabolism in healthy and tumor-bearing rats. Metabolism 1995, 44, 1340–1348. [Google Scholar] [CrossRef]
- Sakai, H.; Sagara, A.; Arakawa, K.; Sugiyama, R.; Hirosaki, A.; Takase, K.; Jo, A.; Sato, K.; Chiba, Y.; Yamazaki, M.; et al. Mechanisms of cisplatin-induced muscle atrophy. Toxicol. Appl. Pharmacol. 2014, 278, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Ikeno, Y.; Tsukimura, Y.; Inomata, M.; Suzuki, Y.; Kon, R.; Ikarashi, N.; Chiba, Y.; Yamada, T.; Kamei, J. Upregulation of ubiquitinated proteins and their degradation pathway in muscle atrophy induced by cisplatin in mice. Toxicol. Appl. Pharmacol. 2020, 403, 115165. [Google Scholar] [CrossRef] [PubMed]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Sirago, G.; Conte, E.; Fracasso, F.; Cormio, A.; Fehrentz, J.A.; Martinez, J.; Musicco, C.; Camerino, G.M.; Fonzino, A.; Rizzi, L.; et al. Growth hormone secretagogues hexarelin and JMV2894 protect skeletal muscle from mitochondrial damages in a rat model of cisplatin-induced cachexia. Sci. Rep. 2017, 7, 13017. [Google Scholar] [CrossRef]
- Sakai, H.; Kimura, M.; Isa, Y.; Yabe, S.; Maruyama, A.; Tsuruno, Y.; Kai, Y.; Sato, F.; Yumoto, T.; Chiba, Y.; et al. Effect of acute treadmill exercise on cisplatin-induced muscle atrophy in the mouse. Pflügers Arch.-Eur. J. Physiol. 2017, 469, 1495–1505. [Google Scholar] [CrossRef]
- Damrauer, J.S.; Stadler, M.E.; Acharyya, S.; Baldwin, A.S.; Couch, M.E.; Guttridge, D.C. Chemotherapy-induced muscle wasting: Association with NF-κB and cancer cachexia. Eur. J. Transl. Myol. 2018, 28, 7590. [Google Scholar] [CrossRef]
- Chen, J.A.; Splenser, A.; Guillory, B.; Luo, J.; Mendiratta, M.; Belinova, B.; Halder, T.; Zhang, G.; Li, Y.P.; Garcia, J.M. Ghrelin prevents tumour- and cisplatin-induced muscle wasting: Characterization of multiple mechanisms involved. J. Cachexia Sarcopenia Muscle 2015, 6, 132–143. [Google Scholar] [CrossRef]
- Berchtold, M.W.; Brinkmeier, H.; Muntener, M. Calcium ion in skeletal muscle: Its crucial role for muscle function, plasticity, and disease. Physiol. Rev. 2000, 80, 1215–1265. [Google Scholar] [CrossRef]
- Conte, E.; Camerino, G.M.; Mele, A.; De Bellis, M.; Pierno, S.; Rana, F.; Fonzino, A.; Caloiero, R.; Rizzi, L.; Bresciani, E.; et al. Growth hormone secretagogues prevent dysregulation of skeletal muscle calcium homeostasis in a rat model of cisplatin-induced cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 386–404. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.W.Y.; Backstrom, I.; Bally, M.B. Sulfonation, an underexploited area: From skeletal development to infectious diseases and cancer. Oncotarget 2016, 7, 55811–55827. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Baines, I.C.; Rhee, S.G. Characterization of a mammalian peroxiredoxin that contains one conserved cysteine. J. Biol. Chem. 1998, 273, 6303–6311. [Google Scholar] [CrossRef] [PubMed]
- Hain, B.A.; Jude, B.; Xu, H.; Smuin, D.M.; Fox, E.J.; Elfar, J.C.; Waning, D.L. Zoledronic Acid Improves Muscle Function in Healthy Mice Treated with Chemotherapy. J. Bone Miner. Res. 2020, 35, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Hain, B.A.; Xu, H.; Wilcox, J.R.; Mutua, D.; Waning, D.L. Chemotherapy-induced loss of bone and muscle mass in a mouse model of breast cancer bone metastases and cachexia. JCSM Rapid Commun. 2019, 2, e00075. [Google Scholar] [CrossRef]
- Sorensen, J.C.; Petersen, A.C.; Timpani, C.A.; Campelj, D.G.; Cook, J.; Trewin, A.J.; Stojanovska, V.; Stewart, M.; Hayes, A.; Rybalka, E. BGP-15 Protects against Oxaliplatin-Induced Skeletal Myopathy and Mitochondrial Reactive Oxygen Species Production in Mice. Front. Pharmacol. 2017, 8, 137. [Google Scholar] [CrossRef]
- Ballaro, R.; Beltra, M.; De Lucia, S.; Pin, F.; Ranjbar, K.; Hulmi, J.J.; Costelli, P.; Penna, F. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 2019, 33, 5482–5494. [Google Scholar] [CrossRef]
- Ballarò, R.; Lopalco, P.; Audrito, V.; Beltrà, M.; Pin, F.; Angelini, R.; Costelli, P.; Corcelli, A.; Bonetto, A.; Szeto, H.H.; et al. Targeting Mitochondria by SS-31 Ameliorates the Whole Body Energy Status in Cancer- and Chemotherapy-Induced Cachexia. Cancers 2021, 13, 850. [Google Scholar] [CrossRef]
- Yi, Y.W.; Bae, I. Effects of solvents on in vitro potencies of platinum compounds. DNA Repair 2011, 10, 1084–1085. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Telma, K.A.; Chang, K.-E.; Lee, T.D.; Madigan, J.P.; Lloyd, J.R.; Goldlust, I.S.; Hoeschele, J.D.; Gottesman, M.M. Say no to DMSO: Dimethylsulfoxide inactivates cisplatin, carboplatin, and other platinum complexes. Cancer Res. 2014, 74, 3913–3922. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- McQuade, R.M.; Stojanovska, V.; Donald, E.; Abalo, R.; Bornstein, J.C.; Nurgali, K. Gastrointestinal dysfunction and enteric neurotoxicity following treatment with anticancer chemotherapeutic agent 5-fluorouracil. Neurogastroenterol. Motil. 2016, 28, 1861–1875. [Google Scholar] [CrossRef] [PubMed]
- Sougiannis, A.T.; VanderVeen, B.N.; Enos, R.T.; Velazquez, K.T.; Bader, J.E.; Carson, M.; Chatzistamou, I.; Walla, M.; Pena, M.M.; Kubinak, J.L.; et al. Impact of 5 fluorouracil chemotherapy on gut inflammation, functional parameters, and gut microbiota. Brain Behav. Immun. 2019, 80, 44–55. [Google Scholar] [CrossRef]
- Stringer, A.M.; Gibson, R.J.; Logan, R.M.; Bowen, J.M.; Yeoh, A.S.; Hamilton, J.; Keefe, D.M. Gastrointestinal microflora and mucins may play a critical role in the development of 5-Fluorouracil-induced gastrointestinal mucositis. Exp. Biol. Med. 2009, 234, 430–441. [Google Scholar] [CrossRef]
- Chen, H.; Xu, C.; Zhang, F.; Liu, Y.; Guo, Y.; Yao, Q. The gut microbiota attenuates muscle wasting by regulating energy metabolism in chemotherapy-induced malnutrition rats. Cancer Chemother. Pharm. 2020. [Google Scholar] [CrossRef]
- VanderVeen, B.N.; Sougiannis, A.T.; Velazquez, K.T.; Carson, J.A.; Fan, D.; Murphy, E.A. The Acute Effects of 5 Fluorouracil on Skeletal Muscle Resident and Infiltrating Immune Cells in Mice. Front. Physiol. 2020, 11, 593468. [Google Scholar] [CrossRef]
- Campelj, D.G.; Timpani, C.A.; Cree, T.; Petersen, A.C.; Hayes, A.; Goodman, C.A.; Rybalka, E. Metronomic 5-Fluorouracil Delivery Primes Skeletal Muscle for Myopathy but Does Not Cause Cachexia. Pharmaceuticals 2021, 14, 478. [Google Scholar] [CrossRef] [PubMed]
- Barreto, R.; Kitase, Y.; Matsumoto, T.; Pin, F.; Colston, K.C.; Couch, K.E.; O’Connell, T.M.; Couch, M.E.; Bonewald, L.F.; Bonetto, A. ACVR2B/Fc counteracts chemotherapy-induced loss of muscle and bone mass. Sci. Rep. 2017, 7, 14470. [Google Scholar] [CrossRef]
- Barreto, R.; Mandili, G.; Witzmann, F.A.; Novelli, F.; Zimmers, T.A.; Bonetto, A. Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways. Front. Physiol. 2016, 7, 472. [Google Scholar] [CrossRef] [PubMed]
- Pin, F.; Barreto, R.; Couch, M.E.; Bonetto, A.; O’Connell, T.M. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J. Cachexia Sarcopenia Muscle 2019, 10, 140–154. [Google Scholar] [CrossRef]
- O’Connell, T.M.; Pin, F.; Couch, M.E.; Bonetto, A. Treatment with Soluble Activin Receptor Type IIB Alters Metabolic Response in Chemotherapy-Induced Cachexia. Cancers 2019, 11, 1222. [Google Scholar] [CrossRef]
- Campelj, D.G.; Timpani, C.A.; Petersen, A.C.; Hayes, A.; Goodman, C.A.; Rybalka, E. The Paradoxical Effect of PARP Inhibitor BGP-15 on Irinotecan-Induced Cachexia and Skeletal Muscle Dysfunction. Cancers 2020, 12, 3810. [Google Scholar] [CrossRef]
- Samuels, S.E.; Knowles, A.L.; Tilignac, T.; Debiton, E.; Madelmont, J.C.; Attaix, D. Higher skeletal muscle protein synthesis and lower breakdown after chemotherapy in cachectic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R133–R139. [Google Scholar] [CrossRef] [PubMed]
- Tilignac, T.; Temparis, S.; Combaret, L.; Taillandier, D.; Pouch, M.N.; Cervek, M.; Cardenas, D.M.; Le Bricon, T.; Debiton, E.; Samuels, S.E.; et al. Chemotherapy inhibits skeletal muscle ubiquitin-proteasome-dependent proteolysis. Cancer Res. 2002, 62, 2771–2777. [Google Scholar] [PubMed]
- Crouch, M.L.; Knowels, G.; Stuppard, R.; Ericson, N.G.; Bielas, J.H.; Marcinek, D.J.; Syrjala, K.L. Cyclophosphamide leads to persistent deficits in physical performance and in vivo mitochondria function in a mouse model of chemotherapy late effects. PLoS ONE 2017, 12, e0181086. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.R.; Dodd, S.L. Xanthine oxidase and activated neutrophils cause oxidative damage to skeletal muscle after contractile claudication. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H252–H256. [Google Scholar] [CrossRef]
- Lee, N.-S.; Byun, J.-H.; Bae, S.-B.; Kim, C.-K.; Lee, K.-T.; Park, S.-K.; Won, J.-H.; Hong, D.-S.; Park, H.-S. Combination of gemcitabine and cisplatin as first-line therapy in advanced non-small-cell lung cancer. Cancer Res. Treat. 2004, 36, 173–177. [Google Scholar] [CrossRef][Green Version]
- Heinemann, V. Gemcitabine plus cisplatin for the treatment of metastatic breast cancer. Clin. Breast Cancer 2002, 3 (Suppl. 1), 24–29. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Rothmann, M. Gemcitabine and cisplatin for advanced, metastatic bladder cancer. J. Clin. Oncol. 2001, 19, 1229–1231. [Google Scholar] [CrossRef]
- Chen, M.-C.; Chen, Y.-L.; Lee, C.-F.; Hung, C.-H.; Chou, T.-C. Supplementation of Magnolol Attenuates Skeletal Muscle Atrophy in Bladder Cancer-Bearing Mice Undergoing Chemotherapy via Suppression of FoxO3 Activation and Induction of IGF-1. PLoS ONE 2015, 10, e0143594. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-C.; Hsu, W.-L.; Hwang, P.-A.; Chen, Y.-L.; Chou, T.-C. Combined administration of fucoidan ameliorates tumor and chemotherapy-induced skeletal muscle atrophy in bladder cancer-bearing mice. Oncotarget 2016, 7, 51608–51618. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.C.; Cramer, M.S.; Mitchell, P.J.; Capen, A.; Huber, L.; Wang, R.; Myers, L.; Jones, B.E.; Eastwood, B.J.; Ballard, D.; et al. Myostatin Neutralization Results in Preservation of Muscle Mass and Strength in Preclinical Models of Tumor-Induced Muscle Wasting. Mol. Cancer Ther. 2015, 14, 1661–1670. [Google Scholar] [CrossRef]
- Kremer, J.M.; Alarcón, G.S.; Lightfoot Jr, R.W.; Willkens, R.F.; Furst, D.E.; Williams, H.J.; Dent, P.B.; Weinblatt, M.E. Methotrexate for rheumatoid arthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 1994, 37, 316–328. [Google Scholar] [CrossRef]
- Pirkmajer, S.; Kulkarni, S.S.; Tom, R.Z.; Ross, F.A.; Hawley, S.A.; Hardie, D.G.; Zierath, J.R.; Chibalin, A.V. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes 2015, 64, 360–369. [Google Scholar] [CrossRef]
- Russo, G.T.; Minutoli, L.; Bitto, A.; Altavilla, D.; Alessi, E.; Giandalia, A.; Romeo, E.L.; Stagno, M.F.; Squadrito, F.; Cucinotta, D.; et al. Methotrexate Increases Skeletal Muscle GLUT4 Expression and Improves Metabolic Control in Experimental Diabetes. J. Nutr. Metab. 2012, 2012, 132056. [Google Scholar] [CrossRef] [PubMed]
- Ramos, S.V.; Hughes, M.C.; Perry, C.G.R. Altered skeletal muscle microtubule-mitochondrial VDAC2 binding is related to bioenergetic impairments after paclitaxel but not vinblastine chemotherapies. Am. J. Physiol.-Cell Physiol. 2019, 316, C449–C455. [Google Scholar] [CrossRef]
- Kauffman, F.C.; Albuquerque, E.X.; Warnick, J.E.; Max, S.R. Effect of vinblastine on neural regulation of metabolism in rat skeletal muscle. Exp. Neurol. 1976, 50, 60–66. [Google Scholar] [CrossRef]
- Chen, X.; Green, P.G.; Levine, J.D. Abnormal muscle afferent function in a model of Taxol chemotherapy-induced painful neuropathy. J. Neurophysiol. 2011, 106, 274–279. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Chaillou, T.; McPeek, A.; Lanner, J.T. Docetaxel does not impair skeletal muscle force production in a murine model of cancer chemotherapy. Physiol. Rep. 2017, 5, e13261. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, T.L.; Hsia, S.; Su, I.L.; Chan, Y.L.; Wu, C.J. Skeletal muscle atrophy is attenuated in tumor-bearing mice under chemotherapy by treatment with fish oil and selenium. Oncotarget 2015, 6, 7758–7773. [Google Scholar] [CrossRef]
- Ju, J.S.; Varadhachary, A.S.; Miller, S.E.; Weihl, C.C. Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy 2010, 6, 929–935. [Google Scholar] [CrossRef]
- Kerr, J.P.; Robison, P.; Shi, G.; Bogush, A.I.; Kempema, A.M.; Hexum, J.K.; Becerra, N.; Harki, D.A.; Martin, S.S.; Raiteri, R.; et al. Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat. Commun. 2015, 6, 8526. [Google Scholar] [CrossRef] [PubMed]
- Parry, S.M.; Puthucheary, Z.A. The impact of extended bed rest on the musculoskeletal system in the critical care environment. Extrem. Physiol. Med. 2015, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Aversa, Z.; Costelli, P.; Muscaritoli, M. Cancer-induced muscle wasting: Latest findings in prevention and treatment. Ther. Adv. Med Oncol. 2017, 9, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Aquila, G.; Re Cecconi, A.D.; Brault, J.J.; Corli, O.; Piccirillo, R. Nutraceuticals and Exercise against Muscle Wasting during Cancer Cachexia. Cells 2020, 9, 2536. [Google Scholar] [CrossRef]
- VanderVeen, B.N.; Fix, D.K.; Carson, J.A. Disrupted Skeletal Muscle Mitochondrial Dynamics, Mitophagy, and Biogenesis during Cancer Cachexia: A Role for Inflammation. Oxidative Med. Cell. Longev. 2017, 2017, 3292087. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Costelli, P. The Redox Balance: A Target for Interventions Against Muscle Wasting in Cancer Cachexia? Antioxid. Redox Signal. 2020, 33, 542–558. [Google Scholar] [CrossRef]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Duarte, J.A.; Le Nguyen, B.; Hyatt, H. Endurance exercise protects skeletal muscle against both doxorubicin-induced and inactivity-induced muscle wasting. Pflugers Arch. 2019, 471, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Smuder, A.J. Exercise stimulates beneficial adaptations to diminish doxorubicin-induced cellular toxicity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R662–R672. [Google Scholar] [CrossRef]
- Bredahl, E.C.; Pfannenstiel, K.B.; Quinn, C.J.; Hayward, R.; Hydock, D.S. Effects of Exercise on Doxorubicin-Induced Skeletal Muscle Dysfunction. Med. Sci. Sports Exerc. 2016, 48, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Kavazis, A.N.; Smuder, A.J.; Powers, S.K. Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J. Appl. Physiol. 2014, 117, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.D.; Marchant, E.D.; Munk, D.J.; Watt, R.K.; Hansen, J.M.; Thomson, D.M.; Hancock, C.R. Multitissue analysis of exercise and metformin on doxorubicin-induced iron dysregulation. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E922–E930. [Google Scholar] [CrossRef]
- Morton, A.B.; Mor Huertas, A.; Hinkley, J.M.; Ichinoseki-Sekine, N.; Christou, D.D.; Smuder, A.J. Mitochondrial accumulation of doxorubicin in cardiac and diaphragm muscle following exercise preconditioning. Mitochondrion 2019, 45, 52–62. [Google Scholar] [CrossRef]
- Huertas, A.M.; Morton, A.B.; Hinkey, J.M.; Ichinoseki-Sekine, N.; Smuder, A.J. Modification of Neuromuscular Junction Protein Expression by Exercise and Doxorubicin. Med. Sci. Sports Exerc. 2020, 52, 1477–1484. [Google Scholar] [CrossRef]
- Jones, L.W.; Alfano, C.M. Exercise-oncology research: Past, present, and future. Acta Oncol. 2013, 52, 195–215. [Google Scholar] [CrossRef]
- Dickinson, J.M.; D’Lugos, A.C.; Mahmood, T.N.; Ormsby, J.C.; Salvo, L.; Dedmon, W.L.; Patel, S.H.; Katsma, M.S.; Mookadam, F.; Gonzales, R.J.; et al. Exercise Protects Skeletal Muscle during Chronic Doxorubicin Administration. Med. Sci. Sports Exerc. 2017, 49, 2394–2403. [Google Scholar] [CrossRef]
- de Lima, E.A.; de Sousa, L.G.O.; de S Teixeira, A.A.; Marshall, A.G.; Zanchi, N.E.; Neto, J.C.R. Aerobic exercise, but not metformin, prevents reduction of muscular performance by AMPk activation in mice on doxorubicin chemotherapy. J. Cell Physiol. 2018, 233, 9652–9662. [Google Scholar] [CrossRef] [PubMed]
- Hojman, P.; Fjelbye, J.; Zerahn, B.; Christensen, J.F.; Dethlefsen, C.; Lonkvist, C.K.; Brandt, C.; Gissel, H.; Pedersen, B.K.; Gehl, J. Voluntary exercise prevents cisplatin-induced muscle wasting during chemotherapy in mice. PLoS ONE 2014, 9, e109030. [Google Scholar] [CrossRef] [PubMed]
- Alves de Lima, E., Jr.; Teixeira, A.A.S.; Biondo, L.A.; Diniz, T.A.; Silveira, L.S.; Coletti, D.; Busquets Rius, S.; Rosa Neto, J.C. Exercise Reduces the Resumption of Tumor Growth and Proteolytic Pathways in the Skeletal Muscle of Mice Following Chemotherapy. Cancers 2020, 12, 3466. [Google Scholar] [CrossRef] [PubMed]
- Kemi, O.J.; Loennechen, J.P.; Wisløff, U.; Ellingsen, Ø. Intensity-controlled treadmill running in mice: Cardiac and skeletal muscle hypertrophy. J. Appl. Physiol. 2002, 93, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Fuller, K.N.Z.; Thyfault, J.P. Barriers in translating preclinical rodent exercise metabolism findings to human health. J. Appl. Physiol. 2021, 130, 182–192. [Google Scholar] [CrossRef]
- Montalvo, R.N.; Doerr, V.; Nguyen, B.L.; Kelley, R.C.; Smuder, A.J. Consideration of Sex as a Biological Variable in the Development of Doxorubicin Myotoxicity and the Efficacy of Exercise as a Therapeutic Intervention. Antioxidants 2021, 10, 343. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.J.; Kim, S.K.; Massett, M.P. Differences in Exercise Capacity and Responses to Training in 24 Inbred Mouse Strains. Front. Physiol. 2017, 8, 974. [Google Scholar] [CrossRef]
- Goh, J.; Ladiges, W. Voluntary Wheel Running in Mice. Curr. Protoc. Mouse Biol. 2015, 5, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kawamoto, H.; Kasaoka, K.; Hitomi, Y.; Kizaki, T.; Sankai, Y.; Ohno, H.; Haga, S.; Takemasa, T. Muscle type-specific response of PGC-1α and oxidative enzymes during voluntary wheel running in mouse skeletal muscle. Acta Physiol. 2006, 188, 217–223. [Google Scholar] [CrossRef]
- Murach, K.A.; McCarthy, J.J.; Peterson, C.A.; Dungan, C.M. Making Mice Mighty: Recent advances in translational models of load-induced muscle hypertrophy. J. Appl. Physiol. 2020, 129, 516–521. [Google Scholar] [CrossRef]
- Huang, S.C.; Wu, J.F.; Saovieng, S.; Chien, W.H.; Hsu, M.F.; Li, X.F.; Lee, S.D.; Huang, C.Y.; Huang, C.Y.; Kuo, C.H. Doxorubicin inhibits muscle inflammation after eccentric exercise. J. Cachexia Sarcopenia Muscle 2017, 8, 277–284. [Google Scholar] [CrossRef]
- Yen, H.C.; Oberley, T.D.; Vichitbandha, S.; Ho, Y.S.; St Clair, D.K. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J. Clin. Investig. 1996, 98, 1253–1260. [Google Scholar] [CrossRef]
- Salvatorelli, E.; Menna, P.; Minotti, G. Managing anthracycline-induced cardiotoxicity: Beginning with the end in mind. Future Cardiol. 2015, 11, 363–366. [Google Scholar] [CrossRef] [PubMed]
- de Lima Junior, E.A.; Yamashita, A.S.; Pimentel, G.D.; De Sousa, L.G.; Santos, R.V.; Gonçalves, C.L.; Streck, E.L.; de Lira, F.S.; Rosa Neto, J.C. Doxorubicin caused severe hyperglycaemia and insulin resistance, mediated by inhibition in AMPk signalling in skeletal muscle. J. Cachexia Sarcopenia Muscle 2016, 7, 615–625. [Google Scholar] [CrossRef]
- Hardee, J.P.; Counts, B.R.; Carson, J.A. Understanding the Role of Exercise in Cancer Cachexia Therapy. Am. J. Lifestyle Med. 2017, 13, 46–60. [Google Scholar] [CrossRef]
- Campbell, K.L.; Winters-Stone, K.M.; Wiskemann, J.; May, A.M.; Schwartz, A.L.; Courneya, K.S.; Zucker, D.S.; Matthews, C.E.; Ligibel, J.A.; Gerber, L.H.; et al. Exercise Guidelines for Cancer Survivors: Consensus Statement from International Multidisciplinary Roundtable. Med. Sci. Sports Exerc. 2019, 51, 2375–2390. [Google Scholar] [CrossRef]
- Battaglini, C.; Bottaro, M.; Dennehy, C.; Rae, L.; Shields, E.; Kirk, D.; Hackney, A.C. The effects of an individualized exercise intervention on body composition in breast cancer patients undergoing treatment. Sao Paulo Med J. 2007, 125, 22–28. [Google Scholar] [CrossRef]
- Courneya, K.S.; Segal, R.J.; Mackey, J.R.; Gelmon, K.; Reid, R.D.; Friedenreich, C.M.; Ladha, A.B.; Proulx, C.; Vallance, J.K.; Lane, K.; et al. Effects of aerobic and resistance exercise in breast cancer patients receiving adjuvant chemotherapy: A multicenter randomized controlled trial. J. Clin. Oncol. 2007, 25, 4396–4404. [Google Scholar] [CrossRef]
- Mijwel, S.; Cardinale, D.A.; Norrbom, J.; Chapman, M.; Ivarsson, N.; Wengström, Y.; Sundberg, C.J.; Rundqvist, H. Exercise training during chemotherapy preserves skeletal muscle fiber area, capillarization, and mitochondrial content in patients with breast cancer. FASEB J. 2018, 32, 5495–5505. [Google Scholar] [CrossRef] [PubMed]
- Lønbro, S.; Farup, J.; Bentsen, S.; Voss, T.; Rittig, N.; Wang, J.; Ørskov, M.; Højris, I.; Mikkelsen, U.R. Lean body mass, muscle fibre size and muscle function in cancer patients during chemotherapy and 10 weeks exercise. JCSM Clin. Rep. 2017, 2, 1–15. [Google Scholar] [CrossRef][Green Version]
- Buckinx, F.; Landi, F.; Cesari, M.; Fielding, R.A.; Visser, M.; Engelke, K.; Maggi, S.; Dennison, E.; Al-Daghri, N.M.; Allepaerts, S.; et al. Pitfalls in the measurement of muscle mass: A need for a reference standard. J. Cachexia Sarcopenia Muscle 2018, 9, 269–278. [Google Scholar] [CrossRef]
- Yao, W.; Jee, W.S.; Chen, J.L.; Li, C.Y.; Frost, H.M. A novel method to ‘exercise’ rats: Making rats rise to erect bipedal stance for feeding-raised cage model. J. Musculoskelet Neuronal Interact 2001, 1, 241–247. [Google Scholar]
- Puppa, M.J.; Gao, S.; Narsale, A.A.; Carson, J.A. Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J. 2014, 28, 998–1009. [Google Scholar] [CrossRef]
- Aulino, P.; Berardi, E.; Cardillo, V.M.; Rizzuto, E.; Perniconi, B.; Ramina, C.; Padula, F.; Spugnini, E.P.; Baldi, A.; Faiola, F.; et al. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer 2010, 10, 363. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Summermatter, S.; Jourdain, M.; Melly, S.; Minetti, G.C.; Lach-Trifilieff, E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti-cancer treatments. Skelet. Muscle 2016, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.M.; Scherer, T.; Chen, J.A.; Guillory, B.; Nassif, A.; Papusha, V.; Smiechowska, J.; Asnicar, M.; Buettner, C.; Smith, R.G. Inhibition of cisplatin-induced lipid catabolism and weight loss by ghrelin in male mice. Endocrinology 2013, 154, 3118–3129. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.P.; Pei, X.M.; Sin, T.K.; Yip, S.P.; Yung, B.Y.; Chan, L.W.; Wong, C.S.; Siu, P.M. Acylated and unacylated ghrelin inhibit doxorubicin-induced apoptosis in skeletal muscle. Acta Physiol. 2014, 211, 201–213. [Google Scholar] [CrossRef]
- Bresciani, E.; Rizzi, L.; Molteni, L.; Ravelli, M.; Liantonio, A.; Ben Haj Salah, K.; Fehrentz, J.A.; Martinez, J.; Omeljaniuk, R.J.; Biagini, G.; et al. JMV2894, a novel growth hormone secretagogue, accelerates body mass recovery in an experimental model of cachexia. Endocrine 2017, 58, 106–114. [Google Scholar] [CrossRef]
- Brierley, D.I.; Harman, J.R.; Giallourou, N.; Leishman, E.; Roashan, A.E.; Mellows, B.A.D.; Bradshaw, H.B.; Swann, J.R.; Patel, K.; Whalley, B.J.; et al. Chemotherapy-induced cachexia dysregulates hypothalamic and systemic lipoamines and is attenuated by cannabigerol. J. Cachexia Sarcopenia Muscle 2019, 10, 844–859. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Hori, S.; Itami, Y.; Oda, Y.; Owari, T.; Fujii, T.; Ohnishi, S.; Morizawa, Y.; Gotoh, D.; Nakai, Y.; et al. Supplementary Oral Anamorelin Mitigates Anorexia and Skeletal Muscle Atrophy Induced by Gemcitabine Plus Cisplatin Systemic Chemotherapy in a Mouse Model. Cancers 2020, 12, 1942. [Google Scholar] [CrossRef]
- Wu, Y.-Q.; Dang, R.-L.; Tang, M.-M.; Cai, H.-L.; Li, H.-D.; Liao, D.-H.; He, X.; Cao, L.-J.; Xue, Y.; Jiang, P. Long Chain Omega-3 Polyunsaturated Fatty Acid Supplementation Alleviates Doxorubicin-Induced Depressive-Like Behaviors and Neurotoxicity in Rats: Involvement of Oxidative Stress and Neuroinflammation. Nutrients 2016, 8, 243. [Google Scholar] [CrossRef]
- Almasud, A.A.; Giles, K.H.; Miklavcic, J.J.; Martins, K.J.B.; Baracos, V.E.; Putman, C.T.; Guan, L.L.; Mazurak, V.C. Fish oil mitigates myosteatosis and improves chemotherapy efficacy in a preclinical model of colon cancer. PLoS ONE 2017, 12, e0183576. [Google Scholar] [CrossRef]
- Wu, C.T.; Liao, J.M.; Ko, J.L.; Lee, Y.L.; Chang, H.Y.; Wu, C.H.; Ou, C.C. D-Methionine Ameliorates Cisplatin-Induced Muscle Atrophy via Inhibition of Muscle Degradation Pathway. Integr. Cancer Ther. 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Torok, Z.A.; Busekrus, R.B.; Hydock, D.S. Effects of Creatine Supplementation on Muscle Fatigue in Rats Receiving Doxorubicin Treatment. Nutr. Cancer 2020, 72, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, A.; Rovetta, F.; Ferroni, M.; Corsetti, G.; Lavazza, A.; Sberveglieri, G.; Aleo, M.F. Taurine rescues cisplatin-induced muscle atrophy in vitro: A morphological study. Oxid. Med. Cell Longev. 2014, 2014, 840951. [Google Scholar] [CrossRef] [PubMed]
- Bredahl, E.C.; Busekrus, R.B.; Hydock, D.S. The combined effect of creatine and resistance training on doxorubicin-induced muscle dysfunction. Nutr. Cancer 2020, 72, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Lobina, C.; Carai, M.A.; Loi, B.; Gessa, G.L.; Riva, A.; Cabri, W.; Petrangolini, G.; Morazzoni, P.; Colombo, G. Protective effect of Panax ginseng in cisplatin-induced cachexia in rats. Future Oncol. 2014, 10, 1203–1214. [Google Scholar] [CrossRef]
- Wang, H.; Chan, Y.-L.; Li, T.-L.; Wu, C.-J. Improving cachectic symptoms and immune strength of tumour-bearing mice in chemotherapy by a combination of Scutellaria baicalensis and Qing-Shu-Yi-Qi-Tang. Eur. J. Cancer 2012, 48, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Hsu, W.L.; Chou, T.C. Anti-cachectic effect of Antrodia cinnamomea extract in lung tumor-bearing mice under chemotherapy. Oncotarget 2018, 9, 19584–19596. [Google Scholar] [CrossRef]
- Chen, J.M.; Yang, T.T.; Cheng, T.S.; Hsiao, T.F.; Chang, P.M.; Leu, J.Y.; Wang, F.S.; Hsu, S.L.; Huang, C.F.; Lai, J.M. Modified Sijunzi decoction can alleviate cisplatin-induced toxicity and prolong the survival time of cachectic mice by recovering muscle atrophy. J. Ethnopharmacol. 2019, 233, 47–55. [Google Scholar] [CrossRef]
- Huang, T.H.; Wu, T.H.; Guo, Y.H.; Li, T.L.; Chan, Y.L.; Wu, C.J. The concurrent treatment of Scutellaria baicalensis Georgi enhances the therapeutic efficacy of cisplatin but also attenuates chemotherapy-induced cachexia and acute kidney injury. J. Ethnopharmacol. 2019, 243, 112075. [Google Scholar] [CrossRef]
- Lee, C.; Jeong, H.; Lee, H.; Hong, M.; Park, S.Y.; Bae, H. Magnolol Attenuates Cisplatin-Induced Muscle Wasting by M2c Macrophage Activation. Front. Immunol. 2020, 11, 77. [Google Scholar] [CrossRef]
- Zhang, H.; Chi, M.; Chen, L.; Sun, X.; Wan, L.; Yang, Q.; Guo, C. Linalool Prevents Cisplatin Induced Muscle Atrophy by Regulating IGF-1/Akt/FoxO Pathway. Front. Pharmacol. 2020, 11, 598166. [Google Scholar] [CrossRef]
- Hong, M.; Han, I.H.; Choi, I.; Cha, N.; Kim, W.; Kim, S.K.; Bae, H. Magnoliae Cortex Alleviates Muscle Wasting by Modulating M2 Macrophages in a Cisplatin-Induced Sarcopenia Mouse Model. Int. J. Mol. Sci. 2021, 22, 3188. [Google Scholar] [CrossRef] [PubMed]
- Abulizi, A.; Hu, L.; Ma, A.; Shao, F.-y.; Zhu, H.-z.; Lin, S.-m.; Shao, G.-y.; Xu, Y.; Ran, J.-h.; Li, J.; et al. Ganoderic acid alleviates chemotherapy-induced fatigue in mice bearing colon tumor. Acta Pharmacol. Sin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hulmi, J.J.; Nissinen, T.A.; Penna, F.; Bonetto, A. Targeting the Activin Receptor Signaling to Counteract the Multi-Systemic Complications of Cancer and Its Treatments. Cells 2021, 10, 516. [Google Scholar] [CrossRef] [PubMed]
- Marceca, G.P.; Londhe, P.; Calore, F. Management of Cancer Cachexia: Attempting to Develop New Pharmacological Agents for New Effective Therapeutic Options. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Temel, J.S.; Abernethy, A.P.; Currow, D.C.; Friend, J.; Duus, E.M.; Yan, Y.; Fearon, K.C. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): Results from two randomised, double-blind, phase 3 trials. Lancet. Oncol. 2016, 17, 519–531. [Google Scholar] [CrossRef]
- van de Worp, W.R.P.H.; Schols, A.M.W.J.; Theys, J.; van Helvoort, A.; Langen, R.C.J. Nutritional Interventions in Cancer Cachexia: Evidence and Perspectives From Experimental Models. Front. Nutr. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Prado, C.M.; Purcell, S.A.; Laviano, A. Nutrition interventions to treat low muscle mass in cancer. J. Cachexia Sarcopenia Muscle 2020, 11, 366–380. [Google Scholar] [CrossRef]
- Prado, C.M.; Anker, S.D.; Coats, A.J.S.; Laviano, A.; von Haehling, S. Nutrition in the spotlight in cachexia, sarcopenia and muscle: Avoiding the wildfire. J. Cachexia Sarcopenia Muscle 2021, 12, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kuchta, K.; Cameron, S. Phytotherapy for Cachexia: Where Do We Stand? Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.S.; Adams, V.; Werner, S.; Fischer, T.; Vinke, P.; Brogger, M.N.; Mangner, N.; Linke, A.; Sehr, P.; Lewis, J.; et al. Small-molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [PubMed]
- Horvath, O.; Ordog, K.; Bruszt, K.; Kalman, N.; Kovacs, D.; Radnai, B.; Gallyas, F.; Toth, K.; Halmosi, R.; Deres, L. Modulation of Mitochondrial Quality Control Processes by BGP-15 in Oxidative Stress Scenarios: From Cell Culture to Heart Failure. Oxidative Med. Cell. Longev. 2021, 2021, 6643871. [Google Scholar] [CrossRef] [PubMed]
- Pető, Á.; Kósa, D.; Fehér, P.; Ujhelyi, Z.; Sinka, D.; Vecsernyés, M.; Szilvássy, Z.; Juhász, B.; Csanádi, Z.; Vígh, L.; et al. Pharmacological Overview of the BGP-15 Chemical Agent as a New Drug Candidate for the Treatment of Symptoms of Metabolic Syndrome. Molecules 2020, 25, 429. [Google Scholar] [CrossRef]
- Literati-Nagy, B.; Peterfai, E.; Kulcsar, E.; Literati-Nagy, Z.; Buday, B.; Tory, K.; Mandl, J.; Sumegi, B.; Fleming, A.; Roth, J.; et al. Beneficial effect of the insulin sensitizer (HSP inducer) BGP-15 on olanzapine-induced metabolic disorders. Brain Res. Bull. 2010, 83, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Racz, I.; Tory, K.; Gallyas, F., Jr.; Berente, Z.; Osz, E.; Jaszlits, L.; Bernath, S.; Sumegi, B.; Rabloczky, G.; Literati-Nagy, P. BGP-15-a novel poly(ADP-ribose) polymerase inhibitor-protects against nephrotoxicity of cisplatin without compromising its antitumor activity. Biochem. Pharmacol. 2002, 63, 1099–1111. [Google Scholar] [CrossRef]
- Henstridge, D.C.; Bruce, C.R.; Drew, B.G.; Tory, K.; Kolonics, A.; Estevez, E.; Chung, J.; Watson, N.; Gardner, T.; Lee-Young, R.S.; et al. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes 2014, 63, 1881–1894. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Sapra, G.; Tham, Y.K.; Cemerlang, N.; Matsumoto, A.; Kiriazis, H.; Bernardo, B.C.; Henstridge, D.C.; Ooi, J.Y.; Pretorius, L.; Boey, E.J.; et al. The small-molecule BGP-15 protects against heart failure and atrial fibrillation in mice. Nat. Commun. 2014, 5, 5705. [Google Scholar] [CrossRef]
- Salah, H.; Li, M.; Cacciani, N.; Gastaldello, S.; Ogilvie, H.; Akkad, H.; Namuduri, A.V.; Morbidoni, V.; Artemenko, K.A.; Balogh, G.; et al. The chaperone co-inducer BGP-15 alleviates ventilation-induced diaphragm dysfunction. Sci. Transl. Med. 2016, 8, 350ra103. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, S.M.; van der Poel, C.; Sayer, T.A.; Schertzer, J.D.; Henstridge, D.C.; Church, J.E.; Lamon, S.; Russell, A.P.; Davies, K.E.; Febbraio, M.A.; et al. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 2012, 484, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Literati-Nagy, B.; Kulcsar, E.; Literati-Nagy, Z.; Buday, B.; Peterfai, E.; Horvath, T.; Tory, K.; Kolonics, A.; Fleming, A.; Mandl, J.; et al. Improvement of insulin sensitivity by a novel drug, BGP-15, in insulin-resistant patients: A proof of concept randomized double-blind clinical trial. Horm. Metab. Res. 2009, 41, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Nguyen, A.K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; et al. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 1739–1744. [Google Scholar] [CrossRef]
- Kourakis, S.; Timpani, C.A.; de Haan, J.B.; Gueven, N.; Fischer, D.; Rybalka, E. Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility? Pharmaceuticals 2020, 13, 306. [Google Scholar] [CrossRef]
- Bhakkiyalakshmi, E.; Dineshkumar, K.; Karthik, S.; Sireesh, D.; Hopper, W.; Paulmurugan, R.; Ramkumar, K.M. Pterostilbene-mediated Nrf2 activation: Mechanistic insights on Keap1:Nrf2 interface. Bioorganic Med. Chem. 2016, 24, 3378–3386. [Google Scholar] [CrossRef]
- Sin, T.K.; Tam, B.T.; Yu, A.P.; Yip, S.P.; Yung, B.Y.; Chan, L.W.; Wong, C.S.; Rudd, J.A.; Siu, P.M. Acute Treatment of Resveratrol Alleviates Doxorubicin-Induced Myotoxicity in Aged Skeletal Muscle Through SIRT1-Dependent Mechanisms. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2016, 71, 730–739. [Google Scholar] [CrossRef]
- Dolinsky, V.W.; Rogan, K.J.; Sung, M.M.; Zordoky, B.N.; Haykowsky, M.J.; Young, M.E.; Jones, L.W.; Dyck, J.R.B. Both aerobic exercise and resveratrol supplementation attenuate doxorubicin-induced cardiac injury in mice. Am. J. Physiol.-Endocrinol. Metab. 2013, 305, E243–E253. [Google Scholar] [CrossRef]
- Cheng, Y.; Di, S.; Fan, C.; Cai, L.; Gao, C.; Jiang, P.; Hu, W.; Ma, Z.; Jiang, S.; Dong, Y.; et al. SIRT1 activation by pterostilbene attenuates the skeletal muscle oxidative stress injury and mitochondrial dysfunction induced by ischemia reperfusion injury. Apoptosis 2016, 21, 905–916. [Google Scholar] [CrossRef]
- Zhang, L.; Cui, L.; Zhou, G.; Jing, H.; Guo, Y.; Sun, W. Pterostilbene, a natural small-molecular compound, promotes cytoprotective macroautophagy in vascular endothelial cells. J. Nutr. Biochem. 2013, 24, 903–911. [Google Scholar] [CrossRef]
- Riche, D.M.; McEwen, C.L.; Riche, K.D.; Sherman, J.J.; Wofford, M.R.; Deschamp, D.; Griswold, M. Analysis of safety from a human clinical trial with pterostilbene. J. Toxicol. 2013, 2013, 463595. [Google Scholar] [CrossRef]
- Lan, X.; Han, X.; Li, Q.; Wang, J. (-)-Epicatechin, a Natural Flavonoid Compound, Protects Astrocytes Against Hemoglobin Toxicity via Nrf2 and AP-1 Signaling Pathways. Mol. Neurobiol. 2017, 54, 7898–7907. [Google Scholar] [CrossRef]
- Taub, P.R.; Ramirez-Sanchez, I.; Ciaraldi, T.P.; Gonzalez-Basurto, S.; Coral-Vazquez, R.; Perkins, G.; Hogan, M.; Maisel, A.S.; Henry, R.R.; Ceballos, G.; et al. Perturbations in skeletal muscle sarcomere structure in patients with heart failure and type 2 diabetes: Restorative effects of (-)-epicatechin-rich cocoa. Clin. Sci. 2013, 125, 383–389. [Google Scholar] [CrossRef]
- McDonald, C.M.; Ramirez-Sanchez, I.; Oskarsson, B.; Joyce, N.; Aguilar, C.; Nicorici, A.; Dayan, J.; Goude, E.; Abresch, R.T.; Villarreal, F.; et al. (-)-Epicatechin induces mitochondrial biogenesis and markers of muscle regeneration in adults with Becker muscular dystrophy. Muscle Nerve 2021, 63, 239–249. [Google Scholar] [CrossRef]
- Acharyya, S.; Butchbach, M.E.; Sahenk, Z.; Wang, H.; Saji, M.; Carathers, M.; Ringel, M.D.; Skipworth, R.J.; Fearon, K.C.; Hollingsworth, M.A.; et al. Dystrophin glycoprotein complex dysfunction: A regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005, 8, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Mena, S.; Rodriguez, M.L.; Ponsoda, X.; Estrela, J.M.; Jaattela, M.; Ortega, A.L. Pterostilbene-induced tumor cytotoxicity: A lysosomal membrane permeabilization-dependent mechanism. PLoS ONE 2012, 7, e44524. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, T.L.; Rupasinghe, H.P.V.; Dellaire, G.; Xu, Z. Regulation of Nrf2/ARE Pathway by Dietary Flavonoids: A Friend or Foe for Cancer Management? Antioxidants 2020, 9, 973. [Google Scholar] [CrossRef]
- Jansson, E.A.; Huang, L.; Malkey, R.; Govoni, M.; Nihlén, C.; Olsson, A.; Stensdotter, M.; Petersson, J.; Holm, L.; Weitzberg, E.; et al. A mammalian functional nitrate reductase that regulates nitrite and nitric oxide homeostasis. Nat. Chem. Biol. 2008, 4, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Carlström, M.; Weitzberg, E. Metabolic Effects of Dietary Nitrate in Health and Disease. Cell Metab. 2018, 28, 9–22. [Google Scholar] [CrossRef]
- Xi, L.; Zhu, S.G.; Hobbs, D.C.; Kukreja, R.C. Identification of protein targets underlying dietary nitrate-induced protection against doxorubicin cardiotoxicity. J. Cell Mol. Med. 2011, 15, 2512–2524. [Google Scholar] [CrossRef]
- Zhu, S.G.; Kukreja, R.C.; Das, A.; Chen, Q.; Lesnefsky, E.J.; Xi, L. Dietary nitrate supplementation protects against Doxorubicin-induced cardiomyopathy by improving mitochondrial function. J. Am. Coll Cardiol. 2011, 57, 2181–2189. [Google Scholar] [CrossRef]
- Campelj, D.G.; Debruin, D.A.; Timpani, C.A.; Hayes, A.; Goodman, C.A.; Rybalka, E. Sodium nitrate co-supplementation does not exacerbate low dose metronomic doxorubicin-induced cachexia in healthy mice. Sci. Rep. 2020, 10, 15044. [Google Scholar] [CrossRef]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef]
- Huot, J.R.; Essex, A.L.; Gutierrez, M.; Barreto, R.; Wang, M.; Waning, D.L.; Plotkin, L.I.; Bonetto, A. Chronic Treatment with Multi-Kinase Inhibitors Causes Differential Toxicities on Skeletal and Cardiac Muscles. Cancers 2019, 11, 571. [Google Scholar] [CrossRef]
- Toledo, M.; Penna, F.; Oliva, F.; Luque, M.; Betancourt, A.; Marmonti, E.; López-Soriano, F.J.; Argilés, J.M.; Busquets, S. A multifactorial anti-cachectic approach for cancer cachexia in a rat model undergoing chemotherapy. J. Cachexia Sarcopenia Muscle 2016, 7, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Cassileth, P.A.; Lusk, E.J.; Torri, S.; DiNubile, N.; Gerson, S.L. Antiemetic efficacy of dexamethasone therapy in patients receiving cancer chemotherapy. Arch. Intern. Med. 1983, 143, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Gouspillou, G.; Scheede-Bergdahl, C.; Spendiff, S.; Vuda, M.; Meehan, B.; Mlynarski, H.; Archer-Lahlou, E.; Sgarioto, N.; Purves-Smith, F.M.; Konokhova, Y.; et al. Anthracycline-containing chemotherapy causes long-term impairment of mitochondrial respiration and increased reactive oxygen species release in skeletal muscle. Sci. Rep. 2015, 5, 8717. [Google Scholar] [CrossRef]
- Sakai, H.; Kimura, M.; Tsukimura, Y.; Yabe, S.; Isa, Y.; Kai, Y.; Sato, F.; Kon, R.; Ikarashi, N.; Narita, M.; et al. Dexamethasone exacerbates cisplatin-induced muscle atrophy. Clin. Exp. Pharmacol. Physiol. 2019, 46, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Betancourt, A.; Busquets, S.; Ponce, M.; Toledo, M.; Guàrdia-Olmos, J.; Peró-Cebollero, M.; López-Soriano, F.J.; Argilés, J.M. The animal cachexia score (ACASCO). Anim. Model. Exp. Med. 2019, 2, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Models Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).