
Oncogenic Kinase Cascades Induce Molecular Mechanisms That Protect Leukemic Cell Models from Lethal Effects of De Novo dNTP Synthesis Inhibition

 , , , ,
, , , , 
Abstract
Simple Summary
Abstract
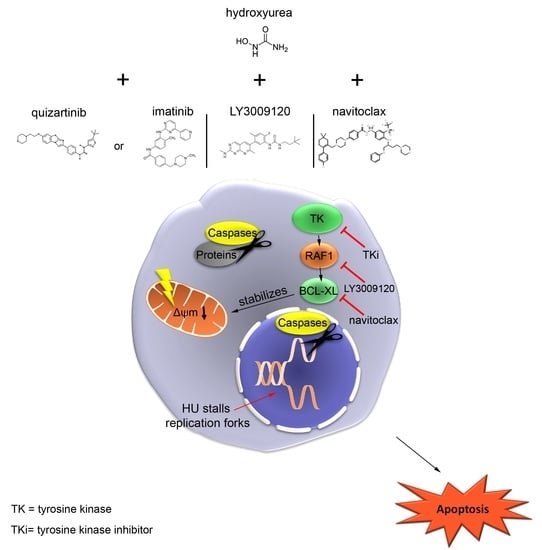
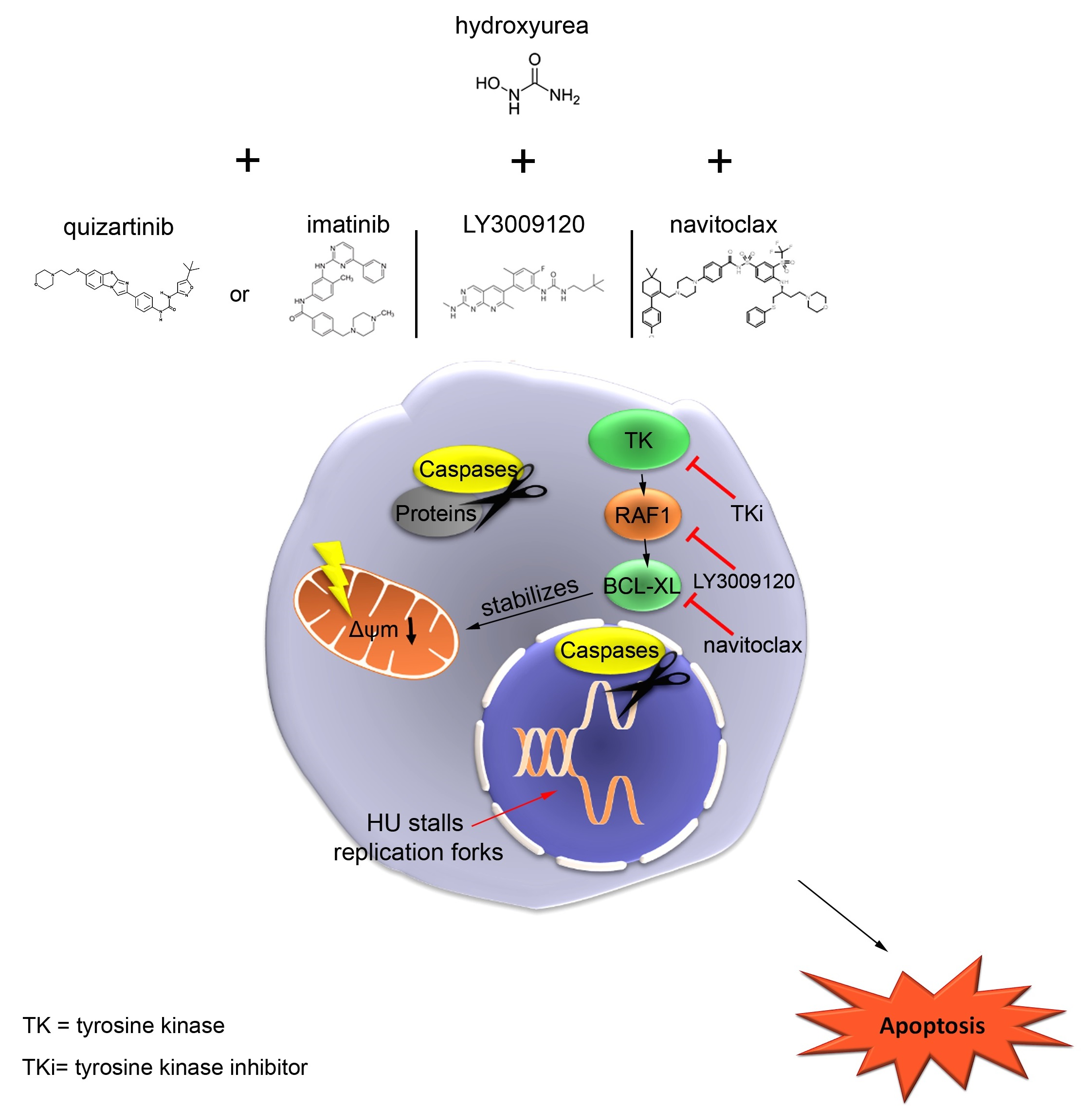{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Proteomics
2.2. Cell Lines
2.3. Drugs and Chemicals
2.4. Transfection of siRNA
2.5. Immunoblot
2.6. Flow Cytometry
2.7. Alkaline Comet (Single-Cell Gel Electrophoresis) Assay
2.8. Statistics
3. Results
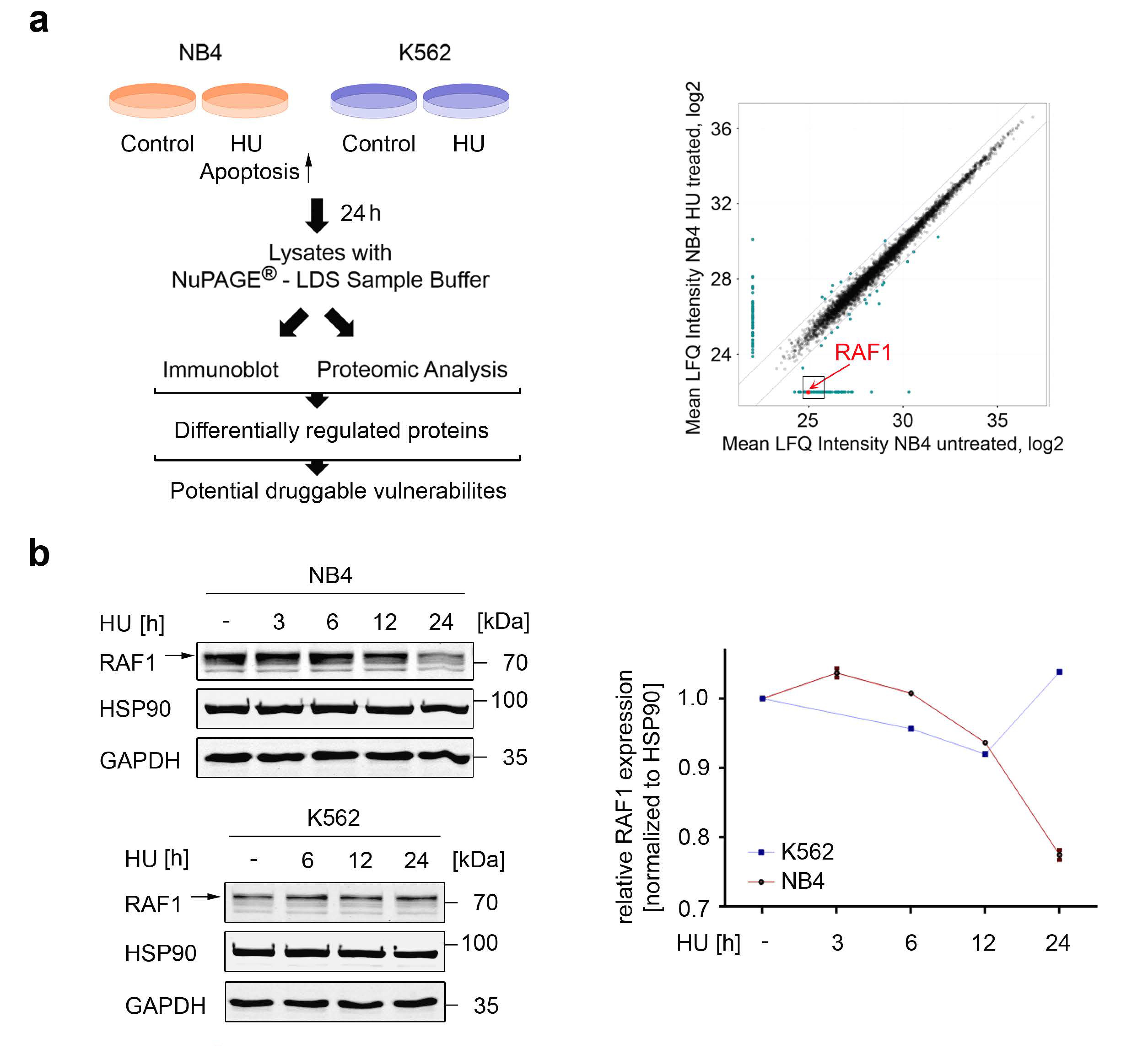
3.1. Global Identification of Factors That Control Apoptosis in Response to Replication Stress
3.2. Assessment of the Biological Relevance of RAF1
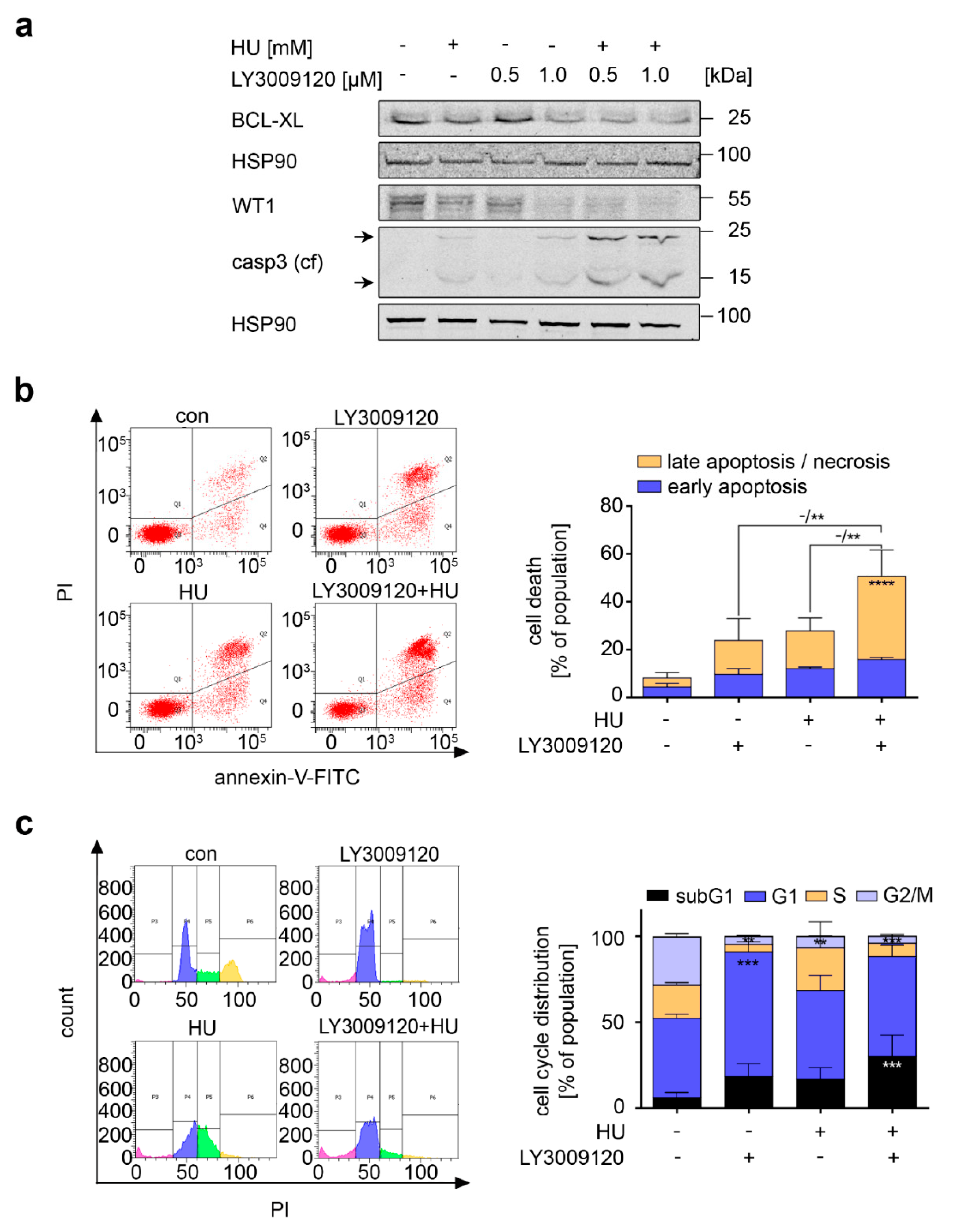
3.3. RAF Promotes Cytoprotective BCL-XL Expression in CML Cells
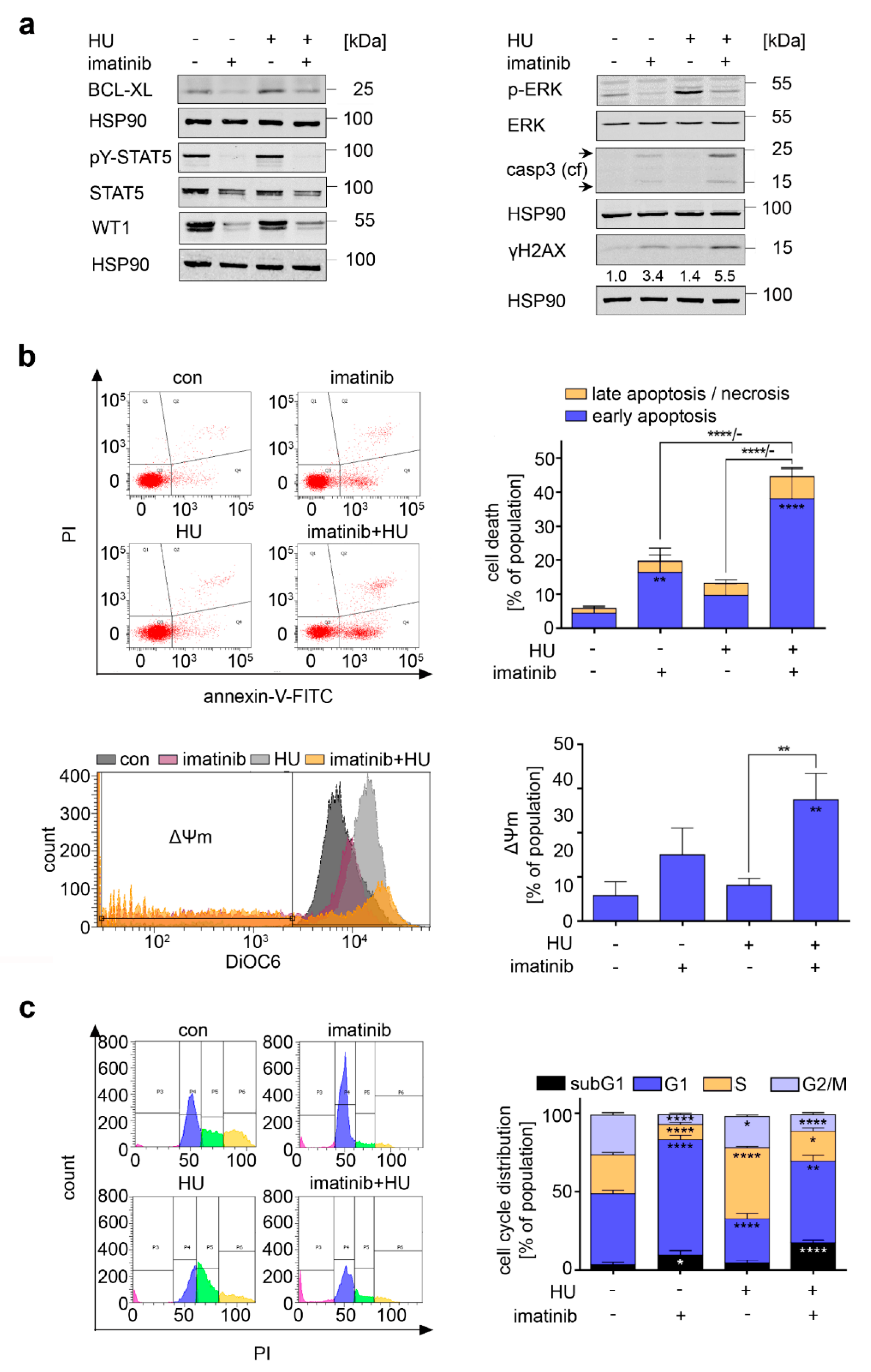
3.4. Consequences of BCR-ABL1 Inhibition in CML Cells
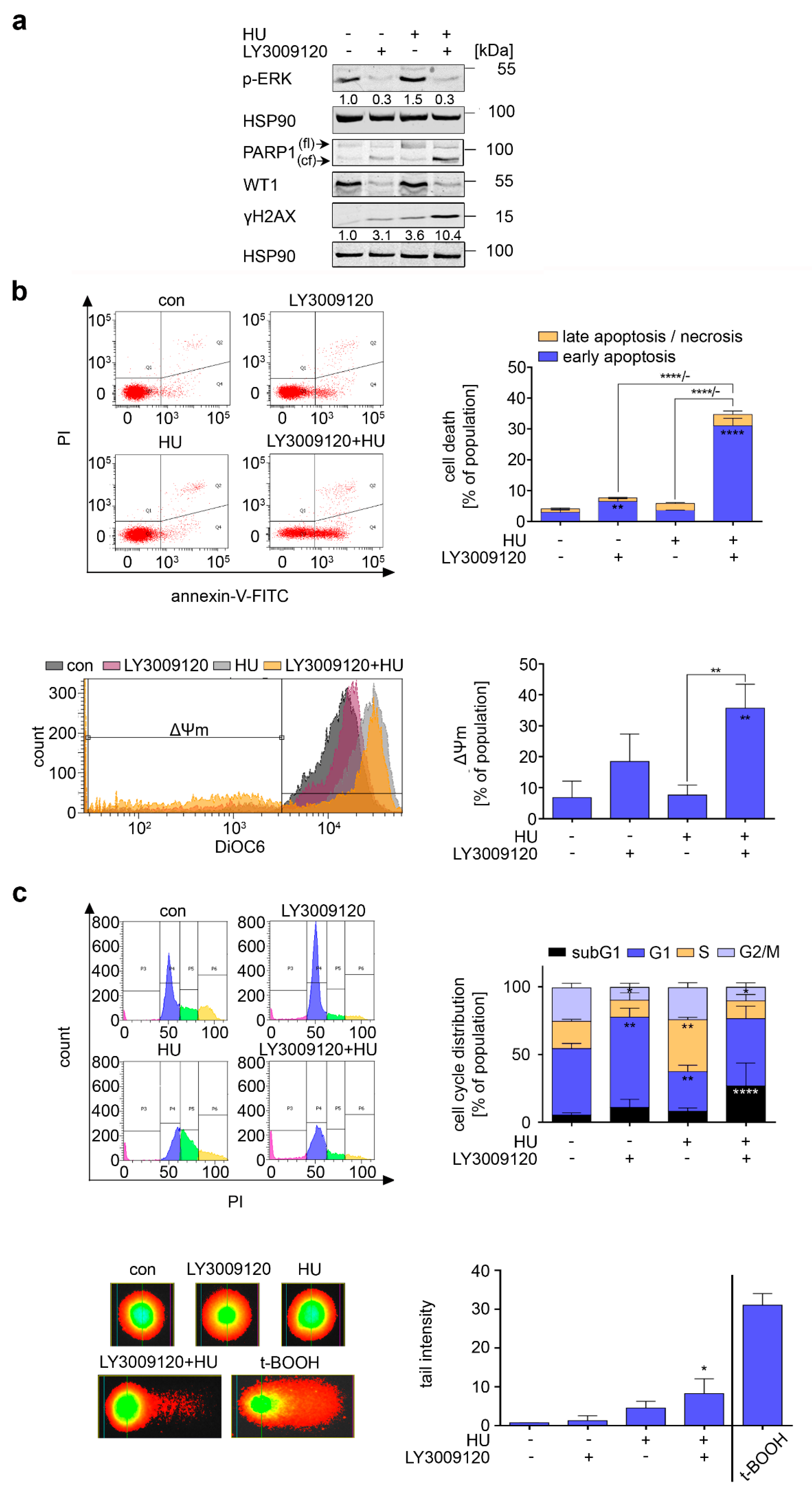
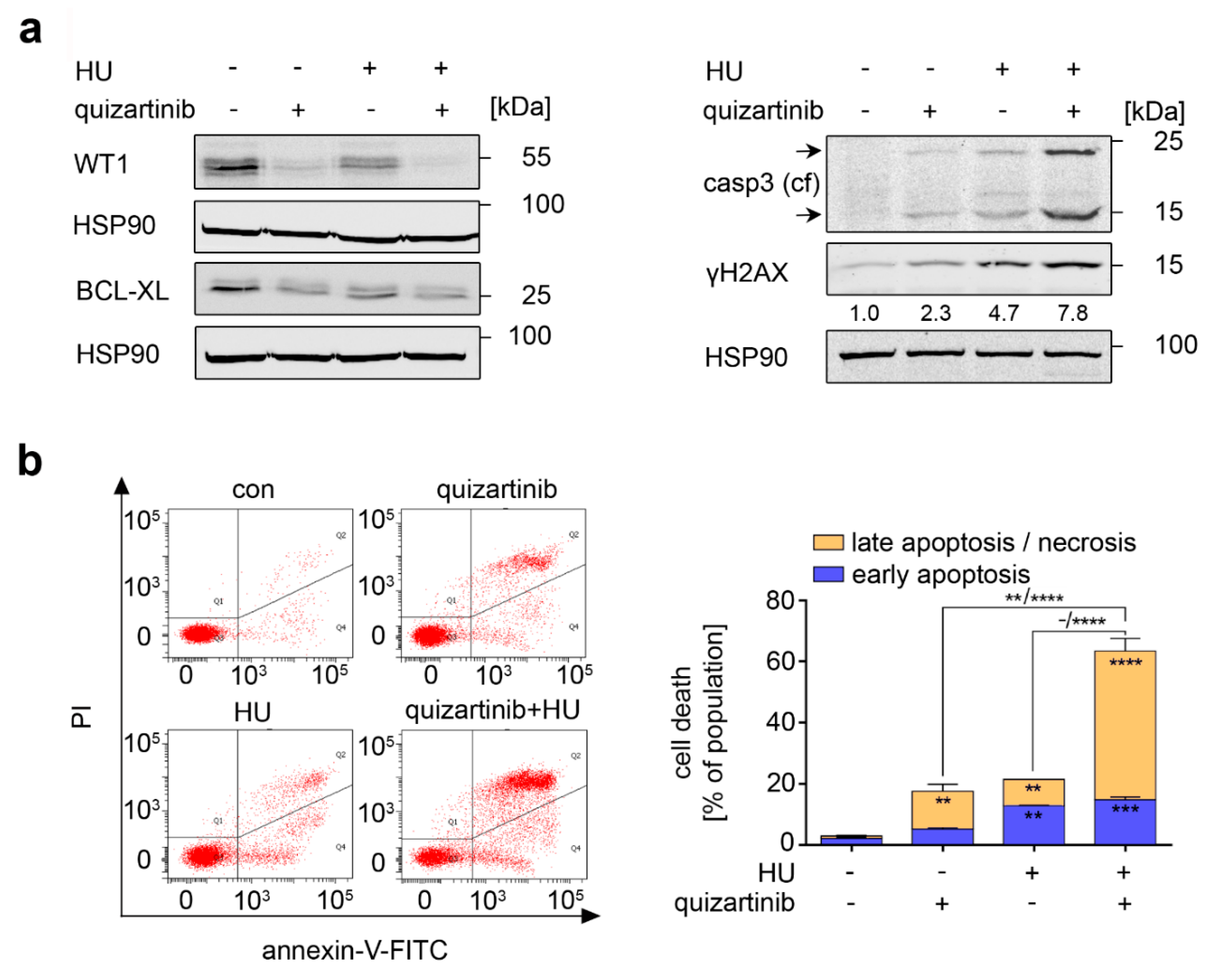
3.5. RAF Is a Target in Hydroxyurea-Treated FLT3-ITD-Positive AML Cells
3.6. Inhibition of FLT3-ITD Sensitizes AML Cells to Hydroxyurea
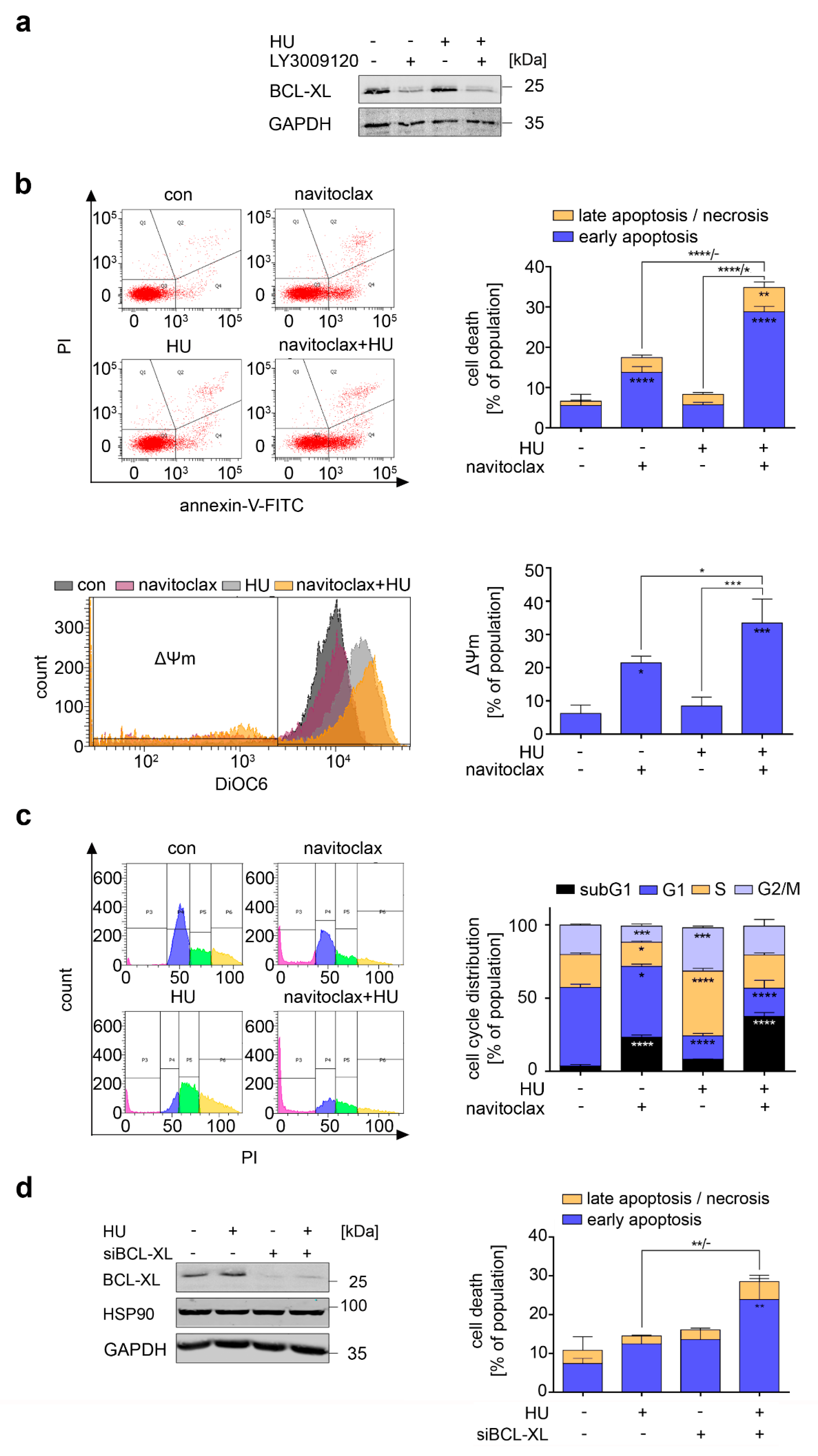
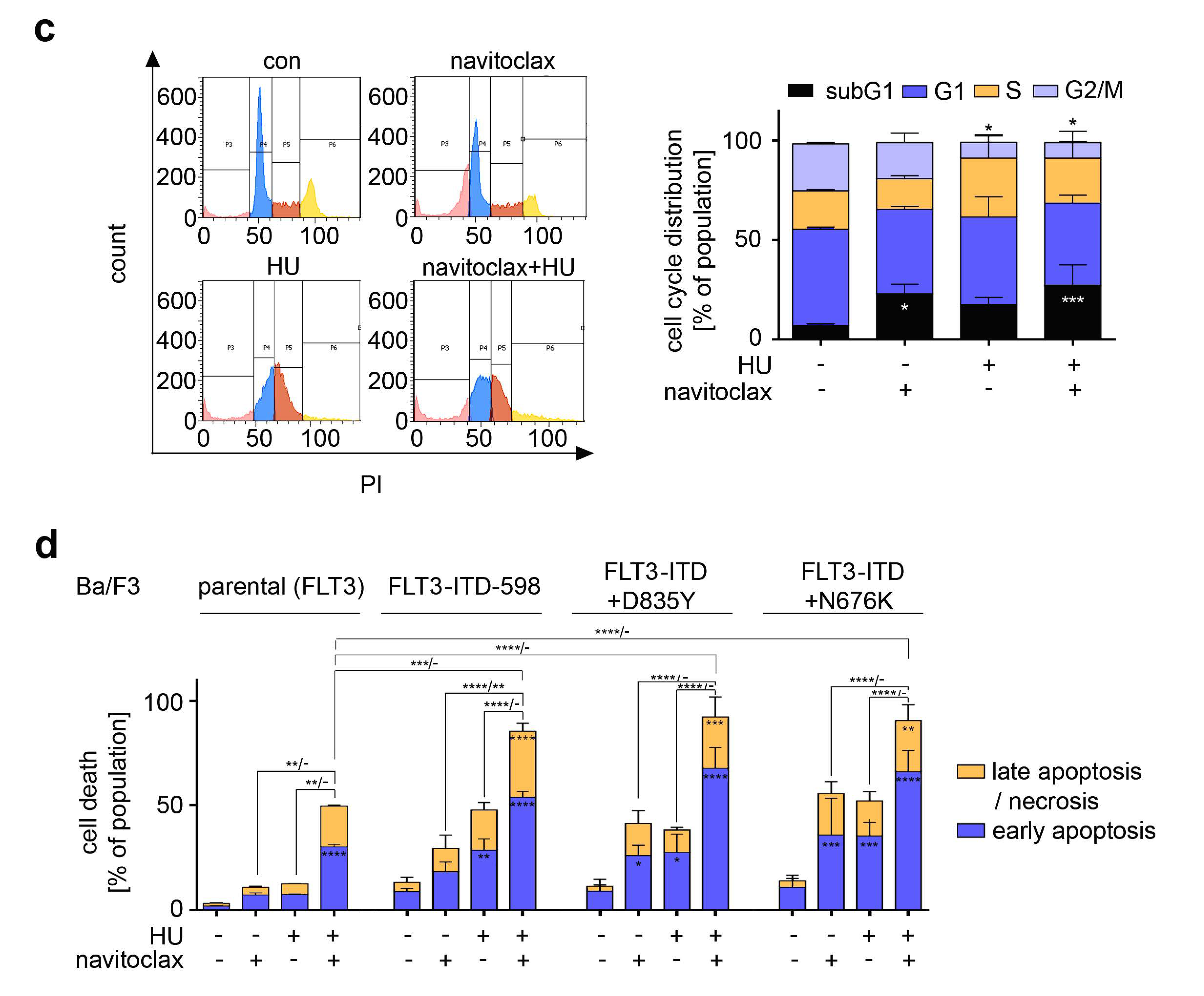
3.7. Navitoclax Combines Favorably with Hydroxyurea against FLT3 Mutant AML Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Sørensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef]
- Mahajan, K.; Mahajan, N.P. Cross talk of tyrosine kinases with the DNA damage signaling pathways. Nucleic Acids Res. 2015, 43, 10588–10601. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Vannucchi, A.M. Current management strategies for polycythemia vera and essential thrombocythemia. Blood Rev. 2020, 42, 100714. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Giri, S.; Tallman, M.S.; Zeidan, A.M.; Stahl, M. Leukapheresis for the management of hyperleukocytosis in acute myeloid leukemia—A systematic review and meta-analysis. Transfusion 2020, 60, 2360–2369. [Google Scholar] [CrossRef]
- Dzulko, M.; Pons, M.; Henke, A.; Schneider, G.; Krämer, O.H. The PP2A subunit PR130 is a key regulator of cell development and oncogenic transformation. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188453. [Google Scholar] [CrossRef] [PubMed]
- Schneeweiss-Gleixner, M.; Byrgazov, K.; Stefanzl, G.; Berger, D.; Eisenwort, G.; Lucini, C.B.; Herndlhofer, S.; Preuner, S.; Obrova, K.; Pusic, P.; et al. CDK4/CDK6 inhibition as a novel strategy to suppress the growth and survival of BCR-ABL1T315I+ clones in TKI-resistant CML. EBioMedicine 2019, 50, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Eklund, H.; Uhlin, U.; Färnegårdh, M.; Logan, D.; Nordlund, P. Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol. 2001, 77, 177–268. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Kopp, B.; Khoury, L.; Audebert, M. Validation of the γH2AX biomarker for genotoxicity assessment: A review. Arch. Toxicol. 2019, 93, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Pons, M.; Reichardt, C.M.; Hennig, D.; Nathan, A.; Kiweler, N.; Stocking, C.; Wichmann, C.; Christmann, M.; Butter, F.; Reichardt, S.; et al. Loss of Wilms tumor 1 protein is a marker for apoptosis in response to replicative stress in leukemic cells. Arch. Toxicol. 2018, 92, 2119–2135. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed]
- Flis, S.; Chojnacki, T. Chronic myelogenous leukemia, a still unsolved problem: Pitfalls and new therapeutic possibilities. Drug Des. Dev. Ther. 2019, 13, 825–843. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, A.; Mian, A.A.; Döring, C.; Metodieva, A.; Oancea, C.; Thalheimer, F.B.; Hansmann, M.L.; Ottmann, O.; Ruthardt, M. The Functional Interplay Between the t(9;22)-Associated Fusion Proteins BCR/ABL and ABL/BCR in Philadelphia Chromosome-Positive Acute Lymphatic Leukemia. PLoS Genet. 2015, 11, e1005144. [Google Scholar] [CrossRef]
- Perl, A.E. Availability of FLT3 inhibitors: How do we use them? Blood 2019, 134, 741–745. [Google Scholar] [CrossRef]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef]
- Eguchi, M.; Minami, Y.; Kuzume, A.; Chi, S. Mechanisms Underlying Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Biomedicines 2020, 8, 245. [Google Scholar] [CrossRef]
- Pons, M.; Nagel, G.; Zeyn, Y.; Beyer, M.; Laguna, T.; Brachetti, T.; Sellmer, A.; Mahboobi, S.; Conradi, R.; Butter, F.; et al. Human platelet lysate as validated replacement for animal serum to assess chemosensitivity. ALTEX 2019, 36, 277–288. [Google Scholar] [CrossRef]
- Freund, P.; Kerenyi, M.A.; Hager, M.; Wagner, T.; Wingelhofer, B.; Pham, H.T.T.; Elabd, M.; Han, X.; Valent, P.; Gouilleux, F.; et al. O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies. Leukemia 2017, 31, 2132–2142. [Google Scholar] [CrossRef]
- Page, B.D.G.; Khoury, H.; Laister, R.C.; Fletcher, S.; Vellozo, M.; Manzoli, A.; Yue, P.; Turkson, J.; Minden, M.D.; Gunning, P.T. Small Molecule STAT5-SH2 Domain Inhibitors Exhibit Potent Antileukemia Activity. J. Med. Chem. 2012, 55, 1047–1055. [Google Scholar] [CrossRef]
- Marx-Blümel, L.; Marx, C.; Kühne, M.; Sonnemann, J. Assessment of HDACi-Induced Cytotoxicity. In HDAC/HAT Function Assessment and Inhibitor Development; Springer: New York, NY, USA, 2017; Volume 1510, pp. 23–45. [Google Scholar] [CrossRef]
- Tewari, M.; Quan, L.T.; O’Rourke, K.; Desnoyers, S.; Zeng, Z.; Beidler, D.R.; Poirier, G.G.; Salvesen, G.S.; Dixit, V.M. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 1995, 81, 801–809. [Google Scholar] [CrossRef]
- Simanshu, D.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Melo, J.V.; Chuah, C. Novel Agents in CML Therapy: Tyrosine Kinase Inhibitors and Beyond. ASH Educ. Program Book 2008, 2008, 427–435. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vachhani, P.; Bose, P.; Rahmani, M.; Grant, S. Rational combination of dual PI3K/mTOR blockade and Bcl-2/-xL inhibition in AML. Physiol. Genom. 2014, 46, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef]
- Jazirehi, A.R.; Vega, M.I.; Chatterjee, D.; Goodglick, L.; Bonavida, B. Inhibition of the Raf–MEK1/2–ERK1/2 Signaling Pathway, Bcl-xL Down-Regulation, and Chemosensitization of Non-Hodgkin’s Lymphoma B Cells by Rituximab. Cancer Res. 2004, 64, 7117–7126. [Google Scholar] [CrossRef]
- Henry, J.R.; Kaufman, M.D.; Peng, S.-B.; Ahn, Y.M.; Caldwell, T.M.; Vogeti, L.; Telikepalli, H.; Lu, W.-P.; Hood, M.M.; Rutkoski, T.J.; et al. Discovery of 1-(3,3-Dimethylbutyl)-3-(2-fluoro-4-methyl-5-(7-methyl-2-(methylamino)pyrido[2,3-d]pyrimidin-6-yl)phenyl)urea (LY3009120) as a Pan-RAF Inhibitor with Minimal Paradoxical Activation and Activity against BRAF or RAS Mutant Tumor Cells. J. Med. Chem. 2015, 58, 4165–4179. [Google Scholar] [CrossRef]
- Khoury, J.D.; Tashakori, M.; Yang, H.; Loghavi, S.; Wang, Y.; Wang, J.; Piya, S.; Borthakur, G. Pan-RAF Inhibition Shows Anti-Leukemic Activity in RAS-Mutant Acute Myeloid Leukemia Cells and Potentiates the Effect of Sorafenib in Cells with FLT3 Mutation. Cancers 2020, 12, 3511. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Nugent, S.M.E.; Mothersill, C.E.; Seymour, C.; McClean, B.; Lyng, F.; Murphy, J.E.J. Increased Mitochondrial Mass in Cells with Functionally Compromised Mitochondria after Exposure to both Direct γ Radiation and Bystander Factors. Radiat. Res. 2007, 168, 134–142. [Google Scholar] [CrossRef]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef]
- Horita, M.; Andreu, E.J.; Benito, A.; Arbona, C.; Sanz, C.; Benet, I.; Prosper, F.; Fernandez-Luna, J.L. Blockade of the Bcr-Abl Kinase Activity Induces Apoptosis of Chronic Myelogenous Leukemia Cells by Suppressing Signal Transducer and Activator of Transcription 5–Dependent Expression of Bcl-XL. J. Exp. Med. 2000, 191, 977–984. [Google Scholar] [CrossRef]
- Zhao, R.; Follows, G.A.; Beer, P.A.; Scott, L.M.; Huntly, B.J.; Green, A.R.; Alexander, D.R. Inhibition of the Bcl-xLDeamidation Pathway in Myeloproliferative Disorders. N. Engl. J. Med. 2008, 359, 2778–2789. [Google Scholar] [CrossRef]
- Anuar, N.N.M.; Hisam, N.S.N.; Liew, S.L.; Ugusman, A. Clinical Review: Navitoclax as a Pro-Apoptotic and Anti-Fibrotic Agent. Front. Pharmacol. 2020, 11, 564108. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Sexl, V.; Valent, P.; Moriggl, R. Inhibition of STAT5: A therapeutic option in BCR-ABL1-driven leukemia. Oncotarget 2014, 5, 9564–9576. [Google Scholar] [CrossRef]
- Slupianek, A.; Schmutte, C.; Tombline, G.; Nieborowska-Skorska, M.; Hoser, G.; Nowicki, M.O.; Pierce, A.J.; Fishel, R.; Skorski, T. BCR/ABL Regulates Mammalian RecA Homologs, Resulting in Drug Resistance. Mol. Cell 2001, 8, 795–806. [Google Scholar] [CrossRef]
- Horne, G.; Jackson, L.; Helgason, V.; Holyoake, T. Stem Cell Guardians—Old and New Perspectives in LSC Biology. Curr. Drug Targets 2017, 18, 405–413. [Google Scholar] [CrossRef]
- Adnan-Awad, S.; Kim, D.; Hohtari, H.; Javarappa, K.K.; Brandstoetter, T.; Mayer, I.; Potdar, S.; Heckman, C.A.; Kytölä, S.; Porkka, K.; et al. Characterization of p190-Bcr-Abl chronic myeloid leukemia reveals specific signaling pathways and therapeutic targets. Leukemia 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mahendrarajah, N.; Paulus, R.; Krämer, O.H. Histone deacetylase inhibitors induce proteolysis of activated CDC42-associated kinase-1 in leukemic cells. J. Cancer Res. Clin. Oncol. 2016, 142, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.; Hopcroft, L.E.M.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.K.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nat. Cell Biol. 2016, 534, 341–346. [Google Scholar] [CrossRef]
- Warsch, W.; Kollmann, K.; Eckelhart, E.; Fajmann, S.; Cerny-Reiterer, S.; Hölbl, A.; Gleixner, K.V.; Dworzak, M.; Mayerhofer, M.; Hoermann, G.; et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood 2011, 117, 3409–3420. [Google Scholar] [CrossRef]
- Brachet-Botineau, M.; Deynoux, M.; Vallet, N.; Polomski, M.; Juen, L.; Hérault, O.; Mazurier, F.; Viaud-Massuard, M.-C.; Prié, G.; Gouilleux, F. A Novel Inhibitor of STAT5 Signaling Overcomes Chemotherapy Resistance in Myeloid Leukemia Cells. Cancers 2019, 11, 2043. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Ge, Z.; Chen, B. Quizartinib (AC220): A promising option for acute myeloid leukemia. Drug Des. Dev. Ther. 2019, 13, 1117–1125. [Google Scholar] [CrossRef]
- Takahashi, S. Mutations of FLT3 receptor affect its surface glycosylation, intracellular localization, and downstream signaling. Leuk. Res. Rep. 2020, 13, 100187. [Google Scholar] [CrossRef]
- Marensi, V.; Keeshan, K.R.; MacEwan, D.J. Pharmacological impact of FLT3 mutations on receptor activity and responsiveness to tyrosine kinase inhibitors. Biochem. Pharmacol. 2021, 183, 114348. [Google Scholar] [CrossRef] [PubMed]
- Limas, J.C.; Cook, J.G. Preparation for DNA replication: The key to a successful S phase. FEBS Lett. 2019, 593, 2853–2867. [Google Scholar] [CrossRef] [PubMed]
- Ovejero, S.; Bueno, A.; Sacristán, M.P. Working on Genomic Stability: From the S-Phase to Mitosis. Genes 2020, 11, 225. [Google Scholar] [CrossRef] [PubMed]
- Tambe, M.; Karjalainen, E.; Vähä-Koskela, M.; Bulanova, D.; Gjertsen, B.T.; Kontro, M.; Porkka, K.; Heckman, C.A.; Wennerberg, K. Pan-RAF inhibition induces apoptosis in acute myeloid leukemia cells and synergizes with BCL2 inhibition. Leukemia 2020, 34, 3186–3196. [Google Scholar] [CrossRef]
- Thomas, X. Acute Promyelocytic Leukemia: A History over 60 Years—From the Most Malignant to the most Curable Form of Acute Leukemia. Oncol. Ther. 2019, 7, 33–65. [Google Scholar] [CrossRef]
- Rocca, S.; Carrà, G.; Poggio, P.; Morotti, A.; Brancaccio, M. Targeting few to help hundreds: JAK, MAPK and ROCK pathways as druggable targets in atypical chronic myeloid leukemia. Mol. Cancer 2018, 17, 1–12. [Google Scholar] [CrossRef]
- Smith, C.C.; Wang, Q.; Chin, C.-S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nat. Cell Biol. 2012, 485, 260–263. [Google Scholar] [CrossRef]
- Von Bubnoff, N.; Engh, R.A.; Åberg, E.; Sänger, J.; Peschel, C.; Duyster, J. FMS-Like Tyrosine Kinase 3–Internal Tandem Duplication Tyrosine Kinase Inhibitors Display a Nonoverlapping Profile of Resistance Mutations In vitro. Cancer Res. 2009, 69, 3032–3041. [Google Scholar] [CrossRef]
- Ouerhani, S.; Bougatef, K.; Soltani, I.; Elgaaied, A.B.A.; Abbes, S.; Menif, S. The prevalence and prognostic significance of KRAS mutation in bladder cancer, chronic myeloid leukemia and colorectal cancer. Mol. Biol. Rep. 2013, 40, 4109–4114. [Google Scholar] [CrossRef]
- Rothenburger, T.; McLaughlin, K.-M.; Herold, T.; Schneider, C.; Oellerich, T.; Rothweiler, F.; Feber, A.; Fenton, T.R.; Wass, M.N.; Keppler, O.T.; et al. SAMHD1 is a key regulator of the lineage-specific response of acute lymphoblastic leukaemias to nelarabine. Commun. Biol. 2020, 3, 1–10. [Google Scholar] [CrossRef]
- Madaan, K.; Kaushik, D.; Verma, T. Hydroxyurea: A key player in cancer chemotherapy. Expert Rev. Anticancer Ther. 2012, 12, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kang, N.; Song, E.; Wie, M.; Lee, E.A.; Hwang, S.; Lee, D.; Ra, J.S.; Park, I.B.; Park, J.; et al. ATAD5 promotes replication restart by regulating RAD51 and PCNA in response to replication stress. Nat. Commun. 2019, 10, 5718. [Google Scholar] [CrossRef]
- Rickman, K.A.; Noonan, R.J.; Lach, F.; Sridhar, S.; Wang, A.; Abhyankar, A.; Huang, A.; Kelly, M.; Auerbach, A.D.; Smogorzewska, A. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 2020, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Su, L.; Hu, S.; Hu, W.; Yip, M.L.R.; Wu, J.; Gaur, S.; Smith, L.; Yuan, Y.-C.; Synold, T.; et al. A Small-Molecule Blocking Ribonucleotide Reductase Holoenzyme Formation Inhibits Cancer Cell Growth and Overcomes Drug Resistance. Cancer Res. 2013, 73, 6484–6493. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pons, M.; Zeyn, Y.; Zahn, S.; Mahendrarajah, N.; Page, B.D.G.; Gunning, P.T.; Moriggl, R.; Brenner, W.; Butter, F.; Krämer, O.H. Oncogenic Kinase Cascades Induce Molecular Mechanisms That Protect Leukemic Cell Models from Lethal Effects of De Novo dNTP Synthesis Inhibition. Cancers 2021, 13, 3464. https://doi.org/10.3390/cancers13143464
Pons M, Zeyn Y, Zahn S, Mahendrarajah N, Page BDG, Gunning PT, Moriggl R, Brenner W, Butter F, Krämer OH. Oncogenic Kinase Cascades Induce Molecular Mechanisms That Protect Leukemic Cell Models from Lethal Effects of De Novo dNTP Synthesis Inhibition. Cancers. 2021; 13(14):3464. https://doi.org/10.3390/cancers13143464
Chicago/Turabian StylePons, Miriam, Yanira Zeyn, Stella Zahn, Nisintha Mahendrarajah, Brent D. G. Page, Patrick T. Gunning, Richard Moriggl, Walburgis Brenner, Falk Butter, and Oliver H. Krämer. 2021. "Oncogenic Kinase Cascades Induce Molecular Mechanisms That Protect Leukemic Cell Models from Lethal Effects of De Novo dNTP Synthesis Inhibition" Cancers 13, no. 14: 3464. https://doi.org/10.3390/cancers13143464
APA StylePons, M., Zeyn, Y., Zahn, S., Mahendrarajah, N., Page, B. D. G., Gunning, P. T., Moriggl, R., Brenner, W., Butter, F., & Krämer, O. H. (2021). Oncogenic Kinase Cascades Induce Molecular Mechanisms That Protect Leukemic Cell Models from Lethal Effects of De Novo dNTP Synthesis Inhibition. Cancers, 13(14), 3464. https://doi.org/10.3390/cancers13143464