
Abnormal Metabolism in the Progression of Nonalcoholic Fatty Liver Disease to Hepatocellular Carcinoma: Mechanistic Insights to Chemoprevention


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Epidemiology and Predictions
2.1. NAFLD
2.2. NAFLD-Related HCC
3. HCC and Metabolic Syndrome
3.1. Metabolic Syndrome
3.2. Obesity and Dyslipidemia
3.3. Insulin Resistance
4. Clinical Approach to NAFLD and NAFLD-Related HCC
4.1. Current Diagnostic and Treatment Strategies of NALFD
4.2. Current Prevention and Treatment Strategies of NAFLD-Related HCC
5. Therapeutic Targets in NAFLD and NAFLD-Related HCC
5.1. PPARs
5.1.1. Thiazolidinediones and PPARγ
5.1.2. Fibrates and PPARα
5.1.3. PPARδ
5.2. Bile Acids
5.3. GLP-1 Receptor Agonists
5.4. ACC Inhibitors
5.5. Statins
6. Future Perspectives
6.1. Gut Microbiome
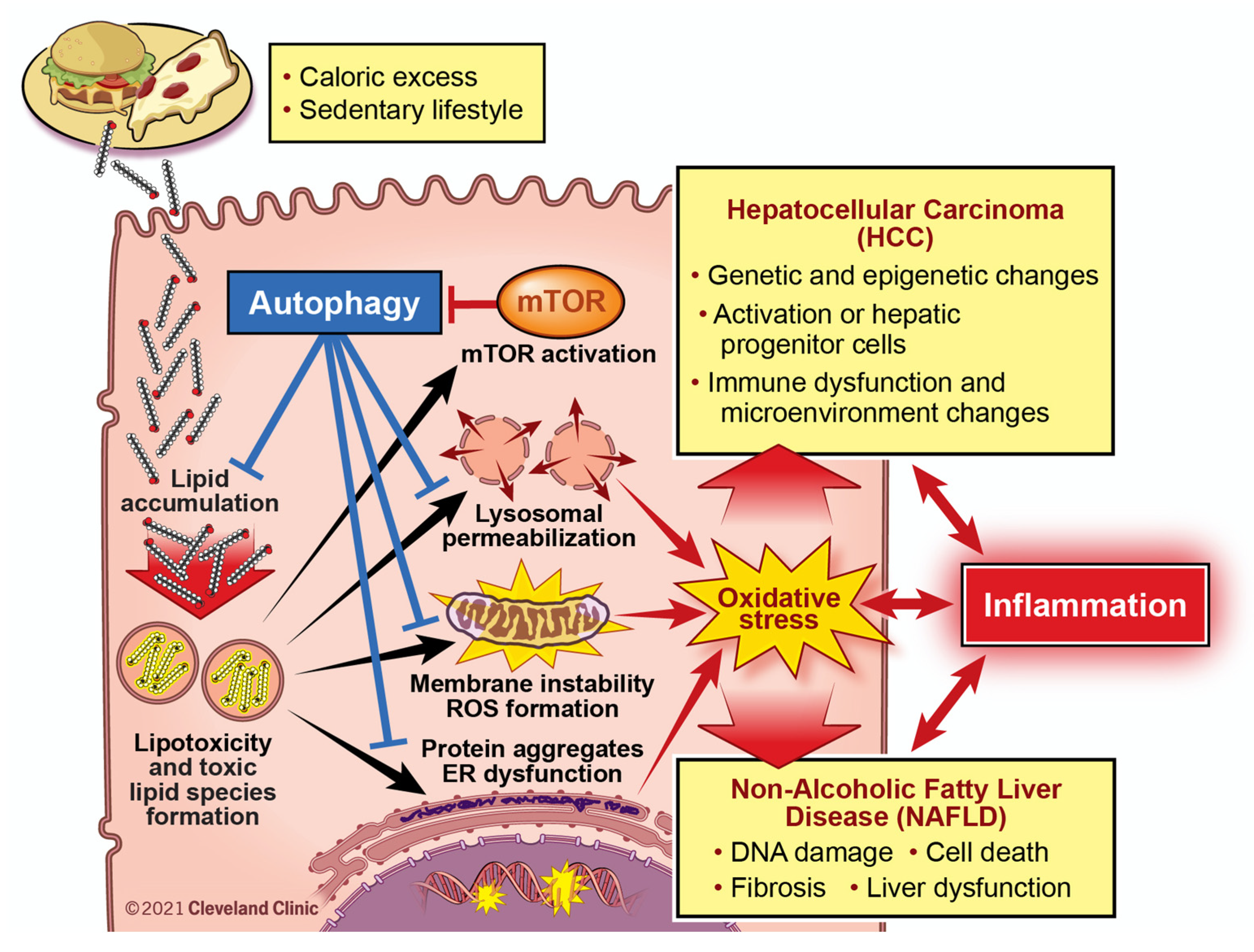
6.2. Lipotoxicity and Autophagy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Dasgupta, P.; Henshaw, C.; Youlden, D.R.; Clark, P.J.; Aitken, J.F.; Baade, P.D. Global Trends in Incidence Rates of Primary Adult Liver Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 171. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of Nonalcoholic Fatty Liver Disease (NAFLD) with Hepatocellular Carcinoma (HCC) in the United States from 2004 to 2009. Hepatol. Baltim. Md 2015, 62, 1723–1730. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2020, 1–16. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatol. Baltim. Md 2021, 73 (Suppl. 1), 4–13. [Google Scholar] [CrossRef]
- Steele, C.B. Vital Signs: Trends in Incidence of Cancers Associated with Overweight and Obesity—United States, 2005–2014. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 1052–1058. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Loomba, R.; Rinella, M.E.; Bugianesi, E.; Marchesini, G.; Neuschwander-Tetri, B.A.; Serfaty, L.; Negro, F.; Caldwell, S.H.; Ratziu, V.; et al. Current and Future Therapeutic Regimens for Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatol. Baltim. Md 2018, 68, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: An Emerging Menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelusi, S.; Baselli, G.; Pietrelli, A.; Dongiovanni, P.; Donati, B.; McCain, M.V.; Meroni, M.; Fracanzani, A.L.; Romagnoli, R.; Petta, S.; et al. Rare Pathogenic Variants Predispose to Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 3682. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. International Consensus Panel MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatol. Baltim. Md 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic Review: The Epidemiology and Natural History of Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis in Adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in NAFLD: State of the Art and Identification of Research Gaps. Hepatol. Baltim. Md 2019, 70, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Khan, M.A.; Yoo, E.R.; Cholankeril, G.; Kim, D.; Ahmed, A. Clinical Epidemiology and Disease Burden of Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2017, 23, 8263–8276. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the Prevalence of the Most Common Causes of Chronic Liver Diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2011, 9, 524–530.e1. [Google Scholar] [CrossRef]
- van der Poorten, D.; Samer, C.F.; Ramezani-Moghadam, M.; Coulter, S.; Kacevska, M.; Schrijnders, D.; Wu, L.E.; McLeod, D.; Bugianesi, E.; Komuta, M.; et al. Hepatic Fat Loss in Advanced Nonalcoholic Steatohepatitis: Are Alterations in Serum Adiponectin the Cause? Hepatol. Baltim. Md 2013, 57, 2180–2188. [Google Scholar] [CrossRef] [Green Version]
- Kogiso, T.; Tokushige, K. The Current View of Nonalcoholic Fatty Liver Disease-Related Hepatocellular Carcinoma. Cancers 2021, 13, 516. [Google Scholar] [CrossRef]
- Brar, G.; Greten, T.F.; Graubard, B.I.; McNeel, T.S.; Petrick, J.L.; McGlynn, K.A.; Altekruse, S.F. Hepatocellular Carcinoma Survival by Etiology: A SEER-Medicare Database Analysis. Hepatol. Commun. 2020, 4, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Kanwal, F. Epidemiology of Hepatocellular Carcinoma in the United States: Where Are We? Where Do We Go? Hepatol. Baltim. Md 2014, 60, 1767–1775. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic Steatohepatitis Is the Most Rapidly Growing Indication for Liver Transplantation in Patients with Hepatocellular Carcinoma in the US. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef]
- Huang, P.L. A Comprehensive Definition for Metabolic Syndrome. Dis. Model. Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yki-Järvinen, H. Non-Alcoholic Fatty Liver Disease as a Cause and a Consequence of Metabolic Syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kahn, S.E. The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [Green Version]
- Scherer, P.E. Adipose Tissue: From Lipid Storage Compartment to Endocrine Organ. Diabetes 2006, 55, 1537–1545. [Google Scholar] [CrossRef] [Green Version]
- Kusminski, C.M.; Bickel, P.E.; Scherer, P.E. Targeting Adipose Tissue in the Treatment of Obesity-Associated Diabetes. Nat. Rev. Drug Discov. 2016, 15, 639–660. [Google Scholar] [CrossRef]
- Fryar, C.; Carroll, M.; Afful, J. Prevalence of Overweight, Obesity, and Severe Obesity among Adults Aged 20 and over: United States, 1960–1962 through 2017–2018. NCHS Health E-Stats; 2020. Available online: https://www.cdc.gov/nchs/data/hestat/obesity-adult-17-18/obesity-adult.htm (accessed on 3 March 2021).
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, Regional, and National Prevalence of Overweight and Obesity in Children and Adults during 1980–2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Wang, J.; Du, J. Association between Body Mass Index and Fatty Liver Risk: A Dose-Response Analysis. Sci. Rep. 2018, 8, 15273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Liu, D.-W.; Yan, H.-Y.; Wang, Z.-Y.; Zhao, S.-H.; Wang, B. Obesity Is an Independent Risk Factor for Non-Alcoholic Fatty Liver Disease: Evidence from a Meta-Analysis of 21 Cohort Studies. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2016, 17, 510–519. [Google Scholar] [CrossRef]
- Koh, J.C.; Loo, W.M.; Goh, K.L.; Sugano, K.; Chan, W.K.; Chiu, W.Y.P.; Choi, M.-G.; Gonlachanvit, S.; Lee, W.-J.; Lee, W.J.J.; et al. Asian Consensus on the Relationship between Obesity and Gastrointestinal and Liver Diseases. J. Gastroenterol. Hepatol. 2016, 31, 1405–1413. [Google Scholar] [CrossRef] [Green Version]
- Welzel, T.M.; Graubard, B.I.; Zeuzem, S.; El-Serag, H.B.; Davila, J.A.; McGlynn, K.A. Metabolic Syndrome Increases the Risk of Primary Liver Cancer in the United States: A Population-Based Case-Control Study. Hepatol. Baltim. Md 2011, 54, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, S.C.; Wolk, A. Overweight, Obesity and Risk of Liver Cancer: A Meta-Analysis of Cohort Studies. Br. J. Cancer 2007, 97, 1005–1008. [Google Scholar] [CrossRef]
- Chen, C.-L.; Yang, H.-I.; Yang, W.-S.; Liu, C.-J.; Chen, P.-J.; You, S.-L.; Wang, L.-Y.; Sun, C.-A.; Lu, S.-N.; Chen, D.-S.; et al. Metabolic Factors and Risk of Hepatocellular Carcinoma by Chronic Hepatitis B/C Infection: A Follow-up Study in Taiwan. Gastroenterology 2008, 135, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Ohki, T.; Tateishi, R.; Sato, T.; Masuzaki, R.; Imamura, J.; Goto, T.; Yamashiki, N.; Yoshida, H.; Kanai, F.; Kato, N.; et al. Obesity Is an Independent Risk Factor for Hepatocellular Carcinoma Development in Chronic Hepatitis C Patients. Clin. Gastroenterol. Hepatol. 2008, 6, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Yang, H.-I.; Su, J.; Brenner, D.; Barrett-Connor, E.; Iloeje, U.; Chen, C.-J. Synergism between Obesity and Alcohol in Increasing the Risk of Hepatocellular Carcinoma: A Prospective Cohort Study. Am. J. Epidemiol. 2013, 177, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Yoo, S.H.; Sohn, W.; Kim, H.W.; Choi, Y.S.; Won, J.H.; Heo, J.Y.; Park, S.J.; Park, Y.M. Obesity and Hepatocellular Carcinoma in Patients Receiving Entecavir for Chronic Hepatitis B. Clin. Mol. Hepatol. 2016, 22, 339–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, B.; Yu, J.H.; Jin, Y.-J.; Suh, Y.J.; Lee, J.-W. Survival Outcomes According to Body Mass Index in Hepatocellular Carcinoma Patient: Analysis of Nationwide Cancer Registry Database. Sci. Rep. 2020, 10, 8347. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-Induced Gut Microbial Metabolite Promotes Liver Cancer through Senescence Secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Zhang, H.E.; Henderson, J.M.; Gorrell, M.D. Animal Models for Hepatocellular Carcinoma. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 993–1002. [Google Scholar] [CrossRef]
- Duan, X.-F.; Tang, P.; Li, Q.; Yu, Z.-T. Obesity, Adipokines and Hepatocellular Carcinoma. Int. J. Cancer 2013, 133, 1776–1783. [Google Scholar] [CrossRef] [Green Version]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-Alcoholic Fatty Liver Disease and Dyslipidemia: An Update. Metab. Clin. Exp. 2016, 65, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-T.; Kuo, P.-L.; Su, S.-B.; Chen, Y.-Y.; Yeh, M.-L.; Huang, C.-I.; Yang, J.-F.; Lin, C.-I.; Hsieh, M.-H.; Hsieh, M.-Y.; et al. Nonalcoholic Fatty Liver Disease Severity Is Associated with the Ratios of Total Cholesterol and Triglycerides to High-Density Lipoprotein Cholesterol. J. Clin. Lipidol. 2016, 10, 420–425.e1. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Wang, X.; Dang, Z.; Jiang, Y.; Wang, X.; Yang, Z. Effect of Serum Triglyceride Level on the Prognosis of Patients with Hepatocellular Carcinoma in the Absence of Cirrhosis. Lipids Health Dis. 2018, 17, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.-H.; Lee, L.-T.; Hung, S.-H.; Lin, W.-Y.; Hung, H.-F.; Yang, W.-S.; Sung, P.-K.; Huang, K.-C. Opposite Association between Diabetes, Dyslipidemia, and Hepatocellular Carcinoma Mortality in the Middle-Aged and Elderly. Hepatology 2014, 59, 2207–2215. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; Newgard, C.B. Molecular and Metabolic Mechanisms of Insulin Resistance and β-Cell Failure in Type 2 Diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatol. Baltim. Md 2019, 70, 711–724. [Google Scholar] [CrossRef]
- Hassan, M.M.; Curley, S.A.; Li, D.; Kaseb, A.; Davila, M.; Abdalla, E.K.; Javle, M.; Moghazy, D.M.; Lozano, R.D.; Abbruzzese, J.L.; et al. Association of Diabetes Duration and Diabetes Treatment with the Risk of Hepatocellular Carcinoma. Cancer 2010, 116, 1938–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davila, J.A.; Morgan, R.O.; Shaib, Y.; McGlynn, K.A.; El-Serag, H.B. Diabetes Increases the Risk of Hepatocellular Carcinoma in the United States: A Population Based Case Control Study. Gut 2005, 54, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Kang, D.; Cao, W.; Wang, Y.; Liu, Z. Diabetes Mellitus and Risk of Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis. Diabetes Metab. Res. Rev. 2012, 28, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; Enjoji, M.; et al. SREBP-1c, Regulated by the Insulin and AMPK Signaling Pathways, Plays a Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, Y.; Kubota, N.; Takamoto, I.; Obata, A.; Iwamoto, M.; Hayashi, T.; Aihara, M.; Kubota, T.; Nishihara, H.; Kadowaki, T. Role of Insulin Receptor Substrates in the Progression of Hepatocellular Carcinoma. Sci. Rep. 2017, 7, 5387. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yang, W.; Zhang, J.; Zheng, X.; Yao, Y.; Tu, K.; Liu, Q. SREBP-1 Has a Prognostic Role and Contributes to Invasion and Metastasis in Human Hepatocellular Carcinoma. Int. J. Mol. Sci. 2014, 15, 7124–7138. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-Term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e10. [Google Scholar] [CrossRef] [Green Version]
- Argo, C.K.; Northup, P.G.; Al-Osaimi, A.M.S.; Caldwell, S.H. Systematic Review of Risk Factors for Fibrosis Progression in Non-Alcoholic Steatohepatitis. J. Hepatol. 2009, 51, 371–379. [Google Scholar] [CrossRef]
- Eguchi, Y.; Wong, G.; Akhtar, O.; Sumida, Y. Non-Invasive Diagnosis of Non-Alcoholic Steatohepatitis and Advanced Fibrosis in Japan: A Targeted Literature Review. Hepatol. Res. 2020, 50, 645–655. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of Current Treatments on Liver Disease, Glucose Metabolism and Cardiovascular Risk in Non-Alcoholic Fatty Liver Disease (NAFLD): A Systematic Review and Meta-Analysis of Randomised Trials. Diabetologia 2012, 55, 885–904. [Google Scholar] [CrossRef]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized Controlled Trial Testing the Effects of Weight Loss on Nonalcoholic Steatohepatitis. Hepatol. Baltim. Md 2010, 51, 121–129. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5, quiz e14–15. [Google Scholar] [CrossRef]
- Keating, S.E.; Hackett, D.A.; George, J.; Johnson, N.A. Exercise and Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. J. Hepatol. 2012, 57, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Murag, S.; Cholankeril, G.; Cheung, A.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Physical Activity, Measured Objectively, Is Associated With Lower Mortality in Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Njei, B.; McCarty, T.R.; Sharma, P.; Lange, A.; Najafian, N.; Ngu, J.N.; Ngomba, V.E.; Echouffo-Tcheugui, J.B. Bariatric Surgery and Hepatocellular Carcinoma: A Propensity Score-Matched Analysis. Obes. Surg. 2018, 28, 3880–3889. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Henson, J.; Osganian, S.; Masia, R.; Chan, A.T.; Chung, R.T.; Corey, K.E. Daily Aspirin Use Associated With Reduced Risk For Fibrosis Progression In Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019, 17, 2776–2784.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.J.; Abdelmalek, M.F.; Suzuki, A.; Cummings, O.W.; Chojkier, M. EPE-A Study Group No Significant Effects of Ethyl-Eicosapentanoic Acid on Histologic Features of Nonalcoholic Steatohepatitis in a Phase 2 Trial. Gastroenterology 2014, 147, 377–384.e1. [Google Scholar] [CrossRef]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases: Marrero et al. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans Is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [Green Version]
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; Bellentani, S. HCC-NAFLD Italian Study Group Clinical Patterns of Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease: A Multicenter Prospective Study. Hepatol. Baltim. Md 2016, 63, 827–838. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between Nonalcoholic Fatty Liver Disease and Risk for Hepatocellular Cancer, Based on Systematic Review. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2012, 10, 1342–1359.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.R.; Nguyen, M.H.; Lim, J.K. Hepatocellular Carcinoma in Patients with Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2016, 22, 8294–8303. [Google Scholar] [CrossRef]
- Liss, K.H.H.; Finck, B.N. PPARs and Nonalcoholic Fatty Liver Disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Tailleux, A.; Wouters, K.; Staels, B. Roles of PPARs in NAFLD: Potential Therapeutic Targets. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2012, 1821, 809–818. [Google Scholar] [CrossRef]
- American Diabetes Association 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2021. Diabetes Care 2021, 44, S111–S124. [Google Scholar] [CrossRef]
- Skat-Rørdam, J.; Højland Ipsen, D.; Lykkesfeldt, J.; Tveden-Nyborg, P. A Role of Peroxisome Proliferator-Activated Receptor γ in Non-Alcoholic Fatty Liver Disease. Basic Clin. Pharmacol. Toxicol. 2019, 124, 528–537. [Google Scholar] [CrossRef]
- Yki-Järvinen, H. Thiazolidinediones. N. Engl. J. Med. 2004, 351, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, E.; Csako, G.; Pucino, F.; Wesley, R.; Loomba, R. Meta-Analysis: Pioglitazone Improves Liver Histology and Fibrosis in Patients with Non-Alcoholic Steatohepatitis. Aliment. Pharmacol. Ther. 2012, 35, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.-W.; Lin, C.-L.; Liao, K.-F. Association of Hepatocellular Carcinoma with Thiazolidinediones Use. Medicine 2020, 99, e19833. [Google Scholar] [CrossRef]
- Chang, C.-H.; Lin, J.-W.; Wu, L.-C.; Lai, M.-S.; Chuang, L.-M.; Chan, K.A. Association of Thiazolidinediones with Liver Cancer and Colorectal Cancer in Type 2 Diabetes Mellitus. Hepatology 2012, 55, 1462–1472. [Google Scholar] [CrossRef]
- Lin, H.C.; Hsu, Y.T.; Kachingwe, B.H.; Hsu, C.Y.; Uang, Y.S.; Wang, L.H. Dose Effect of Thiazolidinedione on Cancer Risk in Type 2 Diabetes Mellitus Patients: A Six-Year Population-Based Cohort Study. J. Clin. Pharm. Ther. 2014, 39, 354–360. [Google Scholar] [CrossRef]
- Huang, M.-Y.; Chung, C.-H.; Chang, W.-K.; Lin, C.-S.; Chen, K.-W.; Hsieh, T.-Y.; Chien, W.-C.; Lin, H.-H. The Role of Thiazolidinediones in Hepatocellular Carcinoma Risk Reduction: A Population-Based Cohort Study in Taiwan. Am. J. Cancer Res. 2017, 7, 1606–1616. [Google Scholar]
- Lai, S.-W.; Chen, P.-C.; Liao, K.-F.; Muo, C.-H.; Lin, C.-C.; Sung, F.-C. Risk of Hepatocellular Carcinoma in Diabetic Patients and Risk Reduction Associated with Anti-Diabetic Therapy: A Population-Based Cohort Study. Am. J. Gastroenterol. 2012, 107, 46–52. [Google Scholar] [CrossRef]
- Yip, T.C.-F.; Wong, V.W.-S.; Chan, H.L.-Y.; Tse, Y.-K.; Hui, V.W.-K.; Liang, L.Y.; Lee, H.W.; Lui, G.C.-Y.; Kong, A.P.-S.; Wong, G.L.-H. Thiazolidinediones Reduce the Risk of Hepatocellular Carcinoma and Hepatic Events in Diabetic Patients with Chronic Hepatitis B. J. Viral Hepat. 2020, 27, 904–914. [Google Scholar] [CrossRef]
- Blanquicett, C.; Roman, J.; Hart, C.M. Thiazolidinediones as Anti-Cancer Agents. Cancer Ther. 2008, 6, 25–34. [Google Scholar]
- Li, S.; Ghoshal, S.; Sojoodi, M.; Arora, G.; Masia, R.; Erstad, D.J.; Lanuti, M.; Hoshida, Y.; Baumert, T.F.; Tanabe, K.K.; et al. Pioglitazone Reduces Hepatocellular Carcinoma Development in Two Rodent Models of Cirrhosis. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2019, 23, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-W.; Farrell, G.C.; Yu, J. Functional Role of Peroxisome-Proliferator-Activated Receptor γ in Hepatocellular Carcinoma. J. Gastroenterol. Hepatol. 2012, 27, 1665–1669. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-T.; Chi, C.-W. Emerging Role of the Peroxisome Proliferator-Activated Receptor-Gamma in Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2014, 1, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Chu, E.S.H.; Zhao, G.; Man, K.; Wu, C.-W.; Cheng, J.T.Y.; Li, G.; Nie, Y.; Lo, C.M.; Teoh, N.; et al. PPARgamma Inhibits Hepatocellular Carcinoma Metastases in Vitro and in Mice. Br. J. Cancer 2012, 106, 1486–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palakurthi, S.S.; Aktas, H.; Grubissich, L.M.; Mortensen, R.M.; Halperin, J.A. Anticancer Effects of Thiazolidinediones Are Independent of Peroxisome Proliferator-Activated Receptor Gamma and Mediated by Inhibition of Translation Initiation. Cancer Res. 2001, 61, 6213–6218. [Google Scholar] [PubMed]
- Feinstein, D.L.; Spagnolo, A.; Akar, C.; Weinberg, G.; Murphy, P.; Gavrilyuk, V.; Dello Russo, C. Receptor-Independent Actions of PPAR Thiazolidinedione Agonists: Is Mitochondrial Function the Key? Biochem. Pharmacol. 2005, 70, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Auboeuf, D.; Rieusset, J.; Fajas, L.; Vallier, P.; Frering, V.; Riou, J.P.; Staels, B.; Auwerx, J.; Laville, M.; Vidal, H. Tissue Distribution and Quantification of the Expression of MRNAs of Peroxisome Proliferator–Activated Receptors and Liver X Receptor-α in Humans: No Alteration in Adipose Tissue of Obese and NIDDM Patients. Diabetes 1997, 46, 1319–1327. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential Expression of Peroxisome Proliferator-Activated Receptors (PPARs): Tissue Distribution of PPAR-Alpha, -Beta, and -Gamma in the Adult Rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.M.; Hennuyer, N.; Staels, B.; Fruchart, J.C.; Fievet, C.; Gonzalez, F.J.; Auwerx, J. Alterations in Lipoprotein Metabolism in Peroxisome Proliferator-Activated Receptor Alpha-Deficient Mice. J. Biol. Chem. 1997, 272, 27307–27312. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome Proliferator-Activated Receptor Alpha Mediates the Adaptive Response to Fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.M.; Doyle, P.J.; Iglesias, M.A.; Watson, D.G.; Cooney, G.J.; Kraegen, E.W. Peroxisome Proliferator-Activated Receptor (PPAR)-Alpha Activation Lowers Muscle Lipids and Improves Insulin Sensitivity in High Fat-Fed Rats: Comparison with PPAR-Gamma Activation. Diabetes 2001, 50, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Stec, D.E.; Gordon, D.M.; Hipp, J.A.; Hong, S.; Mitchell, Z.L.; Franco, N.R.; Robison, J.W.; Anderson, C.D.; Stec, D.F.; Hinds, T.D. Loss of Hepatic PPARα Promotes Inflammation and Serum Hyperlipidemia in Diet-Induced Obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R733–R745. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular Mechanism of PPARα Action and Its Impact on Lipid Metabolism, Inflammation and Fibrosis in Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.J.; Haluzik, M.; Gregory, C.; Dietz, K.R.; Vinson, C.; Gavrilova, O.; Reitman, M.L. WY14,643, a Peroxisome Proliferator-Activated Receptor Alpha (PPARalpha ) Agonist, Improves Hepatic and Muscle Steatosis and Reverses Insulin Resistance in Lipoatrophic A-ZIP/F-1 Mice. J. Biol. Chem. 2002, 277, 24484–24489. [Google Scholar] [CrossRef] [Green Version]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of Long-Term Fenofibrate Therapy on Cardiovascular Events in 9795 People with Type 2 Diabetes Mellitus (the FIELD Study): Randomised Controlled Trial. Lancet Lond. Engl. 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Frick, M.H.; Elo, O.; Haapa, K.; Heinonen, O.P.; Heinsalmi, P.; Helo, P.; Huttunen, J.K.; Kaitaniemi, P.; Koskinen, P.; Manninen, V. Helsinki Heart Study: Primary-Prevention Trial with Gemfibrozil in Middle-Aged Men with Dyslipidemia. Safety of Treatment, Changes in Risk Factors, and Incidence of Coronary Heart Disease. N. Engl. J. Med. 1987, 317, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Balfour, J.A.; McTavish, D.; Heel, R.C. Fenofibrate. A Review of Its Pharmacodynamic and Pharmacokinetic Properties and Therapeutic Use in Dyslipidaemia. Drugs 1990, 40, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.; Odin, J.; Hayashi, P.H.; Chalasani, N.; Fontana, R.J.; Barnhart, H.; Cirulli, E.T.; Kleiner, D.E.; Hoofnagle, J.H. Identification and Characterization of Fenofibrate-Induced Liver Injury. Dig. Dis. Sci. 2017, 62, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.A. Myopathy with Statin–Fibrate Combination Therapy: Clinical Considerations. Nat. Rev. Endocrinol. 2009, 5, 507–518. [Google Scholar] [CrossRef]
- Fernández-Miranda, C.; Pérez-Carreras, M.; Colina, F.; López-Alonso, G.; Vargas, C.; Solís-Herruzo, J.A. A Pilot Trial of Fenofibrate for the Treatment of Non-Alcoholic Fatty Liver Disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef]
- Bajaj, M.; Suraamornkul, S.; Hardies, L.J.; Glass, L.; Musi, N.; DeFronzo, R.A. Effects of Peroxisome Proliferator-Activated Receptor (PPAR)-Alpha and PPAR-Gamma Agonists on Glucose and Lipid Metabolism in Patients with Type 2 Diabetes Mellitus. Diabetologia 2007, 50, 1723–1731. [Google Scholar] [CrossRef] [Green Version]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A Controlled Trial of Gemfibrozil in the Treatment of Patients with Nonalcoholic Steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef]
- Athyros, V.G.; Mikhailidis, D.P.; Didangelos, T.P.; Giouleme, O.I.; Liberopoulos, E.N.; Karagiannis, A.; Kakafika, A.I.; Tziomalos, K.; Burroughs, A.K.; Elisaf, M.S. Effect of Multifactorial Treatment on Non-Alcoholic Fatty Liver Disease in Metabolic Syndrome: A Randomised Study. Curr. Med. Res. Opin. 2006, 22, 873–883. [Google Scholar] [CrossRef]
- Laurin, J.; Lindor, K.D.; Crippin, J.S.; Gossard, A.; Gores, G.J.; Ludwig, J.; Rakela, J.; McGill, D.B. Ursodeoxycholic Acid or Clofibrate in the Treatment of Non-Alcohol-Induced Steatohepatitis: A Pilot Study. Hepatol. Baltim. Md 1996, 23, 1464–1467. [Google Scholar] [CrossRef]
- Ishibashi, S.; Yamashita, S.; Arai, H.; Araki, E.; Yokote, K.; Suganami, H.; Fruchart, J.-C.; Kodama, T. Effects of K-877, a Novel Selective PPARα Modulator (SPPARMα), in Dyslipidaemic Patients: A Randomized, Double Blind, Active- and Placebo-Controlled, Phase 2 Trial. Atherosclerosis 2016, 249, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; Takizawa, T.; Saito, S.; Nagashima, Y.; et al. Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor Alpha Modulator, Improves the Pathogenesis in a Rodent Model of Nonalcoholic Steatohepatitis. Sci. Rep. 2017, 7, 42477. [Google Scholar] [CrossRef]
- Sasaki, Y.; Asahiyama, M.; Tanaka, T.; Yamamoto, S.; Murakami, K.; Kamiya, W.; Matsumura, Y.; Osawa, T.; Anai, M.; Fruchart, J.-C.; et al. Pemafibrate, a Selective PPARα Modulator, Prevents Non-Alcoholic Steatohepatitis Development without Reducing the Hepatic Triglyceride Content. Sci. Rep. 2020, 10, 7818. [Google Scholar] [CrossRef]
- Yamashita, S.; Arai, H.; Yokote, K.; Araki, E.; Matsushita, M.; Nojima, T.; Suganami, H.; Ishibashi, S. Efficacy and Safety of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator (SPPARMα): Pooled Analysis of Phase 2 and 3 Studies in Dyslipidemic Patients with or without Statin Combination. Int. J. Mol. Sci. 2019, 20, 5537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, E.; Yamashita, S.; Arai, H.; Yokote, K.; Satoh, J.; Inoguchi, T.; Nakamura, J.; Maegawa, H.; Yoshioka, N.; Tanizawa, Y.; et al. Effects of Pemafibrate, a Novel Selective PPARα Modulator, on Lipid and Glucose Metabolism in Patients With Type 2 Diabetes and Hypertriglyceridemia: A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial. Diabetes Care 2018, 41, 538–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatanaka, T.; Kakizaki, S.; Saito, N.; Nakano, Y.; Nakano, S.; Hazama, Y.; Yoshida, S.; Hachisu, Y.; Tanaka, Y.; Kashiwabara, K.; et al. Impact of Pemafibrate in Patients with Hypertriglyceridemia and Metabolic Dysfunction-Associated Fatty Liver Disease Pathologically Diagnosed with Non-Alcoholic Steatohepatitis: A Retrospective, Single-Arm Study. Intern. Med. Tokyo Jpn. 2021. [Google Scholar] [CrossRef]
- Holden, P.R.; Tugwood, J.D. Peroxisome Proliferator-Activated Receptor Alpha: Role in Rodent Liver Cancer and Species Differences. J. Mol. Endocrinol. 1999, 22, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.S.; Reddy, J.K. An Overview of Peroxisome Proliferator-Induced Hepatocarcinogenesis. Environ. Health Perspect. 1991, 93, 205–209. [Google Scholar] [CrossRef]
- Rao, M.S.; Reddy, J.K. Hepatocarcinogenesis of Peroxisome Proliferators. Ann. N. Y. Acad. Sci. 1996, 804, 573–587. [Google Scholar] [CrossRef]
- Bonovas, S.; Nikolopoulos, G.K.; Bagos, P.G. Use of Fibrates and Cancer Risk: A Systematic Review and Meta-Analysis of 17 Long-Term Randomized Placebo-Controlled Trials. PLoS ONE 2012, 7, e45259. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.C.; Cunningham, M.L.; Hummer, B.T.; Lau, C.; Meek, B.; Peters, J.M.; Popp, J.A.; Rhomberg, L.; Seed, J.; Klaunig, J.E. Mode of Action Framework Analysis for Receptor-Mediated Toxicity: The Peroxisome Proliferator-Activated Receptor Alpha (PPARα) as a Case Study. Crit. Rev. Toxicol. 2014, 44, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.C.; Peters, J.M.; Klaunig, J.E. The PPARα-Dependent Rodent Liver Tumor Response Is Not Relevant to Humans: Addressing Misconceptions. Arch. Toxicol. 2018, 92, 83–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Chu, E.S.H.; Zhang, J.; Li, X.; Liang, Q.; Chen, J.; Chen, M.; Teoh, N.; Farrell, G.; Sung, J.J.Y.; et al. Peroxisome Proliferator Activated Receptor Alpha Inhibits Hepatocarcinogenesis through Mediating NF-ΚB Signaling Pathway. Oncotarget 2014, 5, 8330–8340. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, D.; Kawabe, N.; Nakamura, H.; Tachibana, K.; Ishimoto, K.; Tanaka, T.; Aburatani, H.; Sakai, J.; Hamakubo, T.; Kodama, T.; et al. Fenofibrate Suppresses Growth of the Human Hepatocellular Carcinoma Cell via PPARα-Independent Mechanisms. Eur. J. Cell Biol. 2011, 90, 657–664. [Google Scholar] [CrossRef]
- Jiao, H.; Zhao, B. Cytotoxic Effect of Peroxisome Proliferator Fenofibrate on Human HepG2 Hepatoma Cell Line and Relevant Mechanisms. Toxicol. Appl. Pharmacol. 2002, 185, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Muzio, G.; Maggiora, M.; Oraldi, M.; Trombetta, A.; Canuto, R.A. PPARalpha and PP2A Are Involved in the Proapoptotic Effect of Conjugated Linoleic Acid on Human Hepatoma Cell Line SK-HEP-1. Int. J. Cancer 2007, 121, 2395–2401. [Google Scholar] [CrossRef]
- You, B.-J.; Hour, M.-J.; Chen, L.-Y.; Luo, S.-C.; Hsu, P.-H.; Lee, H.-Z. Fenofibrate Induces Human Hepatoma Hep3B Cells Apoptosis and Necroptosis through Inhibition of Thioesterase Domain of Fatty Acid Synthase. Sci. Rep. 2019, 9, 3306. [Google Scholar] [CrossRef] [Green Version]
- Girroir, E.E.; Hollingshead, H.E.; He, P.; Zhu, B.; Perdew, G.H.; Peters, J.M. Quantitative Expression Patterns of Peroxisome Proliferator-Activated Receptor-β/δ (PPARβ/δ) Protein in Mice. Biochem. Biophys. Res. Commun. 2008, 371, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandard, S.; Patsouris, D. Nuclear Control of the Inflammatory Response in Mammals by Peroxisome Proliferator-Activated Receptors. Available online: https://www.hindawi.com/journals/ppar/2013/613864/ (accessed on 3 March 2021).
- Escher, P.; Braissant, O.; Basu-Modak, S.; Michalik, L.; Wahli, W.; Desvergne, B. Rat PPARs: Quantitative Analysis in Adult Rat Tissues and Regulation in Fasting and Refeeding. Endocrinology 2001, 142, 4195–4202. [Google Scholar] [CrossRef]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome Proliferator-Activated Receptor Delta Controls Muscle Development and Oxidative Capability. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 2299–2301. [Google Scholar] [CrossRef]
- Gan, Z.; Burkart-Hartman, E.M.; Han, D.-H.; Finck, B.; Leone, T.C.; Smith, E.Y.; Ayala, J.E.; Holloszy, J.; Kelly, D.P. The Nuclear Receptor PPARβ/δ Programs Muscle Glucose Metabolism in Cooperation with AMPK and MEF2. Genes Dev. 2011, 25, 2619–2630. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of Peroxisome Proliferator-Activated Receptor Delta Induces Fatty Acid Beta-Oxidation in Skeletal Muscle and Attenuates Metabolic Syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [Green Version]
- Palomer, X.; Barroso, E.; Pizarro-Delgado, J.; Peña, L.; Botteri, G.; Zarei, M.; Aguilar, D.; Montori-Grau, M.; Vázquez-Carrera, M. PPARβ/δ: A Key Therapeutic Target in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, L.S.; Lione, L. Harnessing the Benefits of PPARβ/δ Agonists. Life Sci. 2013, 93, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.J.; Pearce, G.L.; Jones, N.P.; Sprecher, D.L. Lipid Effects of Peroxisome Proliferator-Activated Receptor-δ Agonist GW501516 in Subjects with Low High-Density Lipoprotein Cholesterol: Characteristics of Metabolic Syndrome. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2289–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.Y.; Choi, R.; Kim, H.M.; Cho, E.J.; Kim, B.H.; Choi, Y.S.; Naowaboot, J.; Lee, E.Y.; Yang, Y.C.; Shin, J.Y.; et al. Peroxisome Proliferator-Activated Receptor δ Agonist Attenuates Hepatic Steatosis by Anti-Inflammatory Mechanism. Exp. Mol. Med. 2012, 44, 578–585. [Google Scholar] [CrossRef]
- Nagasawa, T.; Inada, Y.; Nakano, S.; Tamura, T.; Takahashi, T.; Maruyama, K.; Yamazaki, Y.; Kuroda, J.; Shibata, N. Effects of Bezafibrate, PPAR Pan-Agonist, and GW501516, PPARdelta Agonist, on Development of Steatohepatitis in Mice Fed a Methionine- and Choline-Deficient Diet. Eur. J. Pharmacol. 2006, 536, 182–191. [Google Scholar] [CrossRef]
- Lee, C.-H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.-W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARdelta Regulates Glucose Metabolism and Insulin Sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [Green Version]
- Tong, L.; Wang, L.; Yao, S.; Jin, L.; Yang, J.; Zhang, Y.; Ning, G.; Zhang, Z. PPARδ Attenuates Hepatic Steatosis through Autophagy-Mediated Fatty Acid Oxidation. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Bays, H.E.; Schwartz, S.; Littlejohn, T.; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.-J.; Wang, X.; Naim, S.; Roberts, B.K. MBX-8025, a Novel Peroxisome Proliferator Receptor-Delta Agonist: Lipid and Other Metabolic Effects in Dyslipidemic Overweight Patients Treated with and without Atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef] [Green Version]
- Risérus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of Peroxisome Proliferator-Activated Receptor (PPAR)Delta Promotes Reversal of Multiple Metabolic Abnormalities, Reduces Oxidative Stress, and Increases Fatty Acid Oxidation in Moderately Obese Men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.A.; Wang, D.; Katkuri, S.; Wang, H.; Dey, S.K.; DuBois, R.N. Activation of Nuclear Hormone Receptor Peroxisome Proliferator-Activated Receptor-Delta Accelerates Intestinal Adenoma Growth. Nat. Med. 2004, 10, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Gonzalez, F.J.; Müller, R. Establishing the Role of PPARβ/δ in Carcinogenesis. Trends Endocrinol. Metab. 2015, 26, 595–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barroso, E.; Rodríguez-Calvo, R.; Serrano-Marco, L.; Astudillo, A.M.; Balsinde, J.; Palomer, X.; Vázquez-Carrera, M. The PPARβ/δ Activator GW501516 Prevents the down-Regulation of AMPK Caused by a High-Fat Diet in Liver and Amplifies the PGC-1α-Lipin 1-PPARα Pathway Leading to Increased Fatty Acid Oxidation. Endocrinology 2011, 152, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Cariou, B.; Zaïr, Y.; Staels, B.; Bruckert, E. Effects of the New Dual PPARα/δ Agonist GFT505 on Lipid and Glucose Homeostasis in Abdominally Obese Patients With Combined Dyslipidemia or Impaired Glucose Metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [Green Version]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zaïr, Y.; Sauvinet, V.; Noël, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual Peroxisome Proliferator-Activated Receptor α/δ Agonist GFT505 Improves Hepatic and Peripheral Insulin Sensitivity in Abdominally Obese Subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective Effects of the Dual Peroxisome Proliferator-Activated Receptor Alpha/Delta Agonist, GFT505, in Rodent Models of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Hepatol. Baltim. Md 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Liu, C.; Zhao, M.; Zhang, Q.; Lu, Y.; Liu, P.; Yang, H.; Yang, J.; Chen, X.; Yao, Y. The Pharmacodynamic and Differential Gene Expression Analysis of PPAR α/δ Agonist GFT505 in CDAHFD-Induced NASH Model. PLoS ONE 2020, 15, e0243911. [Google Scholar] [CrossRef]
- Tølbøl, K.S.; Kristiansen, M.N.; Hansen, H.H.; Veidal, S.S.; Rigbolt, K.T.; Gillum, M.P.; Jelsing, J.; Vrang, N.; Feigh, M. Metabolic and Hepatic Effects of Liraglutide, Obeticholic Acid and Elafibranor in Diet-Induced Obese Mouse Models of Biopsy-Confirmed Nonalcoholic Steatohepatitis. World J. Gastroenterol. 2018, 24, 179–194. [Google Scholar] [CrossRef]
- Shen, B.; Li, A.; Wan, Y.-J.Y.; Shen, G.; Zhu, J.; Nie, Y. Lack of PPARβ/δ-Inactivated SGK-1 Is Implicated in Liver Carcinogenesis. BioMed Res. Int. 2020, 2020, 9563851. [Google Scholar] [CrossRef]
- Balandaram, G.; Kramer, L.R.; Kang, B.-H.; Murray, I.A.; Perdew, G.H.; Gonzalez, F.J.; Peters, J.M. Ligand Activation of Peroxisome Proliferator-Activated Receptor-β/δ Suppresses Liver Tumorigenesis in Hepatitis B Transgenic Mice. Toxicology 2016, 363–364, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef] [PubMed]
- Grzych, G.; Chávez-Talavera, O.; Descat, A.; Thuillier, D.; Verrijken, A.; Kouach, M.; Legry, V.; Verkindt, H.; Raverdy, V.; Legendre, B.; et al. NASH-Related Increases in Plasma Bile Acid Levels Depend on Insulin Resistance. JHEP Rep. 2021, 3, 100222. [Google Scholar] [CrossRef]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [Green Version]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile Acids and Nonalcoholic Fatty Liver Disease: Molecular Insights and Therapeutic Perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed Hepatic Bile Acid Signalling despite Elevated Production of Primary and Secondary Bile Acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef]
- Gottlieb, A.; Canbay, A. Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression. Cells 2019, 8, 1358. [Google Scholar] [CrossRef] [PubMed]
- Okushin, K.; Tsutsumi, T.; Enooku, K.; Fujinaga, H.; Kado, A.; Shibahara, J.; Fukayama, M.; Moriya, K.; Yotsuyanagi, H.; Koike, K. The Intrahepatic Expression Levels of Bile Acid Transporters Are Inversely Correlated with the Histological Progression of Nonalcoholic Fatty Liver Disease. J. Gastroenterol. 2016, 51, 808–818. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous Bile Acids Are Ligands for the Nuclear Receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling in Liver Disease and Therapy. Liver Res. 2017, 1, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the Nuclear Receptor FXR Improves Hyperglycemia and Hyperlipidemia in Diabetic Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wang, J.; Liu, Q.; Harnish, D.C. Farnesoid X Receptor Agonist WAY-362450 Attenuates Liver Inflammation and Fibrosis in Murine Model of Non-Alcoholic Steatohepatitis. J. Hepatol. 2009, 51, 380–388. [Google Scholar] [CrossRef]
- Cariou, B.; van Harmelen, K.; Duran-Sandoval, D.; van Dijk, T.H.; Grefhorst, A.; Abdelkarim, M.; Caron, S.; Torpier, G.; Fruchart, J.-C.; Gonzalez, F.J.; et al. The Farnesoid X Receptor Modulates Adiposity and Peripheral Insulin Sensitivity in Mice. J. Biol. Chem. 2006, 281, 11039–11049. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Fu, X.; Van Ness, C.; Meng, Z.; Ma, X.; Huang, W. Bile Acid Receptors and Liver Cancer. Curr. Pathobiol. Rep. 2013, 1, 29–35. [Google Scholar] [CrossRef]
- Wang, Y.-D.; Chen, W.-D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X Receptor Antagonizes Nuclear Factor ΚB in Hepatic Inflammatory Response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Morimura, K.; Shah, Y.; Yang, Q.; Ward, J.M.; Gonzalez, F.J. Spontaneous Hepatocarcinogenesis in Farnesoid X Receptor-Null Mice. Carcinogenesis 2007, 28, 940–946. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ge, X.; Heemstra, L.A.; Chen, W.-D.; Xu, J.; Smith, J.L.; Ma, H.; Kasim, N.; Edwards, P.A.; Novak, C.M. Loss of FXR Protects against Diet-Induced Obesity and Accelerates Liver Carcinogenesis in Ob/Ob Mice. Mol. Endocrinol. Baltim. Md 2012, 26, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.; Thomas, A.; Edwards, G.; Jaseja, R.; Guo, G.L.; Apte, U. Increased Activation of the Wnt/β-Catenin Pathway in Spontaneous Hepatocellular Carcinoma Observed in Farnesoid X Receptor Knockout Mice. J. Pharmacol. Exp. Ther. 2011, 338, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Xu, Z.; Zhang, Y.; Jiang, P.; Huang, G.; Chen, S.; Lyu, X.; Zheng, P.; Zhao, X.; Zeng, Y.; et al. FXR Induces SOCS3 and Suppresses Hepatocellular Carcinoma. Oncotarget 2015, 6, 34606–34616. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Meng, Z.; Lou, G.; Zhou, W.; Wang, X.; Zhang, Y.; Zhang, L.; Liu, X.; Yen, Y.; Lai, L.; et al. Hepatocarcinogenesis in FXR-/- Mice Mimics Human HCC Progression That Operates through HNF1α Regulation of FXR Expression. Mol. Endocrinol. Baltim. Md 2012, 26, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Anfuso, B.; Tiribelli, C.; Adorini, L.; Rosso, N. Obeticholic Acid and INT-767 Modulate Collagen Deposition in a NASH in Vitro Model. Sci. Rep. 2020, 10, 1699. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Palladino, G.; Fiorucci, S. FXR Activation Reverses Insulin Resistance and Lipid Abnormalities and Protects against Liver Steatosis in Zucker (Fa/Fa) Obese Rats. J. Lipid Res. 2010, 51, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, J.D.; Veidal, S.S.; Fensholdt, L.K.D.; Rigbolt, K.T.G.; Papazyan, R.; Nielsen, J.C.; Feigh, M.; Vrang, N.; Young, M.; Jelsing, J.; et al. Combined Obeticholic Acid and Elafibranor Treatment Promotes Additive Liver Histological Improvements in a Diet-Induced Ob/Ob Mouse Model of Biopsy-Confirmed NASH. Sci. Rep. 2019, 9, 9046. [Google Scholar] [CrossRef]
- Goto, T.; Itoh, M.; Suganami, T.; Kanai, S.; Shirakawa, I.; Sakai, T.; Asakawa, M.; Yoneyama, T.; Kai, T.; Ogawa, Y. Obeticholic Acid Protects against Hepatocyte Death and Liver Fibrosis in a Murine Model of Nonalcoholic Steatohepatitis. Sci. Rep. 2018, 8, 8157. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.-U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and Safety of the Farnesoid X Receptor Agonist Obeticholic Acid in Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X Nuclear Receptor Ligand Obeticholic Acid for Non-Cirrhotic, Non-Alcoholic Steatohepatitis (FLINT): A Multicentre, Randomised, Placebo-Controlled Trial. Lancet Lond. Engl. 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic Acid for the Treatment of Non-Alcoholic Steatohepatitis: Interim Analysis from a Multicentre, Randomised, Placebo-Controlled Phase 3 Trial. Lancet Lond. Engl. 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Al-Dury, S.; Wahlström, A.; Panzitt, K.; Thorell, A.; Ståhlman, M.; Trauner, M.; Fickert, P.; Bäckhed, F.; Fändriks, L.; Wagner, M.; et al. Obeticholic Acid May Increase the Risk of Gallstone Formation in Susceptible Patients. J. Hepatol. 2019, 71, 986–991. [Google Scholar] [CrossRef]
- Attia, Y.M.; Tawfiq, R.A.; Ali, A.A.; Elmazar, M.M. The FXR Agonist, Obeticholic Acid, Suppresses HCC Proliferation & Metastasis: Role of IL-6/STAT3 Signalling Pathway. Sci. Rep. 2017, 7, 12502. [Google Scholar] [CrossRef]
- Zhang, R.; Ray, J.W.; Jain, M.K.; Han, S. Ileectomy-Induced Bile Overaccumulation in Mouse Intestine. J. Vis. Exp. JoVE 2017. [Google Scholar] [CrossRef]
- Van de Peppel, I.P.; Verkade, H.J.; Jonker, J.W. Metabolic Consequences of Ileal Interruption of the Enterohepatic Circulation of Bile Acids. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G619–G625. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Jun, H.-S. Anti-Diabetic Actions of Glucagon-like Peptide-1 on Pancreatic Beta-Cells. Metabolism 2014, 63, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.J.; Leech, C.A.; Lin, J.C.; Zulewski, H.; Habener, J.F. Insulinotropic Hormone Glucagon-like Peptide-1 Differentiation of Human Pancreatic Islet-Derived Progenitor Cells into Insulin-Producing Cells. Endocrinology 2002, 143, 3152–3161. [Google Scholar] [CrossRef]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like Peptide-1 Receptor (GLP-1R) Is Present on Human Hepatocytes and Has a Direct Role in Decreasing Hepatic Steatosis in Vitro by Modulating Elements of the Insulin Signaling Pathway. Hepatol. Baltim. Md 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [Green Version]
- Samson, S.L.; Bajaj, M. Potential of Incretin-Based Therapies for Non-Alcoholic Fatty Liver Disease. J. Diabetes Complicat. 2013, 27, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.-M.; Vergès, B. GLP-1 Receptor Agonists in NAFLD. Diabetes Metab. 2017, 43, 2S28–2S33. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; Minicis, S.D.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like Peptide-1 Receptor Activation Stimulates Hepatic Lipid Oxidation and Restores Hepatic Signalling Alteration Induced by a High-Fat Diet in Nonalcoholic Steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Mells, J.E.; Fu, P.P.; Saxena, N.K.; Anania, F.A. GLP-1 Analogs Reduce Hepatocyte Steatosis and Improve Survival by Enhancing the Unfolded Protein Response and Promoting Macroautophagy. PLoS ONE 2011, 6, e25269. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.C.; Lima, K.G.; Levorse, V.; Haute, G.V.; Gassen, R.B.; Garcia, M.C.; Pedrazza, L.; Donadio, M.V.F.; Luft, C.; de Oliveira, J.R. Exenatide Induces Autophagy and Prevents the Cell Regrowth in HepG2 Cells. EXCLI J. 2019, 18, 540–548. [Google Scholar] [CrossRef]
- Krause, G.C.; Lima, K.G.; Dias, H.B.; da Silva, E.F.G.; Haute, G.V.; Basso, B.S.; Gassen, R.B.; Marczak, E.S.; Nunes, R.S.B.; de Oliveira, J.R. Liraglutide, a Glucagon-like Peptide-1 Analog, Induce Autophagy and Senescence in HepG2 Cells. Eur. J. Pharmacol. 2017, 809, 32–41. [Google Scholar] [CrossRef]
- Lu, X.; Xu, C.; Dong, J.; Zuo, S.; Zhang, H.; Jiang, C.; Wu, J.; Wei, J. Liraglutide Activates Nature Killer Cell-Mediated Antitumor Responses by Inhibiting IL-6/STAT3 Signaling in Hepatocellular Carcinoma. Transl. Oncol. 2021, 14, 100872. [Google Scholar] [CrossRef]
- Tong, W.; Ju, L.; Qiu, M.; Xie, Q.; Chen, Y.; Shen, W.; Sun, W.; Wang, W.; Tian, J. Liraglutide Ameliorates Non-Alcoholic Fatty Liver Disease by Enhancing Mitochondrial Architecture and Promoting Autophagy through the SIRT1/SIRT3-FOXO3a Pathway. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2016, 46, 933–943. [Google Scholar] [CrossRef]
- Trevaskis, J.L.; Griffin, P.S.; Wittmer, C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Dolman, C.S.; Erickson, M.R.; Napora, J.; Parkes, D.G.; Roth, J.D. Glucagon-like Peptide-1 Receptor Agonism Improves Metabolic, Biochemical, and Histopathological Indices of Nonalcoholic Steatohepatitis in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G762–G772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Gupta, N.; Anania, F.A. Exendin-4, a Glucagon-like Protein-1 (GLP-1) Receptor Agonist, Reverses Hepatic Steatosis in Ob/Ob Mice. Hepatol. Baltim. Md 2006, 43, 173–181. [Google Scholar] [CrossRef]
- Lv, X.; Dong, Y.; Hu, L.; Lu, F.; Zhou, C.; Qin, S. Glucagon-like Peptide-1 Receptor Agonists (GLP-1 RAs) for the Management of Nonalcoholic Fatty Liver Disease (NAFLD): A Systematic Review. Endocrinol. Diabetes Metab. 2020, 3, e00163. [Google Scholar] [CrossRef]
- Kojima, M.; Takahashi, H.; Kuwashiro, T.; Tanaka, K.; Mori, H.; Ozaki, I.; Kitajima, Y.; Matsuda, Y.; Ashida, K.; Eguchi, Y.; et al. Glucagon-Like Peptide-1 Receptor Agonist Prevented the Progression of Hepatocellular Carcinoma in a Mouse Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 5722. [Google Scholar] [CrossRef] [PubMed]
- Kawakubo, M.; Tanaka, M.; Ochi, K.; Watanabe, A.; Saka-Tanaka, M.; Kanamori, Y.; Yoshioka, N.; Yamashita, S.; Goto, M.; Itoh, M.; et al. Dipeptidyl Peptidase-4 Inhibition Prevents Nonalcoholic Steatohepatitis-Associated Liver Fibrosis and Tumor Development in Mice Independently of Its Anti-Diabetic Effects. Sci. Rep. 2020, 10, 983. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased Lipogenesis, Induced by AKT-MTORC1-RPS6 Signaling, Promotes Development of Human Hepatocellular Carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.E.; Lahiri, S.; Chow, J.D.Y.; Byrne, F.L.; Hargett, S.R.; Breen, D.S.; Olzomer, E.M.; Wu, L.E.; Cooney, G.J.; Turner, N.; et al. Inhibition of Hepatic Lipogenesis Enhances Liver Tumorigenesis by Increasing Antioxidant Defence and Promoting Cell Survival. Nat. Commun. 2017, 8, 14689. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Tan, H.-Y.; Teng, S.; Chan, Y.-T.; Wang, D.; Wang, N. The Role of AMP-Activated Protein Kinase as a Potential Target of Treatment of Hepatocellular Carcinoma. Cancers 2019, 11, 647. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, A.C.; Hidalgo, F.; Tonucci, F.M.; Almada, E.; Pariani, A.; Larocca, M.C.; Favre, C. Metformin and Glucose Starvation Decrease the Migratory Ability of Hepatocellular Carcinoma Cells: Targeting AMPK Activation to Control Migration. Sci. Rep. 2019, 9, 2815. [Google Scholar] [CrossRef]
- Abu-Elheiga, L.; Brinkley, W.R.; Zhong, L.; Chirala, S.S.; Woldegiorgis, G.; Wakil, S.J. The Subcellular Localization of Acetyl-CoA Carboxylase 2. Proc. Natl. Acad. Sci. USA 2000, 97, 1444–1449. [Google Scholar] [CrossRef] [Green Version]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA Carboxylase Inhibition by ND-630 Reduces Hepatic Steatosis, Improves Insulin Sensitivity, and Modulates Dyslipidemia in Rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef] [Green Version]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatol. Baltim. Md 2018, 68, 2197–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.-W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 394–406.e6. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Kayali, Z.; Noureddin, M.; Ruane, P.; Lawitz, E.J.; Bennett, M.; Wang, L.; Harting, E.; Tarrant, J.M.; McColgan, B.J.; et al. GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1463–1473.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawitz, E.J.; Coste, A.; Poordad, F.; Alkhouri, N.; Loo, N.; McColgan, B.J.; Tarrant, J.M.; Nguyen, T.; Han, L.; Chung, C.; et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2018, 16, 1983–1991.e3. [Google Scholar] [CrossRef]
- Stiede, K.; Miao, W.; Blanchette, H.S.; Beysen, C.; Harriman, G.; Harwood, H.J.; Kelley, H.; Kapeller, R.; Schmalbach, T.; Westlin, W.F. Acetyl-Coenzyme A Carboxylase Inhibition Reduces de Novo Lipogenesis in Overweight Male Subjects: A Randomized, Double-Blind, Crossover Study. Hepatol. Baltim. Md 2017, 66, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase (ACC) by Phosphorylation or by the Liver-Specific Inhibitor, ND-654, Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Istvan, E.S.; Deisenhofer, J. Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigler, M.A.; Congdon, L.; Edwards, K.L. An Evidence-Based Review of Statin Use in Patients With Nonalcoholic Fatty Liver Disease. Clin. Med. Insights Gastroenterol. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athyros, V.G.; Boutari, C.; Stavropoulos, K.; Anagnostis, P.; Imprialos, K.P.; Doumas, M.; Karagiannis, A. Statins: An Under-Appreciated Asset for the Prevention and the Treatment of NAFLD or NASH and the Related Cardiovascular Risk. Curr. Vasc. Pharmacol. 2018, 16, 246–253. [Google Scholar] [CrossRef]
- Doumas, M.; Imprialos, K.; Dimakopoulou, A.; Stavropoulos, K.; Binas, A.; Athyros, V.G. The Role of Statins in the Management of Nonalcoholic Fatty Liver Disease. Curr. Pharm. Des. 2018, 24, 4587–4592. [Google Scholar] [CrossRef]
- Singh, S.; Singh, P.P.; Singh, A.G.; Murad, M.H.; Sanchez, W. Statins Are Associated with a Reduced Risk of Hepatocellular Cancer: A Systematic Review and Meta-Analysis. Gastroenterology 2013, 144, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.-J.; Qiu, T.-Y.; Ng, G.-K.; Zheng, Q.; Teo, E.K. Efficacy and Safety of Statin for Hepatocellular Carcinoma Prevention Among Chronic Liver Disease Patients: A Systematic Review and Meta-Analysis. J. Clin. Gastroenterol. 2021. [Google Scholar] [CrossRef]
- Goh, M.J.; Sinn, D.H.; Kim, S.; Woo, S.Y.; Cho, H.; Kang, W.; Gwak, G.-Y.; Paik, Y.-H.; Choi, M.S.; Lee, J.H.; et al. Statin Use and the Risk of Hepatocellular Carcinoma in Patients With Chronic Hepatitis B. Hepatol. Baltim. Md 2020, 71, 2023–2032. [Google Scholar] [CrossRef]
- Li, X.; Sheng, L.; Liu, L.; Hu, Y.; Chen, Y.; Lou, L. Statin and the Risk of Hepatocellular Carcinoma in Patients with Hepatitis B Virus or Hepatitis C Virus Infection: A Meta-Analysis. BMC Gastroenterol. 2020, 20, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.; Liu, Q.; Zhou, Z.; Ding, Y.; Yang, M.; Xu, W.; Chen, K.; Zhang, Q.; Wang, Z.; Li, H. Can Statin Treatment Reduce the Risk of Hepatocellular Carcinoma? A Systematic Review and Meta-Analysis. Technol. Cancer Res. Treat. 2020, 19, 1533033820934881. [Google Scholar] [CrossRef]
- Gu, Y.; Yang, X.; Liang, H.; Li, D. Comprehensive Evaluation of Effects and Safety of Statin on the Progression of Liver Cirrhosis: A Systematic Review and Meta-Analysis. BMC Gastroenterol. 2019, 19, 231. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, K.A.; Divine, G.W.; Sahasrabuddhe, V.V.; Engel, L.S.; VanSlooten, A.; Wells, K.; Yood, M.U.; Alford, S.H. Statin Use and Risk of Hepatocellular Carcinoma in a U.S. Population. Cancer Epidemiol. 2014, 38, 523–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonardo, A.; Loria, P. Potential for Statins in the Chemoprevention and Management of Hepatocellular Carcinoma. J. Gastroenterol. Hepatol. 2012, 27, 1654–1664. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Yasuda, Y.; Sakai, H.; Kubota, M.; Terakura, D.; Baba, A.; Ohno, T.; Kochi, T.; Tsurumi, H.; Tanaka, T.; et al. Pitavastatin Suppresses Diethylnitrosamine-Induced Liver Preneoplasms in Male C57BL/KsJ-Db/Db Obese Mice. BMC Cancer 2011, 11, 281. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Fan-Minogue, H.; Bellovin, D.I.; Yevtodiyenko, A.; Arzeno, J.; Yang, Q.; Gambhir, S.S.; Felsher, D.W. MYC Phosphorylation, Activation, and Tumorigenic Potential in Hepatocellular Carcinoma Are Regulated by HMG-CoA Reductase. Cancer Res. 2011, 71, 2286–2297. [Google Scholar] [CrossRef] [Green Version]
- Pastori, D.; Polimeni, L.; Baratta, F.; Pani, A.; Del Ben, M.; Angelico, F. The Efficacy and Safety of Statins for the Treatment of Non-Alcoholic Fatty Liver Disease. Dig. Liver Dis. 2015, 47, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The Gut Microbiome in Health and in Disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef]
- Brown, J.M.; Hazen, S.L. The Gut Microbial Endocrine Organ: Bacterially-Derived Signals Driving Cardiometabolic Diseases. Annu. Rev. Med. 2015, 66, 343–359. [Google Scholar] [CrossRef] [Green Version]
- Sharpton, S.R.; Ajmera, V.; Loomba, R. Emerging Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease: From Composition to Function. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019, 17, 296–306. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Greten, T.F. Gut Microbiome in HCC—Mechanisms, Diagnosis and Therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpton, S.R.; Schnabl, B.; Knight, R.; Loomba, R. Current Concepts, Opportunities, and Challenges of Gut Microbiome-Based Personalized Medicine in Nonalcoholic Fatty Liver Disease. Cell Metab. 2021, 33, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Gu, X.; Buffa, J.A.; Hurd, A.G.; Wang, Z.; Zhu, W.; Gupta, N.; Skye, S.M.; Cody, D.B.; Levison, B.S.; et al. Development of a Gut Microbe-Targeted Non-Lethal Therapeutic to Inhibit Thrombosis Potential. Nat. Med. 2018, 24, 1407–1417. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Zhou, R.; Chen, X.; Wang, C.; Tan, X.; Wang, L.; Zheng, R.; Zhang, H.; Ling, W.; et al. Associations of Gut-Flora-Dependent Metabolite Trimethylamine-N-Oxide, Betaine and Choline with Non-Alcoholic Fatty Liver Disease in Adults. Sci. Rep. 2016, 6, 19076. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Schaffer, J.E. Lipotoxicity: Many Roads to Cell Dysfunction and Cell Death: Introduction to a Thematic Review Series. J. Lipid Res. 2016, 57, 1327–1328. [Google Scholar] [CrossRef] [Green Version]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic Lethal and Sublethal Stress Signaling in Hepatocytes: Relevance to NASH Pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef] [Green Version]
- Hauck, A.K.; Bernlohr, D.A. Oxidative Stress and Lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaishy, B.; Abel, E.D. Lipids, Lysosomes, and Autophagy. J. Lipid Res. 2016, 57, 1619–1635. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Kaufman, R.J. The Role of ER Stress in Lipid Metabolism and Lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Tan, H.-Y.; Wang, N.; Zhang, Z.-J.; Lao, L.; Wong, C.-W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [Green Version]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular Carcinoma in Non-Alcoholic Steatohepatitis: Current Knowledge and Implications for Management. World J. Hepatol. 2017, 9, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Tummala, K.S.; Brandt, M.; Teijeiro, A.; Graña, O.; Schwabe, R.F.; Perna, C.; Djouder, N. Hepatocellular Carcinomas Originate Predominantly from Hepatocytes and Benign Lesions from Hepatic Progenitor Cells. Cell Rep. 2017, 19, 584–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, S.-Y.; Park, E.-J.; Lee, C.-W. Immunological Distinctions between Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Exp. Mol. Med. 2020, 52, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Wang, B.; Yang, L.; Zhang, Y. The Role of ROS-Induced Autophagy in Hepatocellular Carcinoma. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 306–312. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Mammalian Development and Differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Rubio, N.; Garg, A.D.; Agostinis, P. Autophagy: Shaping the Tumor Microenvironment and Therapeutic Response. Trends Mol. Med. 2013, 19, 428–446. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in Cancer: Moving from Understanding Mechanism to Improving Therapy Responses in Patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the MTOR Pathway in Hepatocellular Carcinoma: Current State and Future Trends. J. Hepatol. 2014, 60, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. MTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12. [Google Scholar] [CrossRef] [Green Version]
- Ferrín, G.; Guerrero, M.; Amado, V.; Rodríguez-Perálvarez, M.; De la Mata, M. Activation of MTOR Signaling Pathway in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 1266. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.-Y.; Huang, S.-L. Current Development of the Second Generation of MTOR Inhibitors as Anticancer Agents. Chin. J. Cancer 2012, 31, 8–18. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orabi, D.; Berger, N.A.; Brown, J.M. Abnormal Metabolism in the Progression of Nonalcoholic Fatty Liver Disease to Hepatocellular Carcinoma: Mechanistic Insights to Chemoprevention. Cancers 2021, 13, 3473. https://doi.org/10.3390/cancers13143473
Orabi D, Berger NA, Brown JM. Abnormal Metabolism in the Progression of Nonalcoholic Fatty Liver Disease to Hepatocellular Carcinoma: Mechanistic Insights to Chemoprevention. Cancers. 2021; 13(14):3473. https://doi.org/10.3390/cancers13143473
Chicago/Turabian StyleOrabi, Danny, Nathan A. Berger, and J. Mark Brown. 2021. "Abnormal Metabolism in the Progression of Nonalcoholic Fatty Liver Disease to Hepatocellular Carcinoma: Mechanistic Insights to Chemoprevention" Cancers 13, no. 14: 3473. https://doi.org/10.3390/cancers13143473
APA StyleOrabi, D., Berger, N. A., & Brown, J. M. (2021). Abnormal Metabolism in the Progression of Nonalcoholic Fatty Liver Disease to Hepatocellular Carcinoma: Mechanistic Insights to Chemoprevention. Cancers, 13(14), 3473. https://doi.org/10.3390/cancers13143473