
Mutated p53 in HGSC—From a Common Mutation to a Target for Therapy

Abstract
Simple Summary
Abstract
1. Epithelial Ovarian Cancer
2. p53 Function in Normal Non-Cancerous Cells
3. TP53 Mutations in Cancer
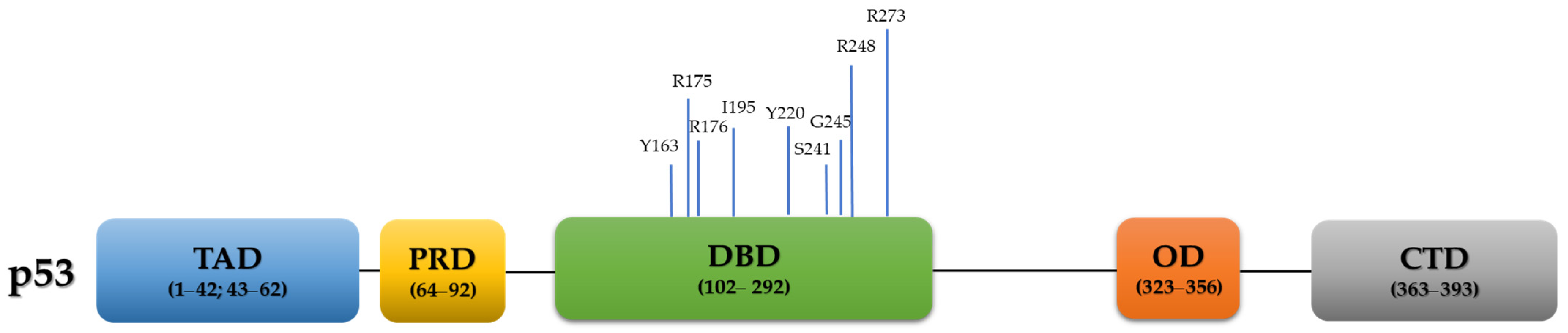
4. TP53 Mutations in HGSC and Their Clinical Relevance
5. Regulation and Function of p53 in HGSC
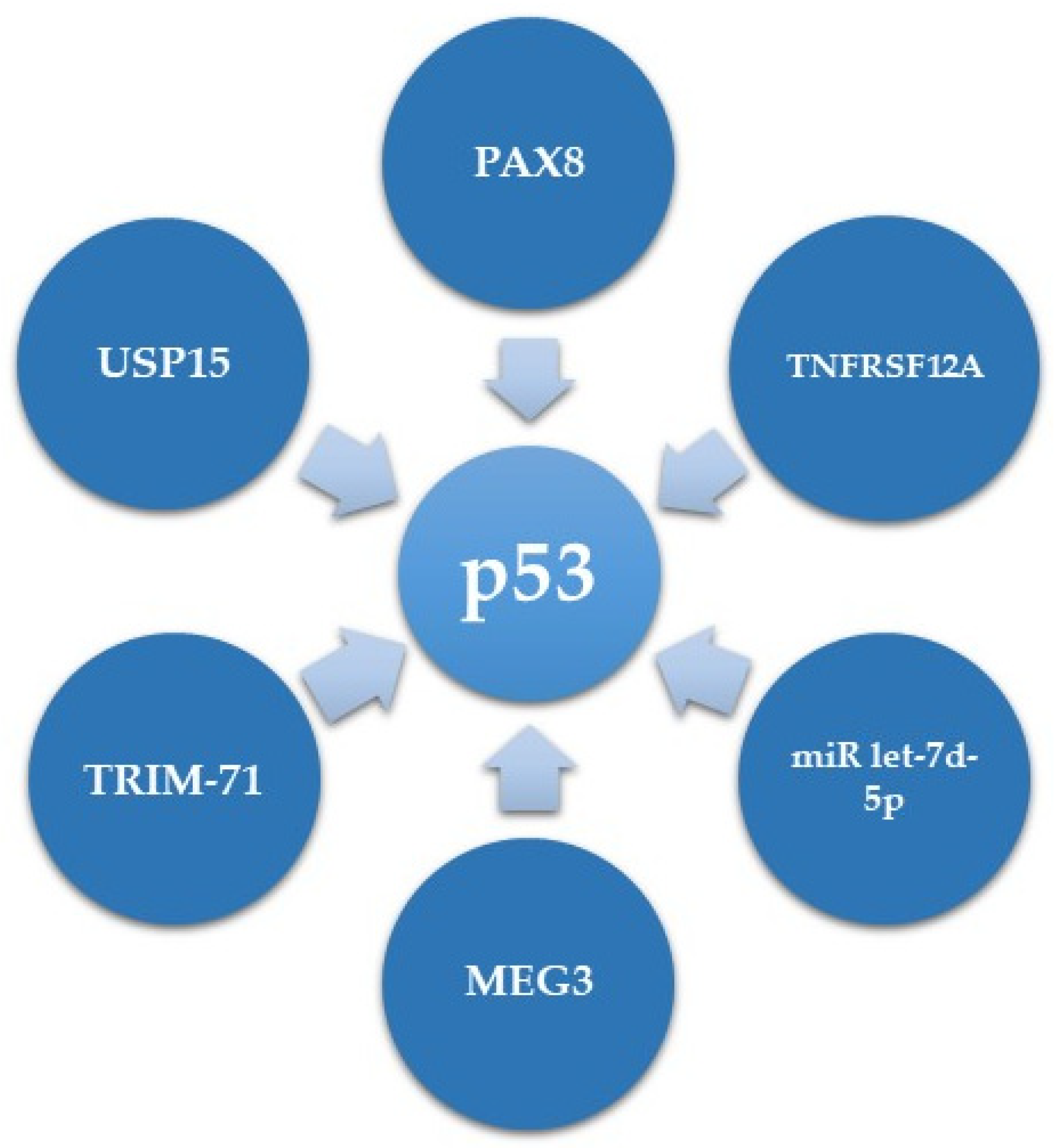
5.1. Upstream Regulators of p53 in HGSC
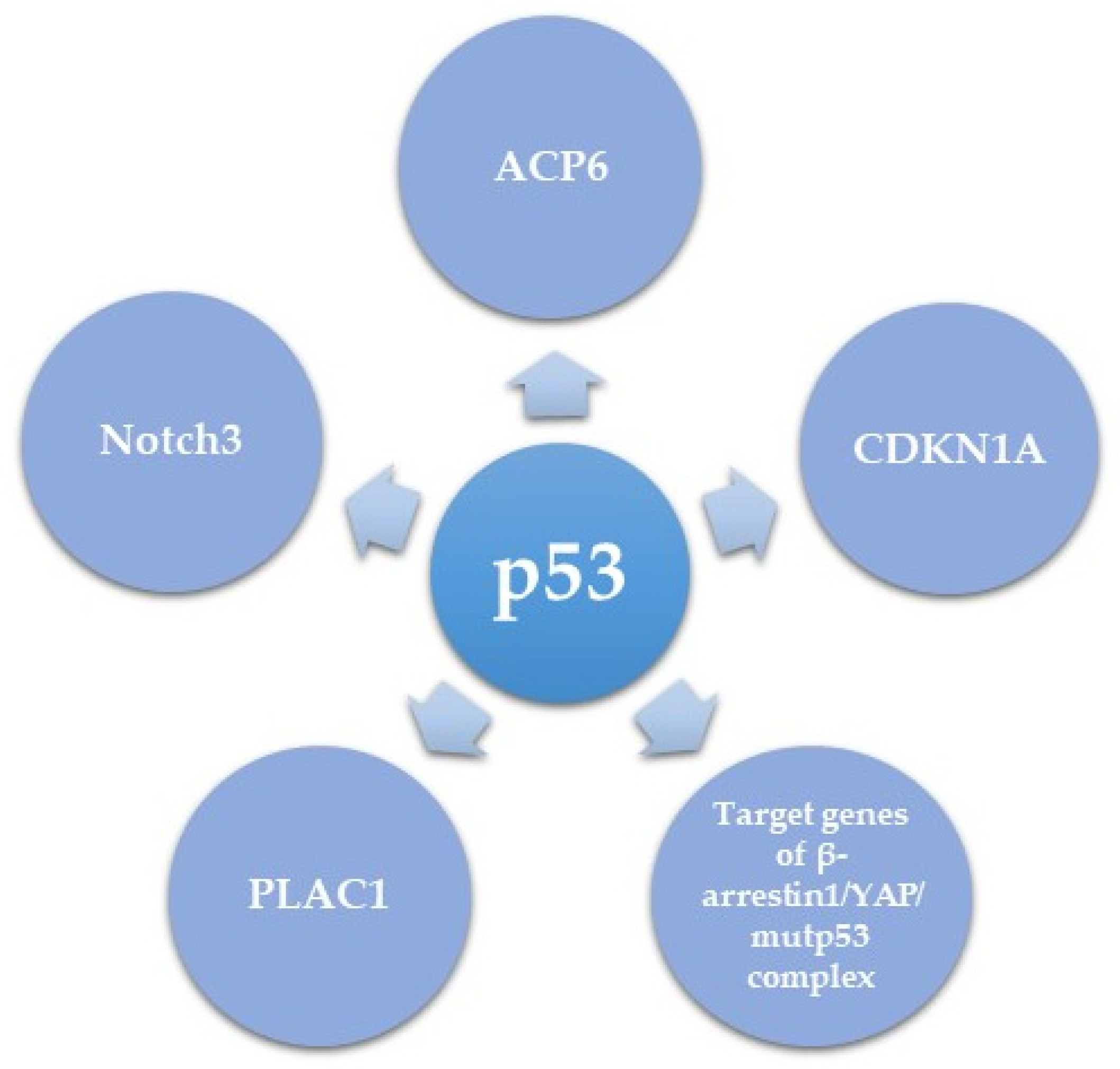
5.2. Downstream Effectors of Mutant p53 in HGSC
6. The Role of p53 in HGSC Pathogenesis
7. The Role of p53 in HGSC Microenvironment
8. p53 as a Target for EOC Therapy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Salehi, F.; Dunfield, L.; Phillips, K.P.; Krewski, D.; Vanderhyden, B.C. Risk Factors for Ovarian Cancer: An Overview with Emphasis on Hormonal Factors. J. Toxicol. Environ. Health Part B 2008, 11, 301–321. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Raja, F.A.; Fotopoulou, C.; Gonzalez-Martin, A.; Colombo, N.; Sessa, C. Newly Diagnosed and Relapsed Epithelial Ovarian Carcinoma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2013, 24, vi24–vi32. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.F.; Hu, W.; Sood, A.K. Microenvironment and Pathogenesis of Epithelial Ovarian Cancer. Horm. Cancer 2010, 1, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.-H.; Bast, R.C. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking Ovarian Cancer: Recommendations for Improving Outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 Mutations in Epithelial Ovarian Cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef]
- Shih, I.-M.; Kurman, R.J. Ovarian Tumorigenesis: A Proposed Model Based on Morphological and Molecular Genetic Analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.-M. Molecular Pathogenesis and Extraovarian Origin of Epithelial Ovarian Cancer. Shifting the Paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.-M. The Dualistic Model of Ovarian Carcinogenesis. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef]
- Jones, P.M.; Drapkin, R. Modeling High-Grade Serous Carcinoma: How Converging Insights into Pathogenesis and Genetics Are Driving Better Experimental Platforms. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer Statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Narod, S. Can Advanced-Stage Ovarian Cancer Be Cured? Nat. Rev. Clin. Oncol. 2016, 13, 255–261. [Google Scholar] [CrossRef]
- Ledermann, J.A. Front-Line Therapy of Advanced Ovarian Cancer: New Approaches. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, viii46–viii50. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab Combined with Chemotherapy for Platinum-Resistant Recurrent Ovarian Cancer: The AURELIA Open-Label Randomized Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.J.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib Combined with Chemotherapy for Recurrent Platinum-Sensitive Ovarian Cancer: A Randomised Phase 2 Trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the P53 Network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.P.; Bourdon, J.-C. The Isoforms of the P53 Protein. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Brosh, R.; Rotter, V. When Mutants Gain New Powers: News from the Mutant P53 Field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Tuna, M.; Ju, Z.; Yoshihara, K.; Amos, C.I.; Tanyi, J.L.; Mills, G.B. Clinical Relevance of TP53 Hotspot Mutations in High-Grade Serous Ovarian Cancers. Br. J. Cancer 2020, 122, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. P53 and Metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Bieging, K.T.; Attardi, L.D. Deconstructing P53 Transcriptional Networks in Tumor Suppression. Trends Cell Biol. 2012, 22, 97–106. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant P53: One Name, Many Proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Jin, S.; Levine, A.J. The P53 Functional Circuit. J. Cell Sci. 2001, 114, 4139–4140. [Google Scholar] [CrossRef]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of P53 Translation and Induction after DNA Damage by Ribosomal Protein L26 and Nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef]
- Comel, A.; Sorrentino, G.; Capaci, V.; Sal, G.D. The Cytoplasmic Side of P53’s Oncosuppressive Activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of P53 Stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 Promotes the Rapid Degradation of P53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 Is a Ubiquitin Ligase E3 for Tumor Suppressor P53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt Enhances Mdm2-Mediated Ubiquitination and Degradation of P53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef]
- Ko, L.J.; Prives, C. P53: Puzzle and Paradigm. Genes Dev. 1996, 10, 1054–1072. [Google Scholar] [CrossRef]
- Giaccia, A.J.; Kastan, M.B. The Complexity of P53 Modulation: Emerging Patterns from Divergent Signals. Genes Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Post-Translational Modification of P53 in Tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. Ubiquitination, Phosphorylation and Acetylation: The Molecular Basis for P53 Regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Scoumanne, A.; Chen, X. Protein Methylation: A New Mechanism of P53 Tumor Suppressor Regulation. Histol. Histopathol. 2008, 23, 1143–1149. [Google Scholar] [CrossRef]
- Kruse, J.-P.; Gu, W. SnapShot: P53 Posttranslational Modifications. Cell 2008, 133, 930-30.e1. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of P53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and Aging: The Critical Roles of P53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. New Insights into P53 Activation. Cell Res. 2010, 20, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A Global Map of P53 Transcription-Factor Binding Sites in the Human Genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Z.; Vogt Sionov, R.; Berger, M.; Zwang, Y.; Perets, R.; Van Etten, R.A.; Oren, M.; Taya, Y.; Haupt, Y. Tyrosine Phosphorylation of Mdm2 by C-Abl: Implications for P53 Regulation. EMBO J. 2002, 21, 3715–3727. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. P53 Ubiquitination: Mdm2 and Beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Wertz, I.; Shimizu, H.; Arnott, D.; Frantz, G.D.; Dowd, P.; O’Rourke, K.; Koeppen, H.; Dixit, V.M. The Ubiquitin Ligase COP1 Is a Critical Negative Regulator of P53. Nature 2004, 429, 86–92. [Google Scholar] [CrossRef]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a P53-Induced Ubiquitin-Protein Ligase, Promotes P53 Degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef]
- Chen, D.; Kon, N.; Li, M.; Zhang, W.; Qin, J.; Gu, W. ARF-BP1/Mule Is a Critical Mediator of the ARF Tumor Suppressor. Cell 2005, 121, 1071–1083. [Google Scholar] [CrossRef]
- Kruse, J.-P.; Gu, W. MSL2 Promotes Mdm2-Independent Cytoplasmic Localization of P53. J. Biol. Chem. 2009, 284, 3250–3263. [Google Scholar] [CrossRef]
- Marine, J.-C.W.; Dyer, M.A.; Jochemsen, A.G. MDMX: From Bench to Bedside. J. Cell Sci. 2007, 120, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The Common Mechanisms of Transformation by the Small DNA Tumor Viruses: The Inactivation of Tumor Suppressor Gene Products: P53. Virology 2009, 384, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 Mutations in Human Cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The First 30 Years of P53: Growing Ever More Complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.-A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing Mutant P53 in Primary High-Grade Serous Ovarian Cancer Using Immunohistochemistry and Massively Parallel Sequencing. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Garziera, M.; Cecchin, E.; Canzonieri, V.; Sorio, R.; Giorda, G.; Scalone, S.; De Mattia, E.; Roncato, R.; Gagno, S.; Poletto, E.; et al. Identification of Novel Somatic TP53 Mutations in Patients with High-Grade Serous Ovarian Cancer (HGSOC) Using Next-Generation Sequencing (NGS). Int. J. Mol. Sci. 2018, 19, 1510. [Google Scholar] [CrossRef]
- Brachova, P.; Thiel, K.W.; Leslie, K.K. The Consequence of Oncomorphic TP53 Mutations in Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 19257–19275. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. Mutant P53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Hashimoto, T.; Tokuchi, Y.; Hayashi, M.; Kobayashi, Y.; Nishida, K.; Hayashi, S.; Ishikawa, Y.; Tsuchiya, S.; Nakagawa, K.; Hayashi, J.; et al. P53 Null Mutations Undetected by Immunohistochemical Staining Predict a Poor Outcome with Early-Stage Non-Small Cell Lung Carcinomas. Cancer Res. 1999, 59, 5572–5577. [Google Scholar]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of Function Mutations in P53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant P53 Gain-of-Function in Cancer. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant P53 Gain of Function in Two Mouse Models of Li-Fraumeni Syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Silwal-Pandit, L.; Langerød, A.; Børresen-Dale, A.-L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef]
- Sturm, I.; Bosanquet, A.G.; Hermann, S.; Güner, D.; Dörken, B.; Daniel, P.T. Mutation of P53 and Consecutive Selective Drug Resistance in B-CLL Occurs as a Consequence of Prior DNA-Damaging Chemotherapy. Cell Death Differ. 2003, 10, 477–484. [Google Scholar] [CrossRef]
- Brachova, P.; Mueting, S.R.; Carlson, M.J.; Goodheart, M.J.; Button, A.M.; Mott, S.L.; Dai, D.; Thiel, K.W.; Devor, E.J.; Leslie, K.K. TP53 Oncomorphic Mutations Predict Resistance to Platinum- and Taxane-Based Standard Chemotherapy in Patients Diagnosed with Advanced Serous Ovarian Carcinoma. Int. J. Oncol. 2014, 46, 607–618. [Google Scholar] [CrossRef]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, 16. [Google Scholar] [CrossRef]
- Sigal, A.; Rotter, V. Oncogenic Mutations of the P53 Tumor Suppressor: The Demons of the Guardian of the Genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar] [PubMed]
- Kang, H.J.; Chun, S.-M.; Kim, K.-R.; Sohn, I.; Sung, C.O. Clinical Relevance of Gain-Of-Function Mutations of P53 in High-Grade Serous Ovarian Carcinoma. PLoS ONE 2013, 8, e72609. [Google Scholar] [CrossRef]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the P53 Tumor Suppressor Gene. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 Mutations in Human Cancers: Functional Selection and Impact on Cancer Prognosis and Outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef]
- Brázdová, M.; Navrátilová, L.; Tichý, V.; Němcová, K.; Lexa, M.; Hrstka, R.; Pečinka, P.; Adámik, M.; Vojtesek, B.; Paleček, E.; et al. Preferential Binding of Hot Spot Mutant P53 Proteins to Supercoiled DNA In Vitro and in Cells. PLoS ONE 2013, 8, e59567. [Google Scholar] [CrossRef]
- Edlund, K.; Larsson, O.; Ameur, A.; Bunikis, I.; Gyllensten, U.; Leroy, B.; Sundström, M.; Micke, P.; Botling, J.; Soussi, T. Data-Driven Unbiased Curation of the TP53 Tumor Suppressor Gene Mutation Database and Validation by Ultradeep Sequencing of Human Tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 9551–9556. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal Structure of a P53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic Mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Attardi, L.D. Not All P53 Gain-of-Function Mutants Are Created Equal. Cell Death Differ. 2013, 20, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor P53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Sasaki, M.; Maki, C.G. Regulation of P53 Nuclear Export through Sequential Changes in Conformation and Ubiquitination *. J. Biol. Chem. 2007, 282, 14616–14625. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional Control of Human P53-Regulated Genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver Mutations in TP53 Are Ubiquitous in High Grade Serous Carcinoma of the Ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef]
- Levanon, K.; Ng, V.; Piao, H.-Y.; Zhang, Y.; Chang, M.C.; Roh, M.H.; Kindelberger, D.W.; Hirsch, M.S.; Crum, C.P.; Marto, J.A.; et al. Primary Ex-Vivo Cultures of Human Fallopian Tube Epithelium as a Model for Serous Ovarian Carcinogenesis. Oncogene 2010, 29, 1103–1113. [Google Scholar] [CrossRef]
- Toyooka, S.; Kiura, K.; Mitsudomi, T. EGFR Mutation and Response of Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 2136, author reply 2136. [Google Scholar] [CrossRef]
- Kim, H.-N.; Woo, H.Y.; Do, S.-I.; Kim, H.-S. Targeted Sequencing of Tubo-Ovarian and Peritoneal High-Grade Serous Carcinoma With Wild-Type P53 Immunostaining Pattern. Vivo Athens Greece 2019, 33, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Yemelyanova, A.; Vang, R.; Kshirsagar, M.; Lu, D.; Marks, M.A.; Shih, I.M.; Kurman, R.J. Immunohistochemical Staining Patterns of P53 Can Serve as a Surrogate Marker for TP53 Mutations in Ovarian Carcinoma: An Immunohistochemical and Nucleotide Sequencing Analysis. Mod. Pathol. 2011, 24, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Garziera, M.; Roncato, R.; Montico, M.; De Mattia, E.; Gagno, S.; Poletto, E.; Scalone, S.; Canzonieri, V.; Giorda, G.; Sorio, R.; et al. New Challenges in Tumor Mutation Heterogeneity in Advanced Ovarian Cancer by a Targeted Next-Generation Sequencing (NGS) Approach. Cells 2019, 8, 584. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C. P53 and Its Isoforms in Cancer. Br. J. Cancer 2007, 97, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Perrier, S.; Aoubala, M.; Ageorges, S.; Groves, M.J.; Diot, A.; Fernandes, K.; Tauro, S.; Bourdon, J.-C. Δ160p53 Is a Novel N-Terminal P53 Isoform Encoded by Δ133p53 Transcript. FEBS Lett. 2010, 584, 4463–4468. [Google Scholar] [CrossRef]
- Senturk, S.; Yao, Z.; Camiolo, M.; Stiles, B.; Rathod, T.; Walsh, A.M.; Nemajerova, A.; Lazzara, M.J.; Altorki, N.K.; Krainer, A.; et al. P53Ψ Is a Transcriptionally Inactive P53 Isoform Able to Reprogram Cells toward a Metastatic-like State. Proc. Natl. Acad. Sci. USA 2014, 111, E3287–E3296. [Google Scholar] [CrossRef]
- Bischof, K.; Knappskog, S.; Hjelle, S.M.; Stefansson, I.; Woie, K.; Salvesen, H.B.; Gjertsen, B.T.; Bjorge, L. Influence of P53 Isoform Expression on Survival in High-Grade Serous Ovarian Cancers. Sci. Rep. 2019, 9, 5244. [Google Scholar] [CrossRef]
- Vang, R.; Levine, D.A.; Soslow, R.A.; Zaloudek, C.; Shih, I.-M.; Kurman, R.J. Molecular Alterations of TP53 Are a Defining Feature of Ovarian High-Grade Serous Carcinoma: A Rereview of Cases Lacking TP53 Mutations in The Cancer Genome Atlas Ovarian Study. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2016, 35, 48–55. [Google Scholar] [CrossRef]
- Chui, M.H.; Momeni Boroujeni, A.; Mandelker, D.; Ladanyi, M.; Soslow, R.A. Characterization of TP53-Wildtype Tubo-Ovarian High-Grade Serous Carcinomas: Rare Exceptions to the Binary Classification of Ovarian Serous Carcinoma. Mod. Pathol. 2021, 34, 490–501. [Google Scholar] [CrossRef]
- Ghannam-Shahbari, D.; Jacob, E.; Kakun, R.R.; Wasserman, T.; Korsensky, L.; Sternfeld, O.; Kagan, J.; Bublik, D.R.; Aviel-Ronen, S.; Levanon, K.; et al. PAX8 Activates a P53-P21-Dependent pro-Proliferative Effect in High Grade Serous Ovarian Carcinoma. Oncogene 2018, 37, 2213–2224. [Google Scholar] [CrossRef]
- Wu, A.-Y.; Gu, L.-Y.; Cang, W.; Cheng, M.-X.; Wang, W.-J.; Di, W.; Huang, L.; Qiu, L.-H. Fn14 Overcomes Cisplatin Resistance of High-Grade Serous Ovarian Cancer by Promoting Mdm2-Mediated P53-R248Q Ubiquitination and Degradation. J. Exp. Clin. Cancer Res. CR 2019, 38. [Google Scholar] [CrossRef]
- Chen, Y.; Hao, Q.; Wang, J.; Li, J.; Huang, C.; Zhang, Y.; Wu, X.; Lu, H.; Zhou, X. Ubiquitin Ligase TRIM71 Suppresses Ovarian Tumorigenesis by Degrading Mutant P53. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Candelaria, N.; Wong, K.-K.; Nikolai, B.C.; Lonard, D.M.; O’Malley, B.W.; Richards, J.S. USP15-Dependent Lysosomal Pathway Controls P53-R175H Turnover in Ovarian Cancer Cells. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Sheng, X.; Li, J.; Yang, L.; Chen, Z.; Zhao, Q.; Tan, L.; Zhou, Y.; Li, J. Promoter Hypermethylation Influences the Suppressive Role of Maternally Expressed 3, a Long Non-Coding RNA, in the Development of Epithelial Ovarian Cancer. Oncol. Rep. 2014, 32, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-N.; Ren, C.-C.; Yang, L.; Nai, M.-M.; Xu, Y.-M.; Zhang, F.; Liu, Y. MicroRNA Let-7d-5p Rescues Ovarian Cancer Cell Apoptosis and Restores Chemosensitivity by Regulating the P53 Signaling Pathway via HMGA1. Int. J. Oncol. 2019, 54, 1771–1784. [Google Scholar] [CrossRef]
- Yin, J.; Kim, T.-H.; Park, N.; Shin, D.; Choi, H.I.; Cho, S.; Park, J.B.; Kim, J.H. TRIM71 Suppresses Tumorigenesis via Modulation of Lin28B-Let-7-HMGA2 Signaling. Oncotarget 2016, 7, 79854–79868. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, P.J.A.; Rodón, L.; Gonzàlez-Juncà, A.; Dirac, A.; Gili, M.; Martínez-Sáez, E.; Aura, C.; Barba, I.; Peg, V.; Prat, A.; et al. USP15 Stabilizes TGF-β Receptor I and Promotes Oncogenesis through the Activation of TGF-β Signaling in Glioblastoma. Nat. Med. 2012, 18, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-C.; Spitale, R.C.; Chang, H.Y. Long Intergenic Non-Coding RNAs–New Links in Cancer Progression. Cancer Res. 2011, 71, 3–7. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The Role of MicroRNAs in Human Cancer. Signal Transduct. Target. Ther. 2016, 1, 1–9. [Google Scholar] [CrossRef]
- Bach, D.-H.; Lee, S.K.; Sood, A.K. Circular RNAs in Cancer. Mol. Ther. Nucleic Acids 2019, 16, 118–129. [Google Scholar] [CrossRef]
- Stein, Y.; Aloni-Grinstein, R.; Rotter, V. Mutant P53 Oncogenicity: Dominant-Negative or Gain-of-Function? Carcinogenesis 2020, 41, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Koslowski, M.; Sahin, U.; Mitnacht-Kraus, R.; Seitz, G.; Huber, C.; Türeci, Ö. A Placenta-Specific Gene Ectopically Activated in Many Human Cancers Is Essentially Involved in Malignant Cell Processes. Cancer Res. 2007, 67, 9528–9534. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lin, X.; Di, X.; Chen, Y.; Zhao, H.; Wang, X. Oncogenic Function of Plac1 on the Proliferation and Metastasis in Hepatocellular Carcinoma Cells. Oncol. Rep. 2017, 37, 465–473. [Google Scholar] [CrossRef]
- Chen, Y.; Schlessinger, D.; Nagaraja, R. T Antigen Transformation Reveals Tp53/RB-Dependent Route to PLAC1 Transcription Activation in Primary Fibroblasts. Oncogenesis 2013, 2, e67. [Google Scholar] [CrossRef] [PubMed]
- Devor, E.J.; Gonzalez-Bosquet, J.; Warrier, A.; Reyes, H.D.; Ibik, N.V.; Schickling, B.M.; Newtson, A.; Goodheart, M.J.; Leslie, K.K. P53 Mutation Status Is a Primary Determinant of Placenta-Specific Protein 1 Expression in Serous Ovarian Cancers. Int. J. Oncol. 2017, 50, 1721–1728. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Q.; Miao, C.; Dongol, S.; Li, Y.; Jin, C.; Dong, R.; Li, Y.; Yang, X.; Kong, B. CCNG1 (Cyclin G1) Regulation by Mutant-P53 via Induction of Notch3 Expression Promotes High-grade Serous Ovarian Cancer (HGSOC) Tumorigenesis and Progression. Cancer Med. 2018, 8, 351–362. [Google Scholar] [CrossRef]
- Tocci, P.; Cianfrocca, R.; Di Castro, V.; Rosanò, L.; Sacconi, A.; Donzelli, S.; Bonfiglio, S.; Bucci, G.; Vizza, E.; Ferrandina, G.; et al. β-Arrestin1/YAP/Mutant P53 Complexes Orchestrate the Endothelin A Receptor Signaling in High-Grade Serous Ovarian Cancer. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Chryplewicz, A.; Tienda, S.M.; Nahotko, D.A.; Peters, P.N.; Lengyel, E.; Eckert, M.A. Mutant P53 Regulates LPA Signaling through Lysophosphatidic Acid Phosphatase Type 6. Sci. Rep. 2019, 9, 5195. [Google Scholar] [CrossRef]
- Nath, A.; Cosgrove, P.A.; Mirsafian, H.; Christie, E.L.; Pflieger, L.; Copeland, B.; Majumdar, S.; Cristea, M.C.; Han, E.S.; Lee, S.J.; et al. Evolution of Core Archetypal Phenotypes in Progressive High Grade Serous Ovarian Cancer. Nat. Commun. 2021, 12, 3039. [Google Scholar] [CrossRef]
- Vang, R.; Shih, I.-M.; Kurman, R.J. Fallopian Tube Precursors of Ovarian Low- and High-Grade Serous Neoplasms. Histopathology 2013, 62, 44–58. [Google Scholar] [CrossRef]
- Meserve, E.E.K.; Brouwer, J.; Crum, C.P. Serous Tubal Intraepithelial Neoplasia: The Concept and Its Application. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2017, 30, 710–721. [Google Scholar] [CrossRef]
- Singh, N.; McCluggage, W.G.; Gilks, C.B. High-Grade Serous Carcinoma of Tubo-Ovarian Origin: Recent Developments. Histopathology 2017, 71, 339–356. [Google Scholar] [CrossRef]
- Perets, R.; Drapkin, R. It’s Totally Tubular Riding the New Wave of Ovarian Cancer Research. Cancer Res. 2016, 76, 10–17. [Google Scholar] [CrossRef]
- Bell, D.A. Origins and Molecular Pathology of Ovarian Cancer. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2005, 18 (Suppl. 2), S19–S32. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.A.; Scully, R.E. Early de Novo Ovarian Carcinoma. A Study of Fourteen Cases. Cancer 1994, 73, 1859–1864. [Google Scholar] [CrossRef]
- Werness, B.A.; Afify, A.M.; Bielat, K.L.; Eltabbakh, G.H.; Piver, M.S.; Paterson, J.M. Altered Surface and Cyst Epithelium of Ovaries Removed Prophylactically from Women with a Family History of Ovarian Cancer. Hum. Pathol. 1999, 30, 151–157. [Google Scholar] [CrossRef]
- Landen, C.N.; Birrer, M.J.; Sood, A.K. Early Events in the Pathogenesis of Epithelial Ovarian Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Piek, J.M.; van Diest, P.J.; Zweemer, R.P.; Jansen, J.W.; Poort-Keesom, R.J.; Menko, F.H.; Gille, J.J.; Jongsma, A.P.; Pals, G.; Kenemans, P.; et al. Dysplastic Changes in Prophylactically Removed Fallopian Tubes of Women Predisposed to Developing Ovarian Cancer. J. Pathol. 2001, 195, 451–456. [Google Scholar] [CrossRef]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial Carcinoma of the Fimbria and Pelvic Serous Carcinoma: Evidence for a Causal Relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef]
- Lee, Y.; Miron, A.; Drapkin, R.; Nucci, M.R.; Medeiros, F.; Saleemuddin, A.; Garber, J.; Birch, C.; Mou, H.; Gordon, R.W.; et al. A Candidate Precursor to Serous Carcinoma That Originates in the Distal Fallopian Tube. J. Pathol. 2007, 211, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Leeper, K.; Garcia, R.; Swisher, E.; Goff, B.; Greer, B.; Paley, P. Pathologic Findings in Prophylactic Oophorectomy Specimens in High-Risk Women. Gynecol. Oncol. 2002, 87, 52–56. [Google Scholar] [CrossRef]
- Levanon, K.; Crum, C.; Drapkin, R. New Insights into the Pathogenesis of Serous Ovarian Cancer and Its Clinical Impact. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5284–5293. [Google Scholar] [CrossRef]
- Medeiros, F.; Muto, M.G.; Lee, Y.; Elvin, J.A.; Callahan, M.J.; Feltmate, C.; Garber, J.E.; Cramer, D.W.; Crum, C.P. The Tubal Fimbria Is a Preferred Site for Early Adenocarcinoma in Women with Familial Ovarian Cancer Syndrome. Am. J. Surg. Pathol. 2006, 30, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Przybycin, C.G.; Kurman, R.J.; Ronnett, B.M.; Shih, I.-M.; Vang, R. Are All Pelvic (Nonuterine) Serous Carcinomas of Tubal Origin? Am. J. Surg. Pathol. 2010, 34, 1407–1416. [Google Scholar] [CrossRef]
- Cass, I.; Walts, A.E.; Barbuto, D.; Lester, J.; Karlan, B. A Cautious View of Putative Precursors of Serous Carcinomas in the Fallopian Tubes of BRCA Mutation Carriers. Gynecol. Oncol. 2014, 134, 492–497. [Google Scholar] [CrossRef]
- Powell, C.B.; Swisher, E.M.; Cass, I.; McLennan, J.; Norquist, B.; Garcia, R.L.; Lester, J.; Karlan, B.Y.; Chen, L. Long Term Follow up of BRCA1 and BRCA2 Mutation Carriers with Unsuspected Neoplasia Identified at Risk Reducing Salpingo-Oophorectomy. Gynecol. Oncol. 2013, 129, 364–371. [Google Scholar] [CrossRef]
- Karst, A.M.; Levanon, K.; Drapkin, R. Modeling High-Grade Serous Ovarian Carcinogenesis from the Fallopian Tube. Proc. Natl. Acad. Sci. USA 2011, 108, 7547–7552. [Google Scholar] [CrossRef]
- Jazaeri, A.A.; Bryant, J.L.; Park, H.; Li, H.; Dahiya, N.; Stoler, M.H.; Ferriss, J.S.; Dutta, A. Molecular Requirements for Transformation of Fallopian Tube Epithelial Cells into Serous Carcinoma. Neoplasia N. Y. N 2011, 13, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.H.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the Fallopian Tube Secretory Epithelium Leads to High-Grade Serous Ovarian Cancer in Brca;Tp53;Pten Models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef]
- Wu, N.-Y.Y.; Fang, C.; Huang, H.-S.; Wang, J.; Chu, T.-Y. Natural History of Ovarian High-Grade Serous Carcinoma from Time Effects of Ovulation Inhibition and Progesterone Clearance of P53-Defective Lesions. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2020, 33, 29–37. [Google Scholar] [CrossRef]
- Karst, A.M.; Drapkin, R. Ovarian Cancer Pathogenesis: A Model in Evolution. J. Oncol. 2010, 2010, 932371. [Google Scholar] [CrossRef] [PubMed]
- Bijron, J.G.; Seldenrijk, C.A.; Zweemer, R.P.; Lange, J.G.; Verheijen, R.H.M.; van Diest, P.J. Fallopian Tube Intraluminal Tumor Spread from Noninvasive Precursor Lesions: A Novel Metastatic Route in Early Pelvic Carcinogenesis. Am. J. Surg. Pathol. 2013, 37, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Crum, C.P. Intercepting Pelvic Cancer in the Distal Fallopian Tube: Theories and Realities. Mol. Oncol. 2009, 3, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.-L.; Shih, I.-M. TP53 Mutations in Serous Tubal Intraepithelial Carcinoma and Concurrent Pelvic High-Grade Serous Carcinoma--Evidence Supporting the Clonal Relationship of the Two Lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef]
- McDaniel, A.S.; Stall, J.N.; Hovelson, D.H.; Cani, A.K.; Liu, C.-J.; Tomlins, S.A.; Cho, K.R. Next-Generation Sequencing of Tubal Intraepithelial Carcinomas. JAMA Oncol. 2015, 1, 1128–1132. [Google Scholar] [CrossRef]
- Bashashati, A.; Ha, G.; Tone, A.; Ding, J.; Prentice, L.M.; Roth, A.; Rosner, J.; Shumansky, K.; Kalloger, S.; Senz, J.; et al. Distinct Evolutionary Trajectories of Primary High-Grade Serous Ovarian Cancers Revealed through Spatial Mutational Profiling. J. Pathol. 2013, 231, 21–34. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High Grade Serous Ovarian Carcinomas Originate in the Fallopian Tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Kohn, E.C.; Ivy, S.P. Whence High-Grade Serous Ovarian Cancer. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2017, 37, 443–448. [Google Scholar] [CrossRef]
- Quartuccio, S.M.; Karthikeyan, S.; Eddie, S.L.; Lantvit, D.D.; Ó hAinmhire, E.; Modi, D.A.; Wei, J.-J.; Burdette, J.E. Mutant P53 Expression in Fallopian Tube Epithelium Drives Cell Migration. Int. J. Cancer 2015, 137, 1528–1538. [Google Scholar] [CrossRef]
- Novak, M.; Lester, J.; Karst, A.M.; Parkash, V.; Hirsch, M.S.; Crum, C.P.; Karlan, B.Y.; Drapkin, R. Stathmin 1 and P16(INK4A) Are Sensitive Adjunct Biomarkers for Serous Tubal Intraepithelial Carcinoma. Gynecol. Oncol. 2015, 139, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Hatano, Y.; Fukuda, S.; Makino, H.; Tomita, H.; Morishige, K.-I.; Hara, A. High-Grade Serous Carcinoma with Discordant P53 Signature: Report of a Case with New Insight Regarding High-Grade Serous Carcinogenesis. Diagn. Pathol. 2018, 13, 24. [Google Scholar] [CrossRef]
- Schildkraut, J.M.; Bastos, E.; Berchuck, A. Relationship between Lifetime Ovulatory Cycles and Overexpression of Mutant P53 in Epithelial Ovarian Cancer. J. Natl. Cancer Inst. 1997, 89, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Fathalla, M.F. Incessant Ovulation--a Factor in Ovarian Neoplasia? Lancet Lond. Engl. 1971, 2, 163. [Google Scholar] [CrossRef]
- Lurie, G.; Wilkens, L.R.; Thompson, P.J.; McDuffie, K.E.; Carney, M.E.; Terada, K.Y.; Goodman, M.T. Combined Oral Contraceptive Use and Epithelial Ovarian Cancer Risk: Time-Related Effects. Epidemiol. Camb. Mass 2008, 19, 237–243. [Google Scholar] [CrossRef]
- Beral, V.; Doll, R.; Hermon, C.; Peto, R.; Reeves, G.; Collaborative Group on Epidemiological Studies of Ovarian Cancer. Ovarian Cancer and Oral Contraceptives: Collaborative Reanalysis of Data from 45 Epidemiological Studies Including 23,257 Women with Ovarian Cancer and 87,303 Controls. Lancet Lond. Engl. 2008, 371, 303–314. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Lantvit, D.D.; Chae, D.H.; Burdette, J.E. Cadherin-6 Type 2, K-Cadherin (CDH6) Is Regulated by Mutant P53 in the Fallopian Tube but Is Not Expressed in the Ovarian Surface. Oncotarget 2016, 7, 69871–69882. [Google Scholar] [CrossRef]
- Kang, M.; Chong, K.Y.; Hartwich, T.M.P.; Bi, F.; Witham, A.K.; Patrick, D.; Morrisson, M.J.; Cady, S.L.; Cerchia, A.P.; Kelk, D.; et al. Ovarian BDNF Promotes Survival, Migration, and Attachment of Tumor Precursors Originated from P53 Mutant Fallopian Tube Epithelial Cells. Oncogenesis 2020, 9, 55. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Akahane, T.; Hirasawa, A.; Tsuda, H.; Kataoka, F.; Nishimura, S.; Tanaka, H.; Tominaga, E.; Nomura, H.; Chiyoda, T.; Iguchi, Y.; et al. The Origin of Stroma Surrounding Epithelial Ovarian Cancer Cells. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2013, 32, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Patocs, A.; Zhang, L.; Xu, Y.; Weber, F.; Caldes, T.; Mutter, G.L.; Platzer, P.; Eng, C. Breast-Cancer Stromal Cells with TP53 Mutations and Nodal Metastases. N. Engl. J. Med. 2007, 357, 2543–2551. [Google Scholar] [CrossRef]
- Lopez-Martinez, M.; Anzola, M.; Cuevas, N.; Aguirre, J.M.; De-Pancorbo, M. Clinical Applications of the Diagnosis of P53 Alterations in Squamous Cell Carcinoma of the Head and Neck. Med. Oral Organo Of. Soc. Espanola Med. Oral Acad. Iberoam. Patol. Med. Bucal 2002, 7, 108–120. [Google Scholar]
- Ganly, I.; Soutar, D.S.; Brown, R.; Kaye, S.B. P53 Alterations in Recurrent Squamous Cell Cancer of the Head and Neck Refractory to Radiotherapy. Br. J. Cancer 2000, 82, 392–398. [Google Scholar] [CrossRef]
- Schauer, I.G.; Zhang, J.; Xing, Z.; Guo, X.; Mercado-Uribe, I.; Sood, A.K.; Huang, P.; Liu, J. Interleukin-1β Promotes Ovarian Tumorigenesis through a P53/NF-ΚB-Mediated Inflammatory Response in Stromal Fibroblasts. Neoplasia N. Y. N 2013, 15, 409–420. [Google Scholar] [CrossRef]
- Trachootham, D.; Chen, G.; Zhang, W.; Lu, W.; Zhang, H.; Liu, J.; Huang, P. Loss of P53 in Stromal Fibroblasts Promotes Epithelial Cell Invasion through Redox-Mediated ICAM1 Signal. Free Radic. Biol. Med. 2013, 58, 1–13. [Google Scholar] [CrossRef][Green Version]
- Hwang, C.-I.; Choi, J.; Zhou, Z.; Flesken-Nikitin, A.; Tarakhovsky, A.; Nikitin, A.Y. MET-Dependent Cancer Invasion May Be Preprogrammed by Early Alterations of P53-Regulated Feedforward Loop and Triggered by Stromal Cell-Derived HGF. Cell Cycle Georget. Tex 2011, 10, 3834–3840. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, Metastasis, Motility and More. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET Signalling: Principles and Functions in Development, Organ Regeneration and Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Ness, R.B.; Cottreau, C. Possible Role of Ovarian Epithelial Inflammation in Ovarian Cancer. J. Natl. Cancer Inst. 1999, 91, 1459–1467. [Google Scholar] [CrossRef]
- Fleming, J.S.; Beaugié, C.R.; Haviv, I.; Chenevix-Trench, G.; Tan, O.L. Incessant Ovulation, Inflammation and Epithelial Ovarian Carcinogenesis: Revisiting Old Hypotheses. Mol. Cell. Endocrinol. 2006, 247, 4–21. [Google Scholar] [CrossRef]
- Jia, D.; Nagaoka, Y.; Katsumata, M.; Orsulic, S. Inflammation Is a Key Contributor to Ovarian Cancer Cell Seeding. Sci. Rep. 2018, 8, 12394. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging Roles of P53 and Other Tumour-Suppressor Genes in Immune Regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Cui, Y. New Perspective on Targeting the Tumor Suppressor P53 Pathway in the Tumor Microenvironment to Enhance the Efficacy of Immunotherapy. J. Immunother. Cancer 2015, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Shatz, M.; Resnick, M.A. Interactions between the Tumor Suppressor P53 and Immune Responses. Curr. Opin. Oncol. 2013, 25, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Ak, P.; Levine, A.J. P53 and NF-ΚB: Different Strategies for Responding to Stress Lead to a Functional Antagonism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3643–3652. [Google Scholar] [CrossRef]
- Mori, T.; Anazawa, Y.; Iiizumi, M.; Fukuda, S.; Nakamura, Y.; Arakawa, H. Identification of the Interferon Regulatory Factor 5 Gene (IRF-5) as a Direct Target for P53. Oncogene 2002, 21, 2914–2918. [Google Scholar] [CrossRef]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. P53-Dependent Chemokine Production by Senescent Tumor Cells Supports NKG2D-Dependent Tumor Elimination by Natural Killer Cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
- Di Minin, G.; Bellazzo, A.; Dal Ferro, M.; Chiaruttini, G.; Nuzzo, S.; Bicciato, S.; Piazza, S.; Rami, D.; Bulla, R.; Sommaggio, R.; et al. Mutant P53 Reprograms TNF Signaling in Cancer Cells through Interaction with the Tumor Suppressor DAB2IP. Mol. Cell 2014, 56, 617–629. [Google Scholar] [CrossRef]
- Pils, D.; Pinter, A.; Reibenwein, J.; Alfanz, A.; Horak, P.; Schmid, B.C.; Hefler, L.; Horvat, R.; Reinthaller, A.; Zeillinger, R.; et al. In Ovarian Cancer the Prognostic Influence of HER2/Neu Is Not Dependent on the CXCR4/SDF-1 Signalling Pathway. Br. J. Cancer 2007, 96, 485–491. [Google Scholar] [CrossRef]
- Popple, A.; Durrant, L.G.; Spendlove, I.; Rolland, P.; Scott, I.V.; Deen, S.; Ramage, J.M. The Chemokine, CXCL12, Is an Independent Predictor of Poor Survival in Ovarian Cancer. Br. J. Cancer 2012, 106, 1306–1313. [Google Scholar] [CrossRef]
- Son, D.-S.; Parl, A.K.; Rice, V.M.; Khabele, D. Keratinocyte Chemoattractant (KC)/Human Growth-Regulated Oncogene (GRO) Chemokines and pro-Inflammatory Chemokine Networks in Mouse and Human Ovarian Epithelial Cancer Cells. Cancer Biol. Ther. 2007, 6, 1302–1312. [Google Scholar] [CrossRef]
- Son, D.-S.; Kabir, S.M.; Dong, Y.-L.; Lee, E.; Adunyah, S.E. Inhibitory Effect of Tumor Suppressor P53 on Proinflammatory Chemokine Expression in Ovarian Cancer Cells by Reducing Proteasomal Degradation of IκB. PLoS ONE 2012, 7, e51116. [Google Scholar] [CrossRef]
- Soussi, T. P53 Antibodies in the Sera of Patients with Various Types of Cancer: A Review. Cancer Res. 2000, 60, 1777–1788. [Google Scholar]
- Lambeck, A.; Leffers, N.; Hoogeboom, B.-N.; Sluiter, W.; Hamming, I.; Klip, H.; ten Hoor, K.; Esajas, M.; van Oven, M.; Drijfhout, J.-W.; et al. P53-Specific T Cell Responses in Patients with Malignant and Benign Ovarian Tumors: Implications for P53 Based Immunotherapy. Int. J. Cancer 2007, 121, 606–614. [Google Scholar] [CrossRef]
- Yang, W.-L.; Gentry-Maharaj, A.; Simmons, A.; Ryan, A.; Fourkala, E.O.; Lu, Z.; Baggerly, K.A.; Zhao, Y.; Lu, K.H.; Bowtell, D.; et al. Elevation of TP53 Autoantibody Before CA125 in Preclinical Invasive Epithelial Ovarian Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 5912–5922. [Google Scholar] [CrossRef]
- Tsai-Turton, M.; Santillan, A.; Lu, D.; Bristow, R.E.; Chan, K.C.; Shih, I.-M.; Roden, R.B.S. P53 Autoantibodies, Cytokine Levels and Ovarian Carcinogenesis. Gynecol. Oncol. 2009, 114, 12–17. [Google Scholar] [CrossRef][Green Version]
- Green, J.A.; Robertson, L.J.; Campbell, I.R.; Jenkins, J. Expression of the P53 Gene and Presence of Serum Autoantibodies in Ovarian Cancer: Correlation with Differentiation. Cancer Detect. Prev. 1995, 19, 151–155. [Google Scholar]
- Angelopoulou, K.; Rosen, B.; Stratis, M.; Yu, H.; Solomou, M.; Diamandis, E.P. Circulating Antibodies against P53 Protein in Patients with Ovarian Carcinoma. Correlation with Clinicopathologic Features and Survival. Cancer 1996, 78, 2146–2152. [Google Scholar] [CrossRef]
- Gadducci, A.; Ferdeghini, M.; Buttitta, F.; Cosio, S.; Fanucchi, A.; Annicchiarico, C.; Gagetti, O.; Bevilacqua, G.; Genazzani, A.R. Assessment of the Prognostic Relevance of Serum Anti-P53 Antibodies in Epithelial Ovarian Cancer. Gynecol. Oncol. 1999, 72, 76–81. [Google Scholar] [CrossRef]
- Goodell, V.; Salazar, L.G.; Urban, N.; Drescher, C.W.; Gray, H.; Swensen, R.E.; McIntosh, M.W.; Disis, M.L. Antibody Immunity to the P53 Oncogenic Protein Is a Prognostic Indicator in Ovarian Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.S.; Wong, J.; Vitonis, A.; Crum, C.P.; Sluss, P.M.; Labaer, J.; Cramer, D. P53 Autoantibodies as Potential Detection and Prognostic Biomarkers in Serous Ovarian Cancer. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2010, 19, 859–868. [Google Scholar] [CrossRef]
- Abendstein, B.; Marth, C.; Müller-Holzner, E.; Widschwendter, M.; Daxenbichler, G.; Zeimet, A.G. Clinical Significance of Serum and Ascitic P53 Autoantibodies in Epithelial Ovarian Carcinoma. Cancer 2000, 88, 1432–1437. [Google Scholar] [CrossRef]
- Vogl, F.D.; Frey, M.; Kreienberg, R.; Runnebaum, I.B. Autoimmunity against P53 Predicts Invasive Cancer with Poor Survival in Patients with an Ovarian Mass. Br. J. Cancer 2000, 83, 1338–1343. [Google Scholar] [CrossRef]
- Katchman, B.A.; Barderas, R.; Alam, R.; Chowell, D.; Field, M.S.; Esserman, L.J.; Wallstrom, G.; LaBaer, J.; Cramer, D.W.; Hollingsworth, M.A.; et al. Proteomic Mapping of P53 Immunogenicity in Pancreatic, Ovarian, and Breast Cancers. Proteomics Clin. Appl. 2016, 10, 720–731. [Google Scholar] [CrossRef]
- Hurley, L.C.; Levin, N.K.; Chatterjee, M.; Coles, J.; Muszkat, S.; Howarth, Z.; Dyson, G.; Tainsky, M.A. Evaluation of Paraneoplastic Antigens Reveals TRIM21 Autoantibodies as Biomarker for Early Detection of Ovarian Cancer in Combination with Autoantibodies to NY-ESO-1 and TP53. Cancer Biomark Sect. Dis. Markers 2020, 27. [Google Scholar] [CrossRef]
- Kobayashi, M.; Katayama, H.; Irajizad, E.; Vykoukal, J.V.; Fahrmann, J.F.; Kundnani, D.L.; Yu, C.-Y.; Cai, Y.; Hsiao, F.C.; Yang, W.-L.; et al. Proteome Profiling Uncovers an Autoimmune Response Signature That Reflects Ovarian Cancer Pathogenesis. Cancers 2020, 12, 485. [Google Scholar] [CrossRef]
- Kaaks, R.; Fortner, R.T.; Hüsing, A.; Barrdahl, M.; Hopper, M.; Johnson, T.; Tjønneland, A.; Hansen, L.; Overvad, K.; Fournier, A.; et al. Tumor-Associated Autoantibodies as Early Detection Markers for Ovarian Cancer? A Prospective Evaluation. Int. J. Cancer 2018, 143, 515–526. [Google Scholar] [CrossRef]
- Lane, D.P.; Hupp, T.R. Drug Discovery and P53. Drug Discov. Today 2003, 8, 347–355. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Walerych, D.; Lisek, K.; Del Sal, G. Mutant P53: One, No One, and One Hundred Thousand. Front. Oncol. 2015, 5, 289. [Google Scholar] [CrossRef]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 Reactivates Mutant P53 by Targeting Cysteines 124 and 277. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Basu, B.; Gourley, C.; Gabra, H.; Vergote, I.B.; Brenton, J.D.; Abrahmsen, L.; Smith, A.; Euler, M.V.; Green, J.A. PISARRO: A EUTROC Phase 1b Study of APR-246 with Carboplatin (C) and Pegylated Liposomal Doxorubicin (PLD) in Relapsed Platinum-Sensitive High Grade Serous Ovarian Cancer (HGSOC). Ann. Oncol. 2016, 27, vi123. [Google Scholar] [CrossRef]
- Aprea Therapeutics PiSARRO-R: P53 Suppressor Activation in Platinum-Resistant High Grade Serous Ovarian Cancer, a Phase II Study of Systemic Pegylated Liposomal Doxorubicin Chemotherapy with APR-246. 2019. Available online: clinicaltrials.gov.
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.-C.; et al. Gain of Function of Mutant P53 by Coaggregation with Multiple Tumor Suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.; Keshamouni, V.G.; Kanapathipillai, M. Polyarginine and Its Analogues Inhibit P53 Mutant Aggregation and Cancer Cell Proliferation in Vitro. Biochem. Biophys. Res. Commun. 2017, 489, 130–134. [Google Scholar] [CrossRef]
- Chen, Z.; Kanapathipillai, M. Inhibition of P53 Mutant Peptide Aggregation In Vitro by Cationic Osmolyte Acetylcholine Chloride. Protein Pept. Lett. 2017, 24, 353–357. [Google Scholar] [CrossRef]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of P53 Aggregation Rescues P53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef]
- Li, Q.; Lozano, G. Molecular Pathways: Targeting Mdm2 and Mdm4 in Cancer Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 34–41. [Google Scholar] [CrossRef]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of P53 Inhibitors: Progress, Challenges and Perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef]
- Zhang, W.-W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-P53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef]
- Cui, H.M.; Guan, C.L.; Liu, Q.; Li, L.Y. Outcome of Patients with Recurrent Epithelial Ovarian Carcinoma Following Treatment with Recombinant Human Adenovirus P53 Combined with Chemotherapy. Chin J. Cancer Biother 2014, 21, 450–454. [Google Scholar]
- Hardwick, N.R.; Frankel, P.; Ruel, C.; Kilpatrick, J.; Tsai, W.; Kos, F.; Kaltcheva, T.; Leong, L.; Morgan, R.; Chung, V.; et al. P53-Reactive T Cells Are Associated with Clinical Benefit in Patients with Platinum-Resistant Epithelial Ovarian Cancer After Treatment with a P53 Vaccine and Gemcitabine Chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1315–1325. [Google Scholar] [CrossRef]
- City of Hope Medical Center P53MVA and Pembrolizumab in Treating Patients with Recurrent Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. 2021. Available online: clinicaltrials.gov.
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.-C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.; Malekzadeh, P.; et al. Immunologic Recognition of a Shared P53 Mutated Neoantigen in a Patient with Metastatic Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen Screening Identifies Broad TP53 Mutant Immunogenicity in Patients with Epithelial Cancers. J. Clin. Invest. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the Development of Personalized Neoantigen-Based Therapeutic Cancer Vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a Neoantigen Derived from a Common TP53 Mutation. Science 2021, 371. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Function | Effect on p53 in HGSC | Reference |
|---|---|---|---|
| PAX8 | Transcription factor | Transcriptionally upregulates GOF p53 | [92] |
| TNFRSF12A | Member of the tumor necrosis factor receptor super-family | Enhances MDM2 mediated ubiquitination and degradation of R248Q mutant p53 | [93] |
| TRIM-71 | E3 ubiquitin ligase | Induces degradation of p53 mutants | [94] |
| USP15 | Deubiquitinase | Elevates levels of the aggregated GOF p53 R175H | [95] |
| MEG3 | lncRNA | Upregulates mutant p53 RNA and protein expression levels | [96] |
| miR let-7d-5p | microRNA | Affects mutant p53 expression in the mRNA and protein level through targeting HMGA | [97] |
| p53 Downstream Effectors | Regulation by p53 | Biological Effect | Reference |
|---|---|---|---|
| CDKN1A (p21) | Direct transcriptional regulation by mutant p53 | Oncogenic (resulting from an oncogenic role of p21) | [92] |
| PLAC1 | Direct transcriptional de-repression by mutant or null p53 of the inhibitory effect of WT p53 | Oncogenic function | [107] |
| Notch3 | Transcriptional upregulation by mutp53 | Activates CCNG1 and leads to promoted HGSC metastasis and cisplatin resistance | [108] |
| Target genes of β-arrestin1/YAP/mutp53 complex | Gene-specific transcriptional regulation | Enhanced proliferation, survival, and invasion of HGSC in vitro and metastasis in vivo | [109] |
| ACP6 | Downregulation by p53 mutants R175H, R249S and R273H | Increased LPA levels and increased migration and proliferation | [110] |
| Pathway | Cell Expressing p53 | Biological Effect | Reference |
|---|---|---|---|
| p53 | Cancer associated fibroblasts | Changes in the transcriptional signature from a tumor suppressive to a tumor supportive signature | [155] |
| IL-1β | Cancer associated fibroblasts | Secretion from epithelial cancer cells leads to down regulation of WT p53 in CAFs | [155] |
| ICAM1 | Cancer associated fibroblasts | Knockdown of WT p53 from CAF induces an ICAM1 mediated invasiveness of immortalized epithelial ovarian cells | [156] |
| MET | Tumor cells | Mutant p53 upregulates Met, that is a receptor to stromal cell derived HGF | [157] |
| NFκB, TNFα | Tumor cells | loss of p53 in EOC leads to a pro-inflammatory response | [172,173] |
| Immune activation | Tumor cells | Mutant p53 in HGSC leads to generation of anti p53 auto-antibodies and p53 specific T-cell immunity | [174,175,176,177,178,179,180,181,182,183,184] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, A.; Perets, R. Mutated p53 in HGSC—From a Common Mutation to a Target for Therapy. Cancers 2021, 13, 3465. https://doi.org/10.3390/cancers13143465
Saleh A, Perets R. Mutated p53 in HGSC—From a Common Mutation to a Target for Therapy. Cancers. 2021; 13(14):3465. https://doi.org/10.3390/cancers13143465
Chicago/Turabian StyleSaleh, Aya, and Ruth Perets. 2021. "Mutated p53 in HGSC—From a Common Mutation to a Target for Therapy" Cancers 13, no. 14: 3465. https://doi.org/10.3390/cancers13143465
APA StyleSaleh, A., & Perets, R. (2021). Mutated p53 in HGSC—From a Common Mutation to a Target for Therapy. Cancers, 13(14), 3465. https://doi.org/10.3390/cancers13143465