
Signed in Blood: Circulating Tumor DNA in Cancer Diagnosis, Treatment and Screening



Abstract
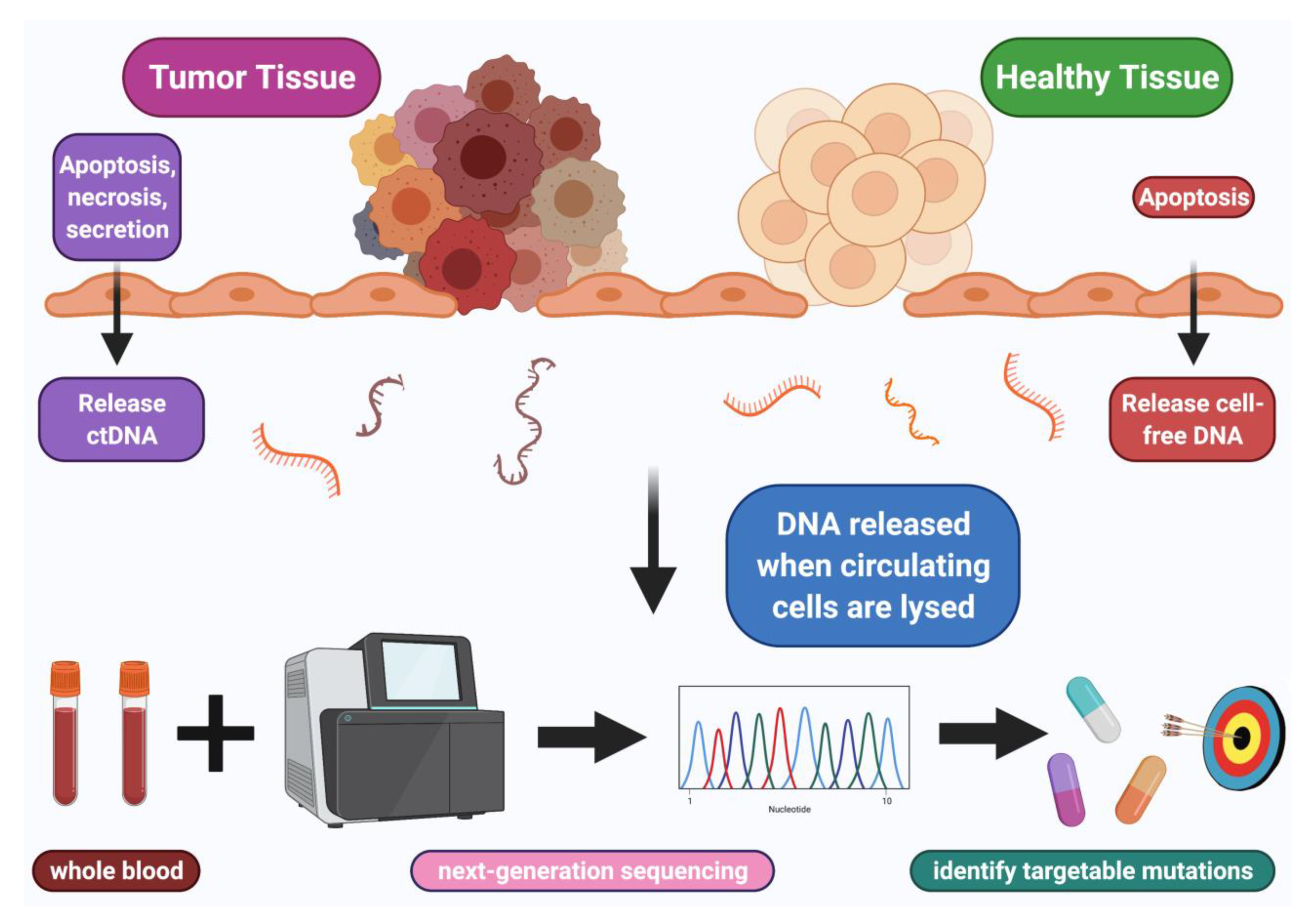
:Simple Summary
Abstract
1. Introduction
- ○
- early diagnosis of cancer;
- ○
- ctDNA as a prognostic variable;
- ○
- measurement of residual disease;
- ○
- discerning molecular alterations that can inform therapeutic decision-making; and
- ○
- monitoring response, resistance, and burden/aggressiveness of disease.
2. Comparison of CTCs, ctDNA, and Tissue DNA
3. Liquid Biopsy and Dynamics of Normal Versus Tumor Cell-Free DNA (cfDNA)
4. How ctDNA Enters and Leaves the Circulation
5. Technological Methods for cfDNA Extraction and Sequencing
6. Clinical Laboratory Improvement Amendments (CLIA) Grade Commercially Available ctDNA Assays
7. Food and Drug Administration (FDA) Approvals for ctDNA Tests
8. Clinical Uses of ctDNA
8.1. ctDNA for Early Diagnosis of Cancer
8.2. ctDNA as a Prognostic Variable
8.3. ctDNA to Measure Residual Disease
8.4. Discerning ctDNA Molecular Alterations That Can Inform Decision Making
9. The Issue of Concordance between ctDNA and Tissue DNA
10. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Husain, H.; Velculescu, V.E. Cancer DNA in the Circulation: The Liquid Biopsy. JAMA 2017, 318, 1272–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): A multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef]
- Khagi, Y.; Goodman, A.M.; Daniels, G.A.; Patel, S.P.; Sacco, A.G.; Randall, J.M.; Bazhenova, L.A.; Kurzrock, R. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor-Based Immunotherapy. Clin. Cancer Res. 2017, 23, 5729–5736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, J.; Lefterova, M.I.; Artyomenko, A.; Kasi, P.M.; Nakamura, Y.; Mody, K.; Catenacci, D.V.T.; Fakih, M.; Barbacioru, C.; Zhao, J.; et al. Validation of Microsatellite Instability Detection Using a Comprehensive Plasma-Based Genotyping Panel. Clin. Cancer Res. 2019, 25, 7035–7045. [Google Scholar] [CrossRef] [PubMed]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Reece, M.; Saluja, H.; Hollington, P.; Karapetis, C.S.; Vatandoust, S.; Young, G.P.; Symonds, E.L. The Use of Circulating Tumor DNA to Monitor and Predict Response to Treatment in Colorectal Cancer. Front. Genet. 2019, 10, 1118. [Google Scholar] [CrossRef] [Green Version]
- Osumi, H.; Shinozaki, E.; Yamaguchi, K.; Zembutsu, H. Early change in circulating tumor DNA as a potential predictor of response to chemotherapy in patients with metastatic colorectal cancer. Sci. Rep. 2019, 9, 17358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.S.; Eun, J.W.; Choi, J.H.; Woo, H.G.; Cho, H.J.; Ahn, H.R.; Suh, C.W.; Baek, G.O.; Cho, S.W.; Cheong, J.Y. MLH1 single-nucleotide variant in circulating tumor DNA predicts overall survival of patients with hepatocellular carcinoma. Sci. Rep. 2020, 10, 17862. [Google Scholar] [CrossRef]
- Sharbatoghli, M.; Vafaei, S.; Aboulkheyr Es, H.; Asadi-Lari, M.; Totonchi, M.; Madjd, Z. Prediction of the treatment response in ovarian cancer: A ctDNA approach. J. Ovarian Res. 2020, 13, 124. [Google Scholar] [CrossRef]
- Husain, H.; Melnikova, V.O.; Kosco, K.; Woodward, B.; More, S.; Pingle, S.C.; Weihe, E.; Park, B.H.; Tewari, M.; Erlander, M.G.; et al. Monitoring Daily Dynamics of Early Tumor Response to Targeted Therapy by Detecting Circulating Tumor DNA in Urine. Clin. Cancer Res. 2017, 23, 4716–4723. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.; Mei, C.; Nan, X.; Hui, L. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine and saliva: A qualitative study. Gene 2016, 590, 142–148. [Google Scholar] [CrossRef]
- Thierry, A.R.; Mouliere, F.; Gongora, C.; Ollier, J.; Robert, B.; Ychou, M.; Del Rio, M.; Molina, F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010, 38, 6159–6175. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef] [Green Version]
- Vu, P.; Khagi, Y.; Riviere, P.; Goodman, A.; Kurzrock, R. Total Number of Alterations in Liquid Biopsies Is an Independent Predictor of Survival in Patients with Advanced Cancers. JCO Precis. Oncol. 2020, 4. [Google Scholar] [CrossRef]
- Ossandon, M.R.; Agrawal, L.; Bernhard, E.J.; Conley, B.A.; Dey, S.M.; Divi, R.L.; Guan, P.; Lively, T.G.; McKee, T.C.; Sorg, B.S.; et al. Circulating Tumor DNA Assays in Clinical Cancer Research. J. Natl. Cancer Inst. 2018, 110, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Saw, R.P.; Thompson, J.F.; Lo, S.; Spillane, A.J.; Shannon, K.F.; Stretch, J.R.; Howle, J.; Menzies, A.M.; Carlino, M.S.; et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann. Oncol. 2019, 30, 815–822. [Google Scholar] [CrossRef]
- Okamura, R.; Piccioni, D.E.; Boichard, A.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kato, S.; Kurzrock, R. High Prevalence of Clonal Hematopoiesis-type Genomic Abnormalities in Cell-free DNA in Invasive Gliomas After Treatment. Int. J. Cancer 2021, 148, 2839–2847. [Google Scholar] [CrossRef]
- Qin, Z.; Ljubimov, V.A.; Zhou, C.; Tong, Y.; Liang, J. Cell-free circulating tumor DNA in cancer. Chin. J. Cancer 2016, 35, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.R.; Zhou, L.; El-Deiry, W.S. Circulating Tumor Cells Versus Circulating Tumor DNA in Colorectal Cancer: Pros and Cons. Curr. Colorectal. Cancer Rep. 2016, 12, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Sharon, E.; Shi, H.; Kharbanda, S.; Koh, W.; Martin, L.R.; Khush, K.K.; Valantine, H.; Pritchard, J.K.; De Vlaminck, I. Quantification of transplant-derived circulating cell-free DNA in absence of a donor genotype. PLoS Comput. Biol. 2017, 13, e1005629. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [Green Version]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.Q.; Raleigh, J.M.; Callahan, J.; Vergara, I.A.; Ftouni, S.; Hatzimihalis, A.; Colebatch, A.J.; Li, J.; Semple, T.; Doig, K.; et al. Circulating Tumor DNA Analysis and Functional Imaging Provide Complementary Approaches for Comprehensive Disease Monitoring in Metastatic Melanoma. JCO Precis. Oncol. 2017, 1, 1–14. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.J.; Pogrebniak, K.; Rueda, O.M.; Provenzano, E.; Grant, J.; Chin, S.F.; Tsui, D.W.Y.; Marass, F.; Gale, D.; et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015, 6, 8760. [Google Scholar] [CrossRef] [Green Version]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Nakahira, K.; Guo, X.; Choi, A.M.; Gu, Z. Very Short Mitochondrial DNA Fragments and Heteroplasmy in Human Plasma. Sci. Rep. 2016, 6, 36097. [Google Scholar] [CrossRef] [Green Version]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagner, T.; Spinelli, C.; Minciacchi, V.R.; Balaj, L.; Zandian, M.; Conley, A.; Zijlstra, A.; Freeman, M.R.; Demichelis, F.; De, S.; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell. Vesicles 2018, 7, 1505403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhanna, N.; Di Grappa, M.A.; Chan, H.H.L.; Khan, T.; Jin, C.S.; Zheng, Y.; Irish, J.C.; Bratman, S.V. Cell-Free DNA Kinetics in a Pre-Clinical Model of Head and Neck Cancer. Sci. Rep. 2017, 7, 16723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, F.; Kulasingam, V.; Diamandis, E.P.; Hoon, D.S.; Kinzler, K.; Pantel, K.; Alix-Panabieres, C. Circulating Tumor DNA as a Cancer Biomarker: Fact or Fiction? Clin. Chem. 2016, 62, 1054–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephan, F.; Marsman, G.; Bakker, L.M.; Bulder, I.; Stavenuiter, F.; Aarden, L.A.; Zeerleder, S. Cooperation of factor VII-activating protease and serum DNase I in the release of nucleosomes from necrotic cells. Arthritis Rheumatol. 2014, 66, 686–693. [Google Scholar] [CrossRef]
- Martin, M.; Leffler, J.; Smolag, K.I.; Mytych, J.; Bjork, A.; Chaves, L.D.; Alexander, J.J.; Quigg, R.J.; Blom, A.M. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ. 2016, 23, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Du Clos, T.W.; Volzer, M.A.; Hahn, F.F.; Xiao, R.; Mold, C.; Searles, R.P. Chromatin clearance in C57Bl/10 mice: Interaction with heparan sulphate proteoglycans and receptors on Kupffer cells. Clin. Exp. Immunol. 1999, 117, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Zhang, B.O.; Xu, C.W.; Shao, Y.; Wang, H.T.; Wu, Y.F.; Song, Y.Y.; Li, X.B.; Zhang, Z.; Wang, W.J.; Li, L.Q.; et al. Comparison of droplet digital PCR and conventional quantitative PCR for measuring EGFR gene mutation. Exp. Ther. Med. 2015, 9, 1383–1388. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Jing, C.; Wu, J.; Ni, J.; Sha, H.; Xu, X.; Du, Y.; Lou, R.; Dong, S.; Feng, J. Circulating tumor DNA detection: A potential tool for colorectal cancer management. Oncol. Lett. 2019, 17, 1409–1416. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Foncillas, J.; Alba, E.; Aranda, E.; Diaz-Rubio, E.; Lopez-Lopez, R.; Tabernero, J.; Vivancos, A. Incorporating BEAMing technology as a liquid biopsy into clinical practice for the management of colorectal cancer patients: An expert taskforce review. Ann. Oncol. 2017, 28, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra168. [Google Scholar] [CrossRef] [PubMed]
- Giroux Leprieur, E.; Helias-Rodzewicz, Z.; Takam Kamga, P.; Costantini, A.; Julie, C.; Corjon, A.; Dumenil, C.; Dumoulin, J.; Giraud, V.; Labrune, S.; et al. Sequential ctDNA whole-exome sequencing in advanced lung adenocarcinoma with initial durable tumor response on immune checkpoint inhibitor and late progression. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Imperial, R.; Nazer, M.; Ahmed, Z.; Kam, A.E.; Pluard, T.J.; Bahaj, W.; Levy, M.; Kuzel, T.M.; Hayden, D.M.; Pappas, S.G.; et al. Matched Whole-Genome Sequencing (WGS) and Whole-Exome Sequencing (WES) of Tumor Tissue with Circulating Tumor DNA (ctDNA) Analysis: Complementary Modalities in Clinical Practice. Cancers 2019, 11, 1399. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Foncillas, J.; Tabernero, J.; Elez, E.; Aranda, E.; Benavides, M.; Camps, C.; Jantus-Lewintre, E.; Lopez, R.; Muinelo-Romay, L.; Montagut, C.; et al. Prospective multicenter real-world RAS mutation comparison between OncoBEAM-based liquid biopsy and tissue analysis in metastatic colorectal cancer. Br. J. Cancer 2018, 119, 1464–1470. [Google Scholar] [CrossRef]
- LB057—Whole Exome Sequencing of Tumor Tissue and Circulating Tumor DNA Ingastrointestinal Stromal Tumors (GIST). Available online: https://www.abstractsonline.com/pp8/#!/9325/presentation/4552 (accessed on 10 January 2021).
- U.S. Food & Drug. FDA Approves Liquid Biopsy NGS Companion Diagnostic Test for Multiple Cancers and Biomarkers. Available online: https://www.fda.gov/drugs/fda-approves-liquid-biopsy-ngs-companion-diagnostic-test-multiple-cancers-and-biomarkers (accessed on 10 January 2021).
- U.S. Food & Drug. Oncology (Cancer)/Hematologic Malignancies Approval Notifications. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/hematologyoncology-cancer-approvals-safety-notifications (accessed on 10 January 2021).
- U.S. Food & Drug. Guardant360 CDx—P200010. Available online: https://www.fda.gov/medical-devices/recently-approved-devices/guardant360-cdx-p200010 (accessed on 10 January 2021).
- U.S. Food & Drug. The Therascreen PIK3CA RGQ PCR Kit-P190001 and P190004. Available online: https://www.fda.gov/medical-devices/recently-approved-devices/therascreen-pik3ca-rgq-pcr-kit-p190001-and-p190004 (accessed on 10 January 2021).
- U.S. Food & Drug. Cobas EGFR Mutation Test v2. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/cobas-egfr-mutation-test-v2 (accessed on 10 January 2021).
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.H.; Wei, W.; Krawczyk, M.; Wang, W.; Luo, H.; Flagg, K.; Yi, S.; Shi, W.; Quan, Q.; Li, K.; et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater. 2017, 16, 1155–1161. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Consortium, C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Cavallone, L.; Aguilar, A.; Aldamry, M.; Lafleur, J.; Brousse, S.; Lan, C.; Alirezaie, N.; Bareke, E.; Majewski, J.; Pelmus, M.; et al. Circulating tumor DNA (ctDNA) during and after neoadjuvant chemotherapy and prior to surgery is a powerful prognostic factor in triple-negative breast cancer (TNBC). J. Clin. Oncol. 2019, 37, 594. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.; Wang, Y.; Li, L.; Kinde, I.; Elsaleh, H.; Wong, R.; Kosmider, S.; Yip, D.; Lee, M.; et al. The potential of circulating tumor DNA (ctDNA) to guide adjuvant chemotherapy decision making in locally advanced rectal cancer (LARC). J. Clin. Oncol. 2017, 35, 3521. [Google Scholar] [CrossRef]
- Cabel, L.; Jeannot, E.; Bieche, I.; Vacher, S.; Callens, C.; Bazire, L.; Morel, A.; Bernard-Tessier, A.; Chemlali, W.; Schnitzler, A.; et al. Prognostic Impact of Residual HPV ctDNA Detection after Chemoradiotherapy for Anal Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 5767–5771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, I.S.; Kato, S.; Fanta, P.T.; Leichman, L.; Okamura, R.; Raymond, V.M.; Lanman, R.B.; Lippman, S.M.; Kurzrock, R. Genomic Profiling of Blood-Derived Circulating Tumor DNA from Patients with Colorectal Cancer: Implications for Response and Resistance to Targeted Therapeutics. Mol. Cancer Ther. 2019, 18, 1852–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatsky, R.; Parker, B.A.; Bui, N.Q.; Helsten, T.; Schwab, R.B.; Boles, S.G.; Kurzrock, R. Next-Generation Sequencing of Tissue and Circulating Tumor DNA: The UC San Diego Moores Center for Personalized Cancer Therapy Experience with Breast Malignancies. Mol. Cancer Ther. 2019, 18, 1001–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Okamura, R.; Baumgartner, J.M.; Patel, H.; Leichman, L.; Kelly, K.; Sicklick, J.K.; Fanta, P.T.; Lippman, S.M.; Kurzrock, R. Analysis of Circulating Tumor DNA and Clinical Correlates in Patients with Esophageal, Gastroesophageal Junction, and Gastric Adenocarcinoma. Clin. Cancer Res. 2018, 24, 6248–6256. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, J.M.; Raymond, V.M.; Lanman, R.B.; Tran, L.; Kelly, K.J.; Lowy, A.M.; Kurzrock, R. Preoperative Circulating Tumor DNA in Patients with Peritoneal Carcinomatosis is an Independent Predictor of Progression-Free Survival. Ann. Surg. Oncol. 2018, 25, 2400–2408. [Google Scholar] [CrossRef] [PubMed]
- Charo, L.M.; Eskander, R.N.; Okamura, R.; Patel, S.P.; Nikanjam, M.; Lanman, R.B.; Piccioni, D.E.; Kato, S.; McHale, M.T.; Kurzrock, R. Clinical implications of plasma circulating tumor DNA in gynecologic cancer patients. Mol. Oncol. 2021, 15, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Okamura, R.; Kurzrock, R.; Mallory, R.J.; Fanta, P.T.; Burgoyne, A.M.; Clary, B.M.; Kato, S.; Sicklick, J.K. Comprehensive genomic landscape and precision therapeutic approach in biliary tract cancers. Int. J. Cancer 2021, 148, 702–712. [Google Scholar] [CrossRef]
- Schwaederle, M.C.; Patel, S.P.; Husain, H.; Ikeda, M.; Lanman, R.B.; Banks, K.C.; Talasaz, A.; Bazhenova, L.; Kurzrock, R. Utility of Genomic Assessment of Blood-Derived Circulating Tumor DNA (ctDNA) in Patients with Advanced Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 5101–5111. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Okamura, R.; Fanta, P.; Patel, C.; Lanman, R.B.; Raymond, V.M.; Kato, S.; Kurzrock, R. Clinical correlates of blood-derived circulating tumor DNA in pancreatic cancer. J. Hematol. Oncol. 2019, 12, 130. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Okamura, R.; Mareboina, M.; Lee, S.; Goodman, A.; Patel, S.P.; Fanta, P.T.; Schwab, R.B.; Vu, P.; Raymond, V.M.; et al. Revisiting Epidermal Growth Factor Receptor (EGFR) Amplification as a Target for Anti-EGFR Therapy: Analysis of Cell-Free Circulating Tumor DNA in Patients with Advanced Malignancies. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Schwaederle, M.; Mohindra, M.; Fontes Jardim, D.L.; Kurzrock, R. MET alterations detected in blood-derived circulating tumor DNA correlate with bone metastases and poor prognosis. J. Hematol. Oncol. 2018, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Li, B.T.; Janku, F.; Jung, B.; Hou, C.; Madwani, K.; Alden, R.; Razavi, P.; Reis-Filho, J.S.; Shen, R.; Isbell, J.M.; et al. Ultra-deep next-generation sequencing of plasma cell-free DNA in patients with advanced lung cancers: Results from the Actionable Genome Consortium. Ann. Oncol. 2019, 30, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Zhang, S.; Waters, J.; Liu, L.; Huang, H.J.; Subbiah, V.; Hong, D.S.; Karp, D.D.; Fu, S.; Cai, X.; et al. Development and Validation of an Ultradeep Next-Generation Sequencing Assay for Testing of Plasma Cell-Free DNA from Patients with Advanced Cancer. Clin. Cancer Res. 2017, 23, 5648–5656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.; Okamura, R.; Kato, S.; Soussi, T.; Kurzrock, R. Survival Implications of the Relationship between Tissue versus Circulating Tumor DNA TP53 Mutations-A Perspective from a Real-World Precision Medicine Cohort. Mol. Cancer Ther. 2020, 19, 2612–2620. [Google Scholar] [CrossRef] [PubMed]
- Mardinian, K.; Okamura, R.; Kato, S.; Kurzrock, R. Temporal and spatial effects and survival outcomes associated with concordance between tissue and blood KRAS alterations in the pan-cancer setting. Int. J. Cancer 2020, 146, 566–576. [Google Scholar] [CrossRef]
- Ricciuti, B.; Jones, G.; Severgnini, M.; Alessi, J.V.; Recondo, G.; Lawrence, M.; Forshew, T.; Lydon, C.; Nishino, M.; Cheng, M.; et al. Early plasma circulating tumor DNA (ctDNA) changes predict response to first-line pembrolizumab-based therapy in non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Janku, F.; Huang, H.J.; Fujii, T.; Shelton, D.N.; Madwani, K.; Fu, S.; Tsimberidou, A.M.; Piha-Paul, S.A.; Wheler, J.J.; Zinner, R.G.; et al. Multiplex KRASG12/G13 mutation testing of unamplified cell-free DNA from the plasma of patients with advanced cancers using droplet digital polymerase chain reaction. Ann. Oncol. 2017, 28, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.J.; Goodman, A.M.; Kato, S.; Ellison, C.K.; Daniels, G.A.; Kim, L.; Nakashe, P.; McCarthy, E.; Mazloom, A.R.; McLennan, G.; et al. Genome-Wide Sequencing of Cell-Free DNA Identifies Copy-Number Alterations That Can Be Used for Monitoring Response to Immunotherapy in Cancer Patients. Mol. Cancer Ther. 2019, 18, 448–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.K.; Oh, M.S. Detection of Minimal Residual Disease Using ctDNA in Lung Cancer: Current Evidence and Future Directions. J. Thorac. Oncol. 2019, 14, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Parkinson, C.A.; Gale, D.; Piskorz, A.M.; Biggs, H.; Hodgkin, C.; Addley, H.; Freeman, S.; Moyle, P.; Sala, E.; Sayal, K.; et al. Exploratory Analysis of TP53 Mutations in Circulating Tumour DNA as Biomarkers of Treatment Response for Patients with Relapsed High-Grade Serous Ovarian Carcinoma: A Retrospective Study. PLoS Med. 2016, 13, e1002198. [Google Scholar] [CrossRef]
- Lipson, E.J.; Velculescu, V.E.; Pritchard, T.S.; Sausen, M.; Pardoll, D.M.; Topalian, S.L.; Diaz, L.A., Jr. Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J. Immunother. Cancer 2014, 2, 42. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.; Wu, Y.L.; Lee, J.S.; Yu, C.J.; Sriuranpong, V.; Sandoval-Tan, J.; Ladrera, G.; Thongprasert, S.; Srimuninnimit, V.; Liao, M.; et al. Detection and Dynamic Changes of EGFR Mutations from Circulating Tumor DNA as a Predictor of Survival Outcomes in NSCLC Patients Treated with First-line Intercalated Erlotinib and Chemotherapy. Clin. Cancer Res. 2015, 21, 3196–3203. [Google Scholar] [CrossRef] [Green Version]
- Anandappa, G.; Starling, N.; Begum, R.; Bryant, A.; Sharma, S.; Renner, D.; Aresu, M.; Peckitt, C.; Sethi, H.; Feber, A.; et al. Minimal residual disease (MRD) detection with circulating tumor DNA (ctDNA) from personalized assays in stage II-III colorectal cancer patients in a U.K. multicenter prospective study (TRACC). J. Clin. Oncol. 2021, 39, 102. [Google Scholar] [CrossRef]
- Cao, H.; Liu, X.; Chen, Y.; Yang, P.; Huang, T.; Song, L.; Xu, R. Circulating Tumor DNA Is Capable of Monitoring the Therapeutic Response and Resistance in Advanced Colorectal Cancer Patients Undergoing Combined Target and Chemotherapy. Front. Oncol. 2020, 10, 466. [Google Scholar] [CrossRef]
- Chen, Z.; Sun, T.; Yang, Z.; Zheng, Y.; Yu, R.; Wu, X.; Yan, J.; Shao, Y.W.; Shao, X.; Cao, W.; et al. Monitoring treatment efficacy and resistance in breast cancer patients via circulating tumor DNA genomic profiling. Mol. Genet. Genom. Med. 2020, 8, e1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz - Cuaran, S.; Mezquita, L.; Swalduz, A.; Aldea, M.; Mazieres, J.; Jovelet, C.; Lacroix, L.; Pradines, A.; Avrillon, V.; MahierAït Oukhatar, C.; et al. 1556P—Circulating tumour DNA (ctDNA) analysis depicts mechanisms of resistance and tumour response to BRAF inhibitors in BRAF-mutant non-small cell lung cancer (NSCLC). Ann. Oncol. 2019, 30, v641. [Google Scholar] [CrossRef]
- Razavi, P.; Dickler, M.N.; Shah, P.D.; Toy, W.; Brown, D.N.; Won, H.H.; Li, B.T.; Shen, R.; Vasan, N.; Modi, S.; et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat. Cancer 2020, 1, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhu, W.; Guan, Y.; Yang, L.; Xia, X.; Chen, S.; Li, Q.; Guan, X.; Yi, Z.; Qian, H.; et al. ctDNA dynamics: A novel indicator to track resistance in metastatic breast cancer treated with anti-HER2 therapy. Oncotarget 2016, 7, 66020–66031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, Y.; Xu, Y.; Li, L.; Gong, Y.; Zhang, K.; Zhang, M.; Guan, Y.; Chang, L.; Xia, X.; et al. Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients. Nat. Commun. 2021, 12, 11. [Google Scholar] [CrossRef]
- Magbanua, M.J.M.; Swigart, L.B.; Wu, H.T.; Hirst, G.L.; Yau, C.; Wolf, D.M.; Tin, A.; Salari, R.; Shchegrova, S.; Pawar, H.; et al. Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response and survival. Ann. Oncol. 2021, 32, 229–239. [Google Scholar] [CrossRef]
- Zhang, Q.; Luo, J.; Wu, S.; Si, H.; Gao, C.; Xu, W.; Abdullah, S.E.; Higgs, B.W.; Dennis, P.A.; van der Heijden, M.S.; et al. Prognostic and Predictive Impact of Circulating Tumor DNA in Patients with Advanced Cancers Treated with Immune Checkpoint Blockade. Cancer Discov. 2020, 10, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Davis, A.A.; Jain, S.; Santa-Maria, C.; Flaum, L.; Beaubier, N.; Platanias, L.C.; Gradishar, W.; Giles, F.J.; Cristofanilli, M. Concordance of Genomic Alterations by Next-Generation Sequencing in Tumor Tissue versus Circulating Tumor DNA in Breast Cancer. Mol. Cancer Ther. 2017, 16, 1412–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Othman, T.; Chen, C.; Sandhu, J.; Ouyang, C.; Fakih, M. Guardant360 Circulating Tumor DNA Assay Is Concordant with FoundationOne Next-Generation Sequencing in Detecting Actionable Driver Mutations in Anti-EGFR Naive Metastatic Colorectal Cancer. Oncologist 2020, 25, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Circulating Tumor Cells (CTC) | Circulating Tumor DNA | Tissue DNA | |
|---|---|---|---|
| Able to be cultured | Able to be cultured | Unable to be cultured | Unable to be cultured |
| Ability to assess genomic, transcriptomic, proteomic data | Able to assess DNA, RNA and protein | Only able to assess DNA | Able to assess DNA, RNA, protein and tumor-infiltrating lymphocytes |
| Influences of collection and interpretation | Potential for sample bias | Minute amounts ctDNA in blood stream | Sample can be from primary or metastatic lesions |
| Ability to predict therapy responses | Serial samples can be predictive of responses to therapy | Serial samples can be predictive of responses to therapy | Serial samples are invasive and have not been shown to be a predictor of response, though new genomic alterations may predict resistance |
| Modifying factors | Heterogeneity within shed cells can be considered an opportunity as tissue biopsies might miss specific clones based on the location/size of the piece of tumor that was taken for analysis | Rate of tumor cell apoptosis, necrosis, and clearance. Possibly size of tumor sites and possibly number and location of metastatic sites can impact ctDNA levels | Tumor heterogeneity within primary and between primary and metastatic sites can occur |
| Technique | Advantages | Limitations | References |
|---|---|---|---|
| Droplet digital PCR (ddPCR) | High sensitivity | Only detects specific genomic sequences within sample | [36,37] |
| Beads, emulsion, amplification and magnetics (BEAMing) | High sensitivity | Only detects known alterations | [38] |
| Cancer Personalized Profiling by deep Sequencing (CAPP-Seq) | High sensitivity | Not fully comprehensive | [39] |
| Tagged-amplicon deep sequencing (TAm-Seq) | High sensitivity | Not fully comprehensive | [40] |
| Whole exome sequencing (WES) | Includes entire exome | Lower sensitivity | [41] |
| Whole genome sequencing (WGS) | Includes entire genome | Lower sensitivity | [42] |
| Cancer Histology | Setting | Results | References |
|---|---|---|---|
| Triple-negative breast cancer | During/after neoadjuvant chemotherapy | After cycle 1, detection of ctDNA was associated with worse DFS (p = 0.027) At the last post-chemotherapy pre-surgery time point, detection of ctDNA was strongly associated with worse pCR and DFS (p = 0.013) and OS (p = 0.006) | [53] |
| Advanced breast cancer | Therapeutic planning and serial testing for treatment response, tumor genomic evolution detection |
68% (42/62) of patients had ≥1 characterized ctDNA alteration (non-VUS) Concordance between tDNA and ctDNA was 48% | [57] |
| Ovarian, uterine, cervical, vulvovaginal, and unknown gynecologic primary | Therapeutic planning and serial testing for treatment response, tumor genomic evolution detection | Therapy matched to ctDNA alterations (n = 33) was associated with improved OS (HR: 0.34, p = 0.007) | [60] |
| Locally advanced rectal cancer | Adjuvant chemotherapy | 122 patients had pre-surgical detectable ctDNA Only 12 of 140 (8.6%) with negative ctDNA (HR 12, p < 0.001) experience recurrence Post-op ctDNA detection predicted recurrence regardless of adjuvant chemotherapy (chemo: HR 10, p < 0.001; no chemo: HR 16, p < 0.001) ctDNA detection predicted recurrence among pts with a pCR (HR 14, p = 0.014) or with pN+ disease (HR 11, p < 0.001) | [54] |
| Local advanced anal squamous cell cancer | Prognostic impact of post chemoradiation ctDNA detection | ctDNA detection after chemoradiation was associated with shorter DFS (p < 0.0001) More ctDNA was associated with higher stage (64% in stage II and 100% in stage III; p = 0.008) baseline ctDNA levels were higher in pN+ (median 85 copies/mL, range = 8–9333) than pN- (median 32 copies/mL, range = 3–1350) p = 0.03 | [55] |
| Advanced colorectal cancer | Therapeutic planning and serial testing for treatment response, tumor genomic evolution detection |
81% (63/78) of patients had ctDNA alteration, with 76% (59/78) having ≥1 characterized (non-VUS) Concordance between tDNA and ctDNA ranged from 62–87% | [56] |
| Biliary tract cancers | Therapeutic planning and serial testing for treatment response, tumor genomic evolution detection |
40 patients with both ctDNA and tDNA sequencing, concordance was higher between ctDNA and metastatic site tDNA than between ctDNA and primary tDNA (78% vs. 65% for TP53, 100% vs. 74% for KRAS and 100% vs. 87% for PIK3CA Therapy matched to genomic alterations (n = 80) had significantly longer PFS (HR 0.60, CI 0.37–0.99; p = 0.047) and higher disease control rate (61% vs. 35%; p = 0.04) | [61] |
| Advanced and resected esophageal, GEJ, and gastric adenocarcinoma | Therapeutic planning and serial testing for treatment response, tumor genomic evolution detection |
76% (42/55) of patients had ctDNA alteration, with 69% (38/55) having ≥1 characterized (non-VUS) Concordance between tDNA and ctDNA ranged from 61 to 87% | [58] |
| Advanced pancreatic ductal adenocarcinoma | Therapeutic planning and serial testing for treatment response, tumor genomic evolution detection |
Concordance between ctDNA and tDNA for TP53 was 61% and for KRAS 52% Concordance for KRAS between ctDNA and tDNA from metastatic sites was significantly higher than between ctDNA and primary tDNA (72% vs. 39%, p = 0.01) Higher levels of total %ctDNA was associated with worse survival (HR, 4.35, CI 1.85–10.24; p = 0.001) | [63] |
| Advanced NSCLC | Changes in VAF were serially measured in patients receiving pembrolizumab and platinum doublet-chemotherapy |
VAF decreased by 90.1% at median 21 days after treatment in patients (n = 18) with radiographic response VAF decreased by 19.9% in patient (n = 15) with stable disease (n = 15) VAF increased by 28.8% in patients (n = 12) with progressive disease; p = 0.003 VAF decrease between the pretreatment and first on-treatment blood draw was associated with higher ORR (60.7% vs. 5.8%; p = 0.0003), VAF decrease between the pretreatment and first on-treatment blood draw was associated longer median PFS (8.3 vs. 3.4 months, HR: 0.29, CI 0.14 to 0.60; p = 0.0007) VAF decrease between the pretreatment and first on-treatment blood draw was associated longer median OS (26.2 vs. 13.2 months, HR: 0.34, 0.15 to 0.75; p = 0.008 | [70] |
| Advanced lung cancers | Ultra-deep cfDNA and matched white blood cells covering 37 lung cancer-related genes |
Sensitivity for plasma NGS to detect de novo oncogenic drivers was 75% (68/91) Specificity for plasma NGS in driver-negative tumors compared to tDNA was 100% (19/19) | [66] |
| Advanced lung adenocarcinoma | Therapeutic planning and serial testing for treatment response, tumor genomic evolution detection |
82% of patients had ≥1 ctDNA alteration(s) Concordance for EGFR alterations in ctDNA vs. tDNA was 80.8%; p = 0.04 | [62] |
| Carcinomatosis (appendix cancer; colorectal; peritoneal mesothelioma; small bowel; cholangiocarcinoma; ovarian; testicular cancer) | Surgical resection of peritoneal metastases |
39% (31/80) of patients had ctDNA alteration Patients with ≥0.25% cfDNA had shorter PFS (7.8 vs. 15.0 months; HR 3.23, 95% CI 1.43–7.28, p = 0.005). | [59] |
| Diverse cancers (including but not limited to: colorectal cancer, non-small cell lung cancer, genitourinary cancers) | EGFR amplification status in 28,584 patients |
8.5% of diverse cancers had a cctDNA EGFR amplification detected Responses were seen in patients with ctDNA EGFR amplification treated with EGFR inhibitors even if no tissue EGFR amplification was detected | [64] |
| Diverse cancers (including but not limited to gastrointestinal, brain, lung) | Clinical associations of MET alterations |
7.1% (31/438) and correlated with bone metastasis (p = 0.007) MET alterations were associated with TP53 co-alterations (p = 0.001) and PTEN co-alterations (p = 0.003) MET alterations were also associated with an increased number of alterations (median, 4 vs. 1, p = 0.001) | [65] |
| Advanced cancers | Ultra-deep cfDNA |
Concordance between ctDNA and tDNA NGS was 82–87% Low VAF vs. high VAF of mutant ctDNA had longer OS (p = 0.018) Decrease in ctDNA VAF was associated with longer time to treatment failure p = 0.03 | [67] |
| Advanced cancers | cfDNA tested with a KRASG12/G13 multiplex assay to detect seven most common mutations in exon 2 hotspot |
Concordance was found in 85% (103/121) patients (kappa, 0.66; ddPCR sensitivity, 84%; ddPCR specificity, 88%) Presence of ≥ 6.2% of KRASG12/G13 cfDNA was associated with shorter overall survival (p = 0.001) | [71] |
| Pan-Cancer | Immune checkpoint blockade | Early changes in copy number alterations predicted response versus resistance | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adashek, J.J.; Janku, F.; Kurzrock, R. Signed in Blood: Circulating Tumor DNA in Cancer Diagnosis, Treatment and Screening. Cancers 2021, 13, 3600. https://doi.org/10.3390/cancers13143600
Adashek JJ, Janku F, Kurzrock R. Signed in Blood: Circulating Tumor DNA in Cancer Diagnosis, Treatment and Screening. Cancers. 2021; 13(14):3600. https://doi.org/10.3390/cancers13143600
Chicago/Turabian StyleAdashek, Jacob J., Filip Janku, and Razelle Kurzrock. 2021. "Signed in Blood: Circulating Tumor DNA in Cancer Diagnosis, Treatment and Screening" Cancers 13, no. 14: 3600. https://doi.org/10.3390/cancers13143600
APA StyleAdashek, J. J., Janku, F., & Kurzrock, R. (2021). Signed in Blood: Circulating Tumor DNA in Cancer Diagnosis, Treatment and Screening. Cancers, 13(14), 3600. https://doi.org/10.3390/cancers13143600