
Deregulation of Transcriptional Enhancers in Cancer

Abstract
Simple Summary
Abstract
1. The Epigenetic Foundation of Tumour Plasticity
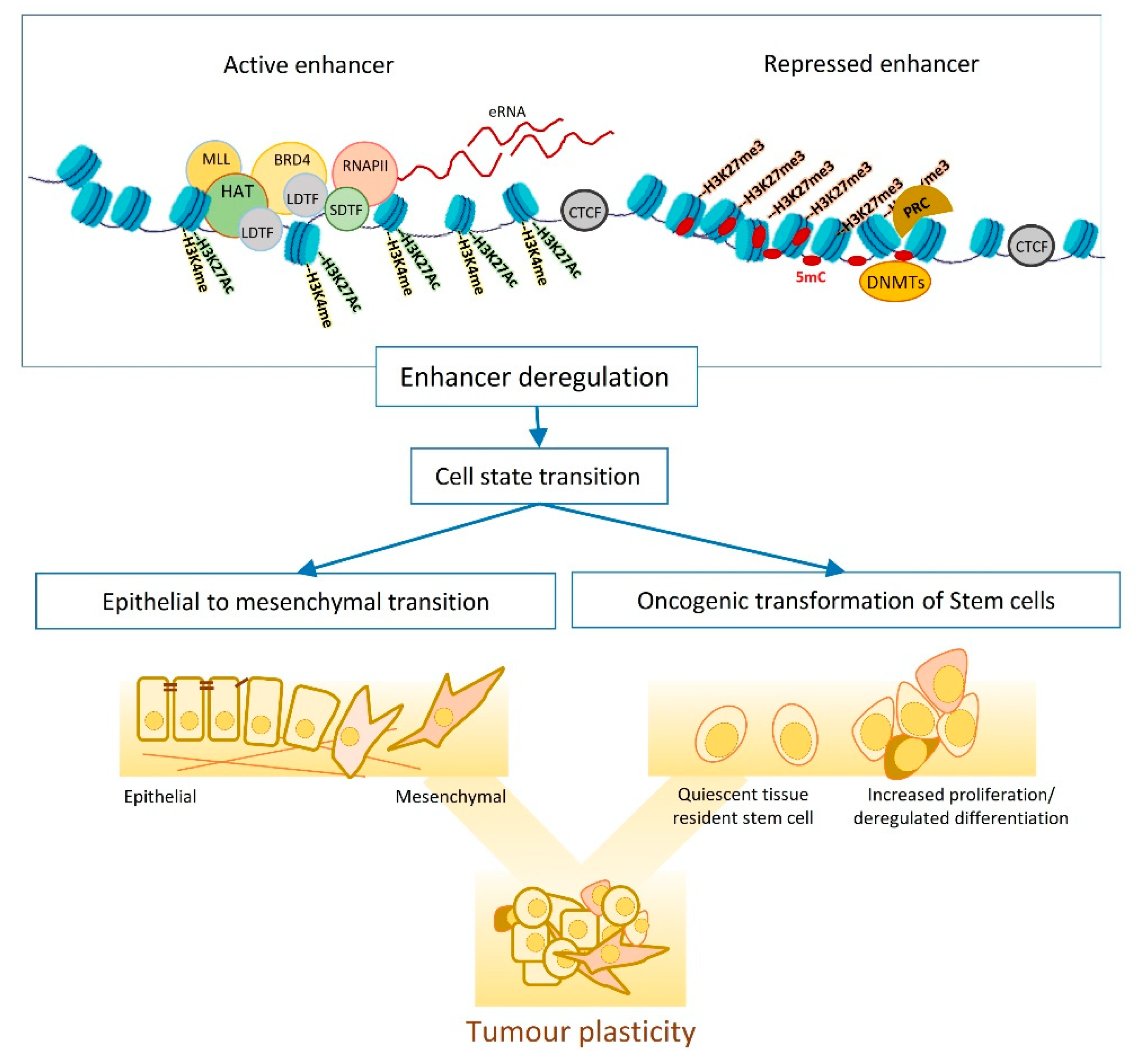
1.1. Tumour Plasticity and Heterogeneity
1.2. Enhancers, the Epigenetic Playground
2. Enhancer Dynamics and Tumour Plasticity:
2.1. Enhancer Dynamics and Adult Stem Cell Differentiation
2.2. Enhancer Dynamics and EMT
3. Cancer-Related Genetic Variations at Enhancers
3.1. Mutations Affecting the Enhancer Sequence
3.2. Mutations at Enhancer Regulators
3.3. Epigenetic Cancer Drugs that Target Enhancers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Suda, K.; Rivard, C.J.; Mitsudomi, T.; Hirsch, F.R. Heterogeneity in Tumors and Resistance to EGFR TKI Therapy—Letter. Cancer Res. 2016, 76, 3109–3110. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gerashchenko, T.S.; Zavyalova, M.V.; Denisov, E.V.; Krakhmal, N.V.; Pautova, D.N.; Litviakov, N.; Vtorushin, S.V.; Cherdyntseva, N.V.; Perelmuter, V.M. Intratumoral Morphological Heterogeneity of Breast Cancer As an Indicator of the Metastatic Potential and Tumor Chemosensitivity. Acta Nat. 2017, 9, 56–67. [Google Scholar] [CrossRef]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [PubMed]
- Geyer, F.C.; Weigelt, B.; Natrajan, R.; Lambros, M.B.; De Biase, D.; Vatcheva, R.; Savage, K.; Mackay, A.; Ashworth, A.; Reis-Filho, J.S. Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J. Pathol. 2009, 220, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Fu, F.; Chen, L.; Lin, Y.; Yang, P.; Wang, C. Single hormone receptor-positive breast cancer patients experienced poor survival outcomes: A systematic review and meta-analysis. Clin. Transl. Oncol. 2019, 22, 474–485. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef]
- Von Loga, K.; Woolston, A.; Punta, M.; Barber, L.J.; Griffiths, B.; Semiannikova, M.; Spain, G.; Challoner, B.; Fenwick, K.; Simon, R.; et al. Extreme intratumour heterogeneity and driver evolution in mismatch repair deficient gastro-oesophageal cancer. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Carpenter, R.; Sirkisoon, S.; Zhu, D.; Rimkus, T.; Harrison, A.; Anderson, A.; Paw, I.; Qasem, S.; Xing, F.; Liu, Y.; et al. Combined inhibition of AKT and HSF1 suppresses breast cancer stem cells and tumor growth. Oncotarget 2017, 8, 73947–73963. [Google Scholar] [CrossRef]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.N.S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5+ human colon cancer stem cells. Nat. Cell Biol. 2017, 545, 187–192. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.K.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef]
- Bhatia, S.; Monkman, J.; Blick, T.; Pinto, C.; Waltham, M.; Nagaraj, S.H.; Thompson, E.W. Interrogation of Phenotypic Plasticity between Epithelial and Mesenchymal States in Breast Cancer. J. Clin. Med. 2019, 8, 893. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Konge, J.; Leteurtre, F.; Goislard, M.; Biard, D.; Morel-Altmeyer, S.; Vaurijoux, A.; Gruel, G.; Chevillard, S.; Lebeau, J. Breast cancer stem cell-like cells generated during TGFβ-induced EMT are radioresistant. Oncotarget 2018, 9, 23519–23531. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Wilde, A.; D’Hérouël, A.F.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.-A.; Huang, S. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, K.; Okazawa, Y.; Haeno, H.; Koyama, Y.; Sulidan, K.; Komiyama, H.; Saeki, H.; Ohtsuji, N.; Ito, Y.; Kojima, Y.; et al. Metastatic seeding of human colon cancer cell clusters expressing the hybrid epithelial/mesenchymal state. Int. J. Cancer 2020, 146, 2547–2562. [Google Scholar] [CrossRef]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef]
- Litviakov, N.; Ibragimova, M.; Tsyganov, M.; Kazantseva, P.; Deryusheva, I.; Pevzner, A.; Doroshenko, A.; Garbukov, E.; Tarabanovskaya, N.; Slonimskaya, E. Amplifications of stemness genes and the capacity of breast tumors for metastasis. Oncotarget 2020, 11, 1988–2001. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.; Knuechel, R.; Kirchner, T. Variable -catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef]
- Walter, R.; Sonnentag, S.; Orian-Rousseau, V.; Munoz-Sagredo, L. Plasticity in Colorectal Cancer: Why Cancer Cells Differentiate. Cancers 2021, 13, 918. [Google Scholar] [CrossRef]
- Vermeulen, L.; Melo, F.D.S.E.; Van Der Heijden, M.; Cameron, K.; De Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.; Rios, A.C.; Pal, B.; Law, C.W.; Jamieson, P.; Liu, R.; Vaillant, F.; Jackling, F.; Liu, K.H.; Smyth, G.K.; et al. Identification of quiescent and spatially restricted mammary stem cells that are hormone responsive. Nat. Cell Biol. 2017, 19, 164–176. [Google Scholar] [CrossRef]
- Yeo, S.K.; Zhu, X.; Okamoto, T.; Hao, M.; Wang, C.; Lu, P.; Lu, L.J.; Guan, J.-L. Single-cell RNA-sequencing reveals distinct patterns of cell state heterogeneity in mouse models of breast cancer. eLife 2020, 9. [Google Scholar] [CrossRef]
- Russnes, H.G.; Navin, N.; Hicks, J.; Borresen-Dale, A.-L. Insight into the heterogeneity of breast cancer through next-generation sequencing. J. Clin. Investig. 2011, 121, 3810–3818. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Hawley, J.R.; Soares, F.; Grillo, G.; Teng, M.; Tonekaboni, S.A.M.; Hua, J.T.; Kron, K.J.; Mazrooei, P.; Ahmed, M.; et al. Noncoding mutations target cis-regulatory elements of the FOXA1 plexus in prostate cancer. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Zarchi, A.; Gerovska, D.; Adachi, K.; Totonchi, M.; Pezeshk, H.; Taft, R.J.; Schöler, H.; Chitsaz, H.; Sadeghi, M.; Baharvand, H.; et al. DNA methylation regulates discrimination of enhancers from promoters through a H3K4me1-H3K4me3 seesaw mechanism. BMC Genom. 2017, 18, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Lesch, B.J. H3K4me1 Distribution Predicts Transcription State and Poising at Promoters. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Pediconi, N.; Salerno, D.; Lupacchini, L.; Angrisani, A.; Peruzzi, G.; De Smaele, E.; Levrero, M.; Belloni, L. EZH2, JMJD3, and UTX epigenetically regulate hepatic plasticity inducing retro-differentiation and proliferation of liver cells. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, X.; Jiang, Y.; Liu, S.; Liu, H.; Sun, X.; Zhang, H.; Liu, Z.; Tao, Y.; Li, C.; et al. Elevating H3K27me3 level sensitizes colorectal cancer to oxaliplatin. J. Mol. Cell Biol. 2019, 12, 125–137. [Google Scholar] [CrossRef]
- Benton, M.L.; Talipineni, S.C.; Kostka, D.; Capra, J.A. Genome-wide enhancer annotations differ significantly in genomic distribution, evolution, and function. BMC Genom. 2019, 20, 511. [Google Scholar] [CrossRef]
- Henriques, T.; Scruggs, B.S.; Inouye, M.O.; Muse, G.W.; Williams, L.H.; Burkholder, A.; Lavender, C.; Fargo, D.C.; Adelman, K. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 2018, 32, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, S.; Cao, Z.; Ouyang, W.; Zhang, Q.; Xie, L.; Zheng, R.; Guo, M.; Ma, M.; Hu, Z.; et al. Chromatin loops associated with active genes and heterochromatin shape rice genome architecture for transcriptional regulation. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.H.; Jena, S.G.; Yamazaki, Y.; Lim, B. Regulation of spatiotemporal limits of developmental gene expression via enhancer grammar. Proc. Natl. Acad. Sci. USA 2020, 117, 15096–15103. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nat. Cell Biol. 2011, 471, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Wang, L.-M.; Luo, Y.; Lai, X.; Li, C.; Shi, J.; Tan, Y.; Fu, S.; Wang, Y.; Zhu, N.; et al. Mutations in epigenetic regulators are involved in acute lymphoblastic leukemia relapse following allogeneic hematopoietic stem cell transplantation. Oncotarget 2015, 7, 2696–2708. [Google Scholar] [CrossRef]
- Ward, R.; Johnson, M.; Shridhar, V.; Van Deursen, J.; Couch, F.J. CBP truncating mutations in ovarian cancer. J. Med. Genet. 2005, 42, 514–518. [Google Scholar] [CrossRef]
- Ogiwara, H.; Sasaki, M.; Mitachi, T.; Oike, T.; Higuchi, S.; Tominaga, Y.; Kohno, T. Targeting p300 Addiction in CBP-Deficient Cancers Causes Synthetic Lethality by Apoptotic Cell Death due to Abrogation of MYC Expression. Cancer Discov. 2016, 6, 430–445. [Google Scholar] [CrossRef]
- Lai, K.; Hu, X.; Chosa, K.; Nguyen, C.; Lin, D.; Lai, K.; Kato, N.; Higuchi, Y.; Highlander, S.; Melendez, E.; et al. p300 Serine 89: A Critical Signaling Integrator and Its Effects on Intestinal Homeostasis and Repair. Cancers 2021, 13, 1288. [Google Scholar] [CrossRef]
- Li, H.-M.; Bi, Y.-R.; Li, Y.; Fu, R.; Lv, W.-C.; Jiang, N.; Xu, Y.; Ren, B.-X.; Chen, Y.-D.; Xie, H.; et al. A potent CBP/p300-Snail interaction inhibitor suppresses tumor growth and metastasis in wild-type p53-expressing cancer. Sci. Adv. 2020, 6, eaaw8500. [Google Scholar] [CrossRef]
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harb. Perspect. Med. 2016, 7, a026534. [Google Scholar] [CrossRef]
- Brooks, N.; Pegg, N.; Worthington, J.; Young, B.; Prosser, A.; Lane, J.; Taddei, D.; Schiewer, M.J.; Gordon, N.; Knudsen, K.E. A novel small molecule inhibitor of p300/CBP for the treatment of castration-resistant prostate cancer: Preclinical evaluation. J. Clin. Oncol. 2017, 35, 168. [Google Scholar] [CrossRef]
- Sanchez, G.J.; A Richmond, P.; Bunker, E.N.; Karman, S.S.; Azofeifa, J.; Garnett, A.T.; Xu, Q.; E Wheeler, G.; Toomey, C.M.; Zhang, Q.; et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2018, 46, 1756–1776. [Google Scholar] [CrossRef]
- Grinat, J.; Heuberger, J.; Vidal, R.O.; Goveas, N.; Kosel, F.; Berenguer-Llergo, A.; Kranz, A.; Wulf-Goldenberg, A.; Behrens, D.; Melcher, B.; et al. The epigenetic regulator Mll1 is required for Wnt-driven intestinal tumorigenesis and cancer stemness. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lindner, P.; Paul, S.; Eckstein, M.; Hampel, C.; Muenzner, J.; Erlenbach-Wuensch, K.; Ahmed, H.P.; Mahadevan, V.; Brabletz, T.; Hartmann, A.; et al. EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Adelman, E.R.; Huang, H.-T.; Roisman, A.; Olsson, A.; Colaprico, A.; Qin, T.; Lindsley, R.C.; Bejar, R.; Salomonis, N.; Grimes, H.L.; et al. Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia. Cancer Discov. 2019, 9, 1080–1101. [Google Scholar] [CrossRef] [PubMed]
- Javaid, S.; Zhang, J.; Anderssen, E.; Black, J.; Wittner, B.S.; Tajima, K.; Ting, D.; Smolen, G.A.; Zubrowski, M.; Desai, R.; et al. Dynamic Chromatin Modification Sustains Epithelial-Mesenchymal Transition following Inducible Expression of Snail-1. Cell Rep. 2013, 5, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Schulz, W.A.; Lang, A.; Koch, J.; Greife, A. The histone demethylase UTX/KDM6A in cancer: Progress and puzzles. Int. J. Cancer 2019, 145, 614–620. [Google Scholar] [CrossRef]
- Ler, L.D.; Ghosh, S.; Chai, X.; Thike, A.A.; Heng, H.L.; Siew, E.Y.; Dey, S.; Koh, L.K.; Lim, J.Q.; Lim, W.K.; et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci. Transl. Med. 2017, 9, eaai8312. [Google Scholar] [CrossRef]
- Knutson, S.K.; Wigle, T.J.; Warholic, N.M.; Sneeringer, C.J.; Allain, C.J.; Klaus, C.R.; Sacks, J.D.; Raimondi, A.; Majer, C.R.; Song, J.; et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA Hypomethylation Contributes to Genomic Instability and Intestinal Cancer Initiation. Cancer Prev. Res. 2016, 9, 534–546. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, A.; Sottile, J.; Sánchez-Tejada, L.; Fajardo, C.; Cámara, R.; Lamas, C.; Barberá, V.M.; Picó, A. DNA Methylation of Tumor Suppressor Genes in Pituitary Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2019, 104, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Dailey, L. High throughput technologies for the functional discovery of mammalian enhancers: New approaches for understanding transcriptional regulatory network dynamics. Genomics 2015, 106, 151–158. [Google Scholar] [CrossRef]
- Davie, K.; Jacobs, J.; Atkins, M.; Potier, D.; Christiaens, V.; Halder, G.; Aerts, S. Discovery of Transcription Factors and Regulatory Regions Driving In Vivo Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling. PLoS Genet. 2015, 11, e1004994. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.; Zhang, X.; Wang, L.; Engreitz, J.; Melnikov, A.; Rogov, P.; Tewhey, R.; Isakova, A.; Deplancke, B.; Bernstein, B.E.; et al. Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc. Natl. Acad. Sci. USA 2017, 114, E1291–E1300. [Google Scholar] [CrossRef]
- Askovich, P.S.; Ramsey, S.A.; Diercks, A.H.; Kennedy, K.A.; Knijnenburg, T.A.; Aderem, A. Identifying novel transcription factors involved in the inflammatory response by using binding site motif scanning in genomic regions defined by histone acetylation. PLoS ONE 2017, 12, e0184850. [Google Scholar] [CrossRef]
- Lee, J.-E.; Park, Y.-K.; Park, S.; Jang, Y.; Waring, N.; Dey, A.; Ozato, K.; Lai, B.; Peng, W.; Ge, K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Mullen, A.C.; Orlando, D.A.; Newman, J.J.; Lovén, J.; Kumar, R.M.; Bilodeau, S.; Reddy, J.; Guenther, M.G.; DeKoter, R.; Young, R.A. Master Transcription Factors Determine Cell-Type-Specific Responses to TGF-β Signaling. Cell 2011, 147, 565–576. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Chiang, H.-C.; Hsieh, Y.-P.; Lu, C.; Park, B.H.; Jatoi, I.; Jin, V.X.; Hu, Y.; Li, R. BRCA1 mutations attenuate super-enhancer function and chromatin looping in haploinsufficient human breast epithelial cells. Breast Cancer Res. 2019, 21, 1–15. [Google Scholar] [CrossRef]
- Carico, Z.M.; Stefan, H.C.; Justice, M.; Yimit, A.; Dowen, J.M. A cohesin cancer mutation reveals a role for the hinge domain in genome organization and gene expression. PLoS Genet. 2021, 17, e1009435. [Google Scholar] [CrossRef]
- Collier, M.D.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Katanayeva, N.; Battistello, E.; Mina, M.; Douglass, K.; Rey, T.; Raynaud, F.; et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 2019, 51, 517–528. [Google Scholar] [CrossRef]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stütz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.H.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nat. Cell Biol. 2014, 511, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Rhie, S.K.; Perez, A.; Lay, F.D.; Schreiner, S.; Shi, J.; Polin, J.; Farnham, P.J. A high-resolution 3D epigenomic map reveals insights into the creation of the prostate cancer transcriptome. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Achinger-Kawecka, J.; Stirzaker, C.; Chia, K.-M.; Portman, N.; Campbell, E.; Du, Q.; Laven-Law, G.; Nair, S.S.; Yong, A.; Wilkinson, A.; et al. Epigenetic therapy suppresses endocrine-resistant breast tumour growth by re-wiring ER-mediated 3D chromatin interactions. bioRxiv 2021. [Google Scholar] [CrossRef]
- Guilhamon, P.; Chesnelong, C.; Kushida, M.M.; Nikolic, A.; Singhal, D.; MacLeod, G.; Tonekaboni, S.A.M.; Cavalli, F.M.; Arlidge, C.; Rajakulendran, N.; et al. Single-cell chromatin accessibility profiling of glioblastoma identifies an invasive cancer stem cell population associated with lower survival. eLife 2021, 10, 64090. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Paucek, R.D.; Gooding, A.R.; Brown, Z.Z.; Ge, E.J.; Muir, T.W.; Cech, T.R. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat. Struct. Mol. Biol. 2017, 24, 1028–1038. [Google Scholar] [CrossRef]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Kushwaha, G.; Dozmorov, M.; Wren, J.D.; Qiu, J.; Shi, H.; Xu, D. Hypomethylation coordinates antagonistically with hypermethylation in cancer development: A case study of leukemia. Hum. Genom. 2016, 10, 83–102. [Google Scholar] [CrossRef]
- Gao, X.; Sheng, Y.; Yang, J.; Wang, C.; Zhang, R.; Zhu, Y.; Zhang, Z.; Zhang, K.; Yan, S.; Sun, H.; et al. Osteopontin alters DNA methylation through up-regulating DNMT1 and sensitizes CD133+/CD44+ cancer stem cells to 5 azacytidine in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 1–14. [Google Scholar] [CrossRef]
- Pathania, R.; Ramachandran, S.; Elangovan, S.; Padia, R.; Yang, P.; Cinghu, S.; Veeranan-Karmegam, R.; Arjunan, P.; Gnana-Prakasam, J.P.; Sadanand, F.; et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Huang, J.; Tang, Y.; Luo, X.; Ge, L.; Sheng, X.; Sun, X.; Chen, Y.; Zhu, D. Regional methylome profiling reveals dynamic epigenetic heterogeneity and convergent hypomethylation of stem cell quiescence-associated genes in breast cancer following neoadjuvant chemotherapy. Cell Biosci. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.; Assenov, Y.; Minner, S.; Bogatyrova, O.; Simon, R.; Koop, C.; Oakes, C.; Zucknick, M.; Lipka, D.B.; Weischenfeldt, J.; et al. Intratumor DNA Methylation Heterogeneity Reflects Clonal Evolution in Aggressive Prostate Cancer. Cell Rep. 2014, 8, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.K.; Goode, D.; Iwasaki, M.; Wei, M.C.; Kuo, H.-P.; Zhu, L.; Schneidawind, D.; Duque-Afonso, J.; Weng, Z.; Cleary, M.L. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell 2015, 28, 198–209. [Google Scholar] [CrossRef]
- Luyten, A.; Zang, C.; Liu, X.S.; Shivdasani, R.A. Active enhancers are delineated de novo during hematopoiesis, with limited lineage fidelity among specified primary blood cells. Genes Dev. 2014, 28, 1827–1839. [Google Scholar] [CrossRef]
- Gao, P.; Chen, C.; Howell, E.D.; Li, Y.; Tober, J.; Uzun, Y.; He, B.; Gao, L.; Zhu, Q.; Siekmann, A.F.; et al. Transcriptional regulatory network controlling the ontogeny of hematopoietic stem cells. Genes Dev. 2020, 34, 950–964. [Google Scholar] [CrossRef]
- Cabal-Hierro, L.; Van Galen, P.; Prado, M.A.; Higby, K.J.; Togami, K.; Mowery, C.T.; Paulo, J.A.; Xie, Y.; Cejas, P.; Furusawa, T.; et al. Chromatin accessibility promotes hematopoietic and leukemia stem cell activity. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Peng, L.; Guo, H.; Ma, P.; Sun, Y.; Dennison, L.; Aplan, P.D.; Hess, J.L.; Friedman, A.D. HoxA9 binds and represses the Cebpa +8 kb enhancer. PLoS ONE 2019, 14, e0217604. [Google Scholar] [CrossRef]
- Friedman, A.D. C/EBPα in normal and malignant myelopoiesis. Int. J. Hematol. 2015, 101, 330–341. [Google Scholar] [CrossRef]
- Benetatos, L.; Benetatou, A.; Vartholomatos, G. Enhancers and MYC interplay in hematopoiesis. J. Mol. Med. 2020, 98, 471–481. [Google Scholar] [CrossRef]
- Bahr, C.; Von Paleske, L.; Uslu, V.V.; Remeseiro, S.; Takayama, N.; Ng, S.W.; Murison, A.; Langenfeld, K.; Petretich, M.; Scognamiglio, R.; et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nat. Cell Biol. 2018, 553, 515–520. [Google Scholar] [CrossRef]
- Mehta, C.; Johnson, K.D.; Gao, X.; Ong, I.M.; Katsumura, K.R.; McIver, S.C.; Ranheim, E.A.; Bresnick, E.H. Integrating Enhancer Mechanisms to Establish a Hierarchical Blood Development Program. Cell Rep. 2017, 20, 2966–2979. [Google Scholar] [CrossRef]
- Hirsch, D.; Barker, N.; McNeil, N.; Hu, Y.; Camps, J.; McKinnon, K.; Clevers, H.; Ried, T.; Gaiser, T. LGR5 positivity defines stem-like cells in colorectal cancer. Carcinogenesis 2014, 35, 849–858. [Google Scholar] [CrossRef]
- Goto, N.; Fukuda, A.; Yamaga, Y.; Yoshikawa, T.; Maruno, T.; Maekawa, H.; Inamoto, S.; Kawada, K.; Sakai, Y.; Miyoshi, H.; et al. Lineage tracing and targeting of IL17RB+tuft cell-like human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2019, 116, 12996–13005. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.; Clevers, H. Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell Biol. 2021, 22, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Tong, K.; Zhao, Y.; Balasubramanian, I.; Yap, G.S.; Ferraris, R.P.; Bonder, E.M.; Verzi, M.P.; Gao, N. Paneth Cell Multipotency Induced by Notch Activation following Injury. Cell Stem Cell 2018, 23, 46–59.e5. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Schewe, M.; Sacchetti, A.; Feijtel, D.; van de Geer, W.S.; Teeuwssen, M.; Sleddens, H.F.; Joosten, R.; van Royen, M.E.; van de Werken, H.J.; et al. Paneth Cells Respond to Inflammation and Contribute to Tissue Regeneration by Acquiring Stem-like Features through SCF/c-Kit Signaling. Cell Rep. 2018, 24, 2312–2328.e7. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-H.; Li, F.; Ferreiro-Neira, I.; Ho, L.-L.; Luyten, A.; Nalapareddy, K.; Long, H.; Verzi, M.; Shivdasani, R.A. Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nat. Cell Biol. 2014, 506, 511–515. [Google Scholar] [CrossRef]
- Kaaij, L.T.J.; Van De Wetering, M.; Fang, F.; Decato, B.; Molaro, A.; Van De Werken, H.J.G.; Van Es, J.H.; Schuijers, J.; De Wit, E.; De Laat, W.; et al. DNA methylation dynamics during intestinal stem cell differentiation reveals enhancers driving gene expression in the villus. Genome Biol. 2013, 14, R50. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Garnier, L.; Cleroux, E.; Giordano, A.; Dumas, M.; Bardet, A.F.; Kergrohen, T.; Quesada, S.; Cesses, P.; Weber, M.; et al. Loss of Apc Rapidly Impairs DNA Methylation Programs and Cell Fate Decisions in Lgr5+ Intestinal Stem Cells. Cancer Res. 2020, 80, 2101–2113. [Google Scholar] [CrossRef]
- Baba, Y.; Yagi, T.; Sawayama, H.; Hiyoshi, Y.; Ishimoto, T.; Iwatsuki, M.; Miyamoto, Y.; Yoshida, N.; Baba, H. Long Interspersed Element-1 Methylation Level as a Prognostic Biomarker in Gastrointestinal Cancers. Digestion 2018, 97, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Pandiyan, K.; Eun, J.W.; Yang, X.; Hong, S.H.; Nam, S.W.; Jones, P.A.; Liang, G.; You, J.S. Epigenetic landscape change analysis during human EMT sheds light on a key EMT mediator TRIM29. Oncotarget 2017, 8, 98322–98335. [Google Scholar] [CrossRef][Green Version]
- Galle, E.; Thienpont, B.; Cappuyns, S.; Venken, T.; Busschaert, P.; Van Haele, M.; Van Cutsem, E.; Roskams, T.; Van Pelt, J.; Verslype, C.; et al. DNA methylation-driven EMT is a common mechanism of resistance to various therapeutic agents in cancer. Clin. Epigenetics 2020, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. TGF-β induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 2014, 9, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; De Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The Snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene 2016, 36, 942–955. [Google Scholar] [CrossRef]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail Mediates E-Cadherin Repression by the Recruitment of the Sin3A/Histone Deacetylase 1 (HDAC1)/HDAC2 Complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef]
- Navandar, M.; Garding, A.; Sahu, S.K.; Pataskar, A.; Schick, S.; Tiwari, V.K. ERK signalling modulates epigenome to drive epithelial to mesenchymal transition. Oncotarget 2017, 8, 29269–29281. [Google Scholar] [CrossRef]
- Shin, S.; Buel, G.R.; Nagiec, M.J.; Han, M.-J.; Roux, P.P.; Blenis, J.; Yoon, S.-O. ERK2 regulates epithelial-to-mesenchymal plasticity through DOCK10-dependent Rac1/FoxO1 activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2967–2976. [Google Scholar] [CrossRef]
- Chang, H.; Liu, Y.; Xue, M.; Liu, H.; Du, S.; Zhang, L.; Wang, P. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res. 2016, 44, 2514–2527. [Google Scholar] [CrossRef]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.-O.; Heidecke, C.-D.; Friess, H.; Büchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, C.; Jiang, H.; Liang, L.; Shi, W.; Zhang, Q.; Sun, P.; Xiang, R.; Wang, Y.; Yang, S. ZEB1 induces ER-α promoter hypermethylation and confers antiestrogen resistance in breast cancer. Cell Death Dis. 2017, 8, e2732. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-T.; Zhong, H.-T.; Li, G.-W.; Shen, J.-X.; Ye, Q.-Q.; Zhang, M.-L.; Liu, J. Oncogenic functions of the EMT-related transcription factor ZEB1 in breast cancer. J. Transl. Med. 2020, 18, 1–10. [Google Scholar] [CrossRef]
- Jägle, S.; Busch, H.; Freihen, V.; Beyes, S.; Schrempp, M.; Boerries, M.; Hecht, A. SNAIL1-mediated downregulation of FOXA proteins facilitates the inactivation of transcriptional enhancer elements at key epithelial genes in colorectal cancer cells. PLoS Genet. 2017, 13, e1007109. [Google Scholar] [CrossRef] [PubMed]
- BenAyed-Guerfali, D.; Dabbèche-Bouricha, E.; Ayadi, W.; Trifa, F.; Charfi, S.; Khabir, A.; Sellami-Boudawara, T.; Mokdad-Gargouri, R. Association of FOXA1 and EMT markers (Twist1 and E-cadherin) in breast cancer. Mol. Biol. Rep. 2019, 46, 3247–3255. [Google Scholar] [CrossRef]
- Batlle, E.; Henderson, J.T.; Beghtel, H.; Born, M.M.V.D.; Sancho, E.; Huls, G.; Meeldijk, J.; Robertson, J.; van de Wetering, M.; Pawson, T.; et al. β-Catenin and TCF Mediate Cell Positioning in the Intestinal Epithelium by Controlling the Expression of EphB/EphrinB. Cell 2002, 111, 251–263. [Google Scholar] [CrossRef]
- Schnappauf, O.; Beyes, S.; Dertmann, A.; Freihen, V.; Frey, P.; Jägle, S.; Rose, K.; Michoel, T.; Grosschedl, R.; Hecht, A. Enhancer decommissioning by Snail1-induced competitive displacement of TCF7L2 and down-regulation of transcriptional activators results in EPHB2 silencing. Biochim. et Biophys. Acta (BBA) Bioenerg. 2016, 1859, 1353–1367. [Google Scholar] [CrossRef]
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C.; et al. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Vidakovic, A.T.; Rueda, O.M.; Vervoort, S.J.; Batra, A.S.; Goldgraben, M.; Uribe-Lewis, S.; Greenwood, W.; Coffer, P.J.; Bruna, A.; Caldas, C. Context-Specific Effects of TGF-β/SMAD3 in Cancer Are Modulated by the Epigenome. Cell Rep. 2015, 13, 2480–2490. [Google Scholar] [CrossRef]
- Tuupanen, S.; Turunen, M.; Lehtonen, R.J.; Hallikas, O.; Vanharanta, S.; Kivioja, T.; Bjorklund, M.; Wei, G.-H.; Yan, J.; Niittymäki, I.; et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 2009, 41, 885–890. [Google Scholar] [CrossRef]
- Cuykendall, T.N.; Rubin, M.; Khurana, E. Non-coding genetic variation in cancer. Curr. Opin. Syst. Biol. 2017, 1, 9–15. [Google Scholar] [CrossRef]
- Wu, S.; Ou, T.; Xing, N.; Lu, J.; Wan, S.; Wang, C.; Zhang, X.; Yang, F.; Huang, Y.; Cai, Z. Whole-genome sequencing identifies ADGRG6 enhancer mutations and FRS2 duplications as angiogenesis-related drivers in bladder cancer. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Mansour, M.; Abraham, B.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef]
- Rheinbay, E.; Nielsen, M.M.; Abascal, F.; Wala, J.A.; Shapira, O.; Tiao, G.; Hornshøj, H.; Hess, J.M.; Juul, R.I.; Lin, Z.; et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 2020, 578, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; PCAWG Transcriptome Working Group; Chen, F.; Fonseca, N.A.; He, Y.; Fujita, M.; Nakagawa, H.; Zhang, Z.; Brazma, A.; Creighton, C.J.; et al. High-coverage whole-genome analysis of 1220 cancers reveals hundreds of genes deregulated by rearrangement-mediated cis-regulatory alterations. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- De Matos, M.R.; Posa, I.; Carvalho, F.; Morais, V.A.; Grosso, A.R.; De Almeida, S.F. A Systematic Pan-Cancer Analysis of Genetic Heterogeneity Reveals Associations with Epigenetic Modifiers. Cancers 2019, 11, 391. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, A.; Dugué, P.-A.; Nøst, T.H.; Southey, M.C.; Buchanan, D.D.; Schmidt, D.F.; Makalic, E.; Hodge, A.M.; English, D.R.; Doo, N.W.; et al. Stochastic Epigenetic Mutations Are Associated with Risk of Breast Cancer, Lung Cancer, and Mature B-cell Neoplasms. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2026–2037. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Kaiser, M.F.; Heuck, C.; Melchor, L.; Wardell, C.; Murison, A.; Chavan, S.S.; Johnson, D.C.; Begum, D.B.; Dahir, N.M.; et al. The Spectrum and Clinical Impact of Epigenetic Modifier Mutations in Myeloma. Clin. Cancer Res. 2016, 22, 5783–5794. [Google Scholar] [CrossRef]
- Nagaraja, S.; Quezada, M.; Gillespie, S.M.; Arzt, M.; Lennon, J.J.; Woo, P.J.; Hovestadt, V.; Kambhampati, M.; Filbin, M.G.; Suva, M.L.; et al. Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Mol. Cell 2019, 76, 965–980.e12. [Google Scholar] [CrossRef]
- Ichise, T.; Yoshida, N.; Ichise, H. CBP/p300 antagonises EGFR-Ras-Erk signalling and suppresses increased Ras-Erk signalling-induced tumour formation in mice. J. Pathol. 2019, 249, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; DeCarolis, P.L.; Angeles, C.V.; Brenet, F.; Schultz, N.; Antonescu, C.R.; Scandura, J.; Sander, C.; Viale, A.J.; Socci, N.D.; et al. Frequent Alterations and Epigenetic Silencing of Differentiation Pathway Genes in Structurally Rearranged Liposarcomas. Cancer Discov. 2011, 1, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Fraga, M.; Ballestar, E.; Hamelin, R.; Yamamoto, H.; Boix-Chornet, M.; Caballero, R.T.; Alaminos, M.; Setien, F.; Paz, M.F.; et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 2006, 38, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Mosaab, A.; El-Ayadi, M.; Khorshed, E.N.; Amer, N.; Refaat, A.; El-Beltagy, M.; Hassan, Z.; Soror, S.H.; Zaghloul, M.S.; El-Naggar, S. Histone H3K27M Mutation Overrides Histological Grading in Pediatric Gliomas. Sci. Rep. 2020, 10, 8368. [Google Scholar] [CrossRef]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef]
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016, 7, 11316. [Google Scholar] [CrossRef]
- Orta, M.L.; Montaño, J.M.C.; Domínguez, I.; Pastor, N.; Morón, E.B.; López-Lázaro, M.; Cortés, F.; Mateos, S.; Helleday, T. 5-Aza-2′-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Res. 2013, 41, 5827–5836. [Google Scholar] [CrossRef]
- Ou, Y.; Zhang, Q.; Tang, Y.; Lu, Z.; Lu, X.; Zhou, X.; Liu, C. DNA methylation enzyme inhibitor RG108 suppresses the radioresistance of esophageal cancer. Oncol. Rep. 2018, 39, 993–1002. [Google Scholar] [CrossRef]
- Nakagawa, M.; Fujita, S.; Katsumoto, T.; Yamagata, K.; Ogawara, Y.; Hattori, A.; Kagiyama, Y.; Honma, D.; Araki, K.; Inoue, T.; et al. Dual inhibition of enhancer of zeste homolog 1/2 overactivates WNT signaling to deplete cancer stem cells in multiple myeloma. Cancer Sci. 2019, 110, 194–208. [Google Scholar] [CrossRef]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.-C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective Inhibition of EZH2 by EPZ-6438 Leads to Potent Antitumor Activity in EZH2-Mutant Non-Hodgkin Lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.M.; Tan, P.B.O.; Liu, E.T.; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.E.; Dowling, D.P.; Boone, M.A.; Phillips, A.J.; Christianson, D.W. Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.-Q.; Zhang, X.; Zhen, Y.; He, X.-Y.; Zhao, S.; Li, X.-F.; Yang, B.; Gao, F.; Guo, F.-Y.; Fu, L.; et al. A novel small-molecule activator of Sirtuin-1 induces autophagic cell death/mitophagy as a potential therapeutic strategy in glioblastoma. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Morrison-Smith, C.D.; Knox, T.M.; Filic, I.; Soroko, K.M.; Eschle, B.K.; Wilkens, M.K.; Gokhale, P.C.; Giles, F.; Griffin, A.; Brown, B.; et al. Combined Targeting of the BRD4–NUT–p300 Axis in NUT Midline Carcinoma by Dual Selective Bromodomain Inhibitor, NEO2734. Mol. Cancer Ther. 2020, 19, 1406–1414. [Google Scholar] [CrossRef]
- Wiese, M.; Hamdan, F.H.; Kubiak, K.; Diederichs, C.; Gielen, G.H.; Nussbaumer, G.; Carcaboso, A.M.; Hulleman, E.; Johnsen, S.A.; Kramm, C.M. Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Gryder, B.E.; Yohe, M.E.; Chou, H.-C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3–FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Liao, W.S.-L.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Rosner, G.L.; Yu, Y.; Ahmed, A.A.; Bast, R.C., Jr. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef]
- Engelke, C.G.; Chinnaiyan, A.M. aBETting therapeutic resistance by Wnt signaling. Cell Res. 2015, 25, 1187–1188. [Google Scholar] [CrossRef]
- Tögel, L.; Nightingale, R.; Chueh, A.; Jayachandran, A.; Tran, H.; Phesse, T.; Wu, R.; Sieber, O.; Arango, D.; Dhillon, A.S.; et al. Dual Targeting of Bromodomain and Extraterminal Domain Proteins, and WNT or MAPK Signaling, Inhibits c-MYC Expression and Proliferation of Colorectal Cancer Cells. Mol. Cancer Ther. 2016, 15, 1217–1226. [Google Scholar] [CrossRef]
- Prager, B.C.; Vasudevan, H.N.; Dixit, D.; Bernatchez, J.A.; Wu, Q.; Wallace, L.C.; Bhargava, S.; Lee, D.; King, B.H.; Morton, A.R.; et al. The Meningioma Enhancer Landscape Delineates Novel Subgroups and Drives Druggable Dependencies. Cancer Discov. 2020, 10, 1722–1741. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and Characterization of Super-Enhancer-Associated Dependencies in Diffuse Large B Cell Lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholic, N.M.; Johnston, L.D.; Klaus, C.R.; Wigle, T.J.; Iwanowicz, D.; Littlefield, B.A.; Porter-Scott, M.; Smith, J.J.; Moyer, M.P.; et al. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS ONE 2014, 9, e111840. [Google Scholar] [CrossRef] [PubMed]
- McDowell, I.C.; Barrera, A.; D’Ippolito, A.M.; Vockley, C.M.; Hong, L.K.; Leichter, S.M.; Bartelt, L.C.; Majoros, W.H.; Song, L.; Safi, A.; et al. Glucocorticoid receptor recruits to enhancers and drives activation by motif-directed binding. Genome Res. 2018, 28, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Hossan, T.; Alawi, M.; Najafova, Z.; Indenbirken, D.; Bedi, U.; Taipaleenmäki, H.; Ben-Batalla, I.; Scheller, M.; Loges, S.; et al. Bromodomain Protein BRD4 Is Required for Estrogen Receptor-Dependent Enhancer Activation and Gene Transcription. Cell Rep. 2014, 8, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Droog, M.; Nevedomskaya, E.; Dackus, G.M.; Fles, R.; Kim, Y.; Hollema, H.; Mourits, M.J.; Nederlof, P.M.; van Boven, H.H.; Linn, S.C.; et al. Estrogen receptor α wields treatment-specific enhancers between morphologically similar endometrial tumors. Proc. Natl. Acad. Sci. USA 2017, 114, E1316–E1325. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Modification | Location | Modifiers | Examples of Modifier’s Contribution to Cancer | |
|---|---|---|---|---|
| H3K27ac | Active enhancers and promoters | Writer | P300, CBP | Inactivating mutations [36,37,38], deregulated interactions [39,40], exploitation downstream of oncogenic signallings for EMT [41], expression deregulation [42,43,44] |
| Eraser | HDAC1,2 | |||
| H3K4me3 | Active promoters | Writer | MLL1, SETD1B | Deregulations downstream of oncogenic signalling pathways and EMT [45,46] |
| Eraser | KDM2B | |||
| H3K4me1 | Poised and active enhancers | Writer | MLL3/4 | Inactivating mutations [47], hijacked by oncogenic signallings [48] |
| Eraser | KDM1B | |||
| H3K27me3 | Repressed and poised CpG-rich promoters and enhancers | Writer | PRC2 | Inactivating mutation [49,50], deregulated expression [51] |
| Eraser | KDM6 | |||
| DNA methylation | CpG-enriched promoters and genebody | Writer | DNMTs | Inactivating mutation [52], expression deregulation [53] |
| Eraser | TET | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirzadeh Azad, F.; Atlasi, Y. Deregulation of Transcriptional Enhancers in Cancer. Cancers 2021, 13, 3532. https://doi.org/10.3390/cancers13143532
Mirzadeh Azad F, Atlasi Y. Deregulation of Transcriptional Enhancers in Cancer. Cancers. 2021; 13(14):3532. https://doi.org/10.3390/cancers13143532
Chicago/Turabian StyleMirzadeh Azad, Fatemeh, and Yaser Atlasi. 2021. "Deregulation of Transcriptional Enhancers in Cancer" Cancers 13, no. 14: 3532. https://doi.org/10.3390/cancers13143532
APA StyleMirzadeh Azad, F., & Atlasi, Y. (2021). Deregulation of Transcriptional Enhancers in Cancer. Cancers, 13(14), 3532. https://doi.org/10.3390/cancers13143532